Introduction
Oral carcinoma is an aggressive malignant disease,
with ~640,000 new cases being detected annually worldwide; in
addition, it is responsible for ~145,000 cases of mortality every
year (1). Five-year survival rates of
patients with early oral cancer are between 55 and 60%, and
decrease to 30–40% in cases of advanced oral cancer. The
development of oral cancer is mainly associated with the
consumption of areca nut, tobacco and alcohol (2–4). There are
various types of oral cancer; however, >90% are squamous cell
carcinomas. Because it is very invasive, the prognosis of oral
squamous cell carcinoma (OSCC) is poor (5). Currently, surgical removal, chemotherapy
and radiotherapy are the most common and effective treatments for
patients with oral carcinoma. However, because of distant
metastasis, chemotherapeutic resistance and poor tolerance, these
treatments can fail. Therefore, the development of novel
chemotherapeutics with low toxicity and high efficiency for oral
carcinoma is crucial.
Interest has grown in natural compounds for the
development of anticancer drugs, due to their low toxicity and
higher tolerance in patients (6).
Pristimerin, a quininemethide triterpenoid compound, is isolated
from several plant species belonging to the Celastraceae and
Hippocrateaceae families. It is commonly used as an
antioxidant, anti-malarial, insecticidal, anti-inflammatory and
anti-fungal agent (7–9). Pristimerin has also been reported to
induce apoptosis of various human cancer cells, including in
multiple myeloma (10), breast
(11), liver (12), pancreatic (13) and prostate cancer (14). In addition to apoptosis induction
(11), the mechanisms involved in the
anticancer effects of pristimerin include stimulation of reactive
oxygen species generation (15),
blocking of nuclear factor-κB (16)
and proteasome inhibition (10).
To the best of our knowledge, the anticancer effects
of pristimerin on OSCC have rarely been reported. In the present
study, the potent antitumor effects of pristimerin on OSCC cells
were investigated. Pristimerin exhibited potent anti-proliferative
and apoptosis-inducing effects on the OSCC cell lines CAL-27 and
SCC-25. The underlying mechanisms of these effects were primarily
mediated by G1 phase cell cycle arrest and inhibition of
the mitogen-activated protein kinase (MAPK)/extracellular
signal-regulate kinase 1/2 (Erk 1/2) and protein kinase B (Akt)
signaling pathways.
Materials and methods
Reagents and cell culture
Pristimerin, 5-fluorouracil, cisplatin and propidium
iodide (PI) were purchased from Sigma-Aldrich (Merck KGaA,
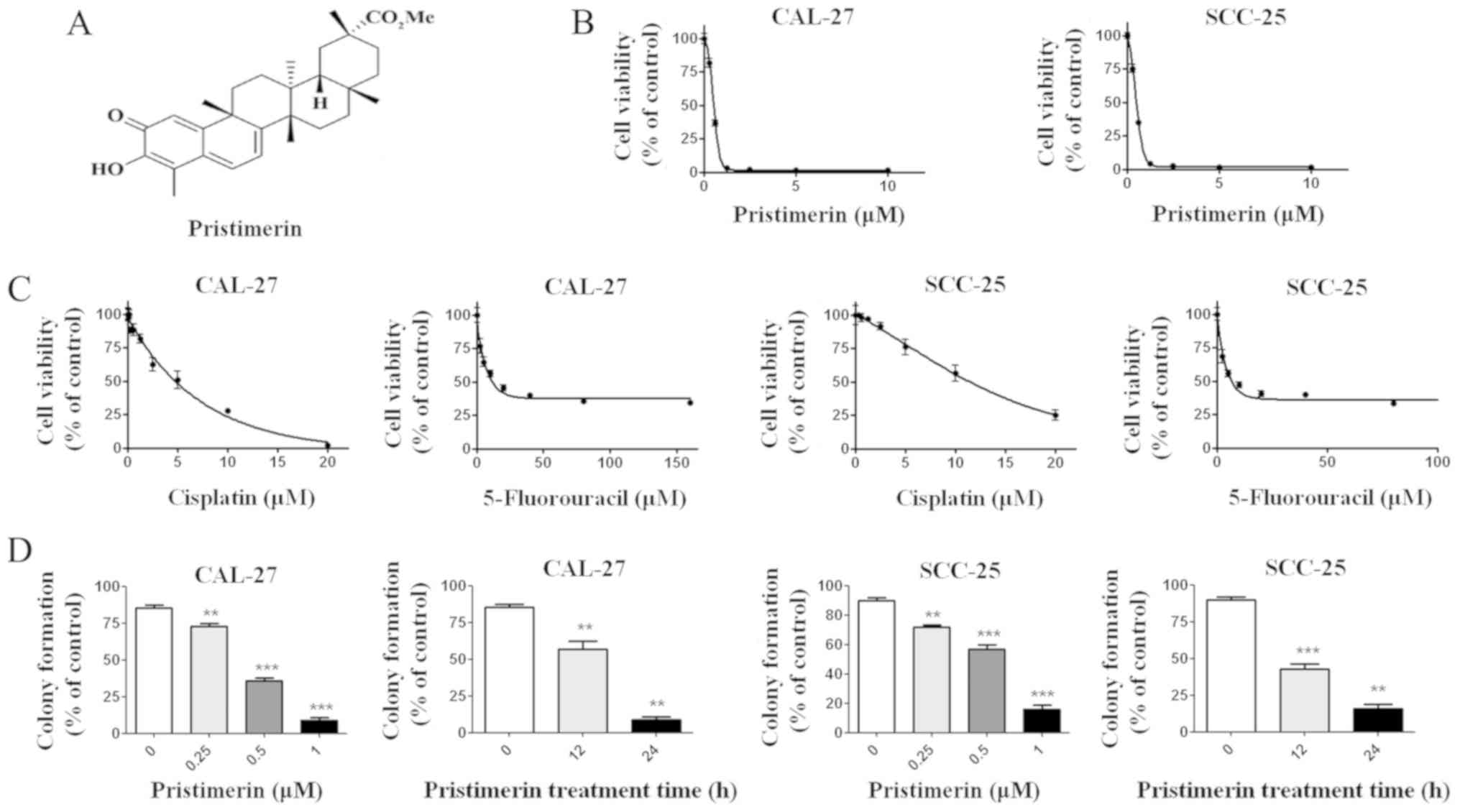
Darmstadt, Germany). Pristimerin (molecular structure shown in
Fig. 1A) was prepared as a 20 mM
stock solution in dimethyl sulfoxide. The CAL-27 and SCC-25 cells,
which were initially isolated from the epidermal tongue tissue of
patients with OSCC, were kindly donated by Professor Hongzhang
Huang (Department Oral & Maxillofacial Surgery, Sun Yat Sen
University, Guangzhou, China). The cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) F-12 (Biological
Industries, Shanghai, China) supplemented with 10% fetal bovine
serum (FBS; Biological Industries), at 37°C in a humidified
atmosphere containing 5% CO2.
Cell viability assay
Cell viability was measured by MTS assay (CellTiter
96 AQueous MTS Reagent; Promega Corporation, Madison, WI, USA),
according to the manufacturer's protocol. Briefly, CAL-27 and
SCC-25 cells (5×103 cells/well) were seeded into a
96-well plate for 12 h to allow cell attachment, and were then
treated with increasing concentrations of pristimerin (0, 0.15625,
0.3125, 0.625, 1.25, 2.5, 5, 10 µM), cisplatin (0, 0.3125, 0.625,
1.25, 2.5, 5, 10, 20 µM) or 5-fluorouracil (0, 1.25, 2.5, 5, 10,
20, 40, 80, 160 µM) at 37°C for 68 h. Thereafter, 20 µl MTS/PMS
(20:1 in volume) was added and incubated for an additional 4 h.
Cell viability was finally determined using a microplate reader
(Detie, Nanjing, China) at 490 nm. The half maximal inhibitory
concentration (IC50) of pristimerin was calculated.
Clonogenicity assay
CAL-27 and SCC-25 cells (2×105/ml) were
treated with increasing concentrations of pristimerin (0, 0.25,
0.5, 1 µM) at 37°C for 12 or 24 h. They were then collected, washed
three times with PBS and seeded in a 12-well plate
(103/well) in DMEM F-12 medium containing 0.3% agar and
20% FBS. After a further 10–14 days of culture at 37°C, colonies
containing >50 cells were counted under an inverted
phase-contrast microscope.
Flow cytometric analysis of cell
apoptosis
CAL-27 and SCC-25 cells (2×105/ml) were
treated with various concentrations of pristimerin (0, 0.25, 0.5, 1
µM) at 37°C for 12 or 24 h. After collection, cells were washed
with PBS and stained with Annexin V-fluorescein isothiocyanate
(FITC)/PI (Annexin V-FITC Apoptosis Detection kit; Sigma-Aldrich;
Merck KGaA) according to the manufacturer's protocol. The number of
viable, necrotic and apoptotic cells were assessed by flow
cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Assessment of cell cycle
distribution
CAL-27 and SCC-25 cells (2×105/ml) were
treated with various concentrations of pristimerin (0, 0.25, 0.5, 1
µM) at 37°C for 12 or 24 h. After collection, they were washed
twice with cold PBS and fixed with cold 75% ethanol at 4°C
overnight. Ethanol was eventually discarded, cells were resuspended
in PBS containing PI (50 µg/ml) and were incubated in a water bath
(37°C) for 1 h. The cell cycle distribution was finally examined by
flow cytometry.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following treatment with pristimerin (0, 0.25, 0.5,
1 µM for 24 h and 1 µM for 12 h), total cellular RNA was extracted
from CAL-27 and SCC-25 cells with TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's protocol. RNA was reverse
transcribed into cDNA (MMLV reverse transcriptase; Promega
Corporation), according to the manufacturer's protocol, and the
mRNA expression levels were measured by GoTaq qPCR Master Mix
(Promega, Corporation) using the ABI7000 cycler (Applied
Biosystems; Thermo Fisher Scientific Inc.). The primers for RT-qPCR
were designed as follows: p21, forward 5′-TCTTGTACCCTTGTGCCTCG-3′,
reverse 5′-GAAGATCAGCCGGCGTTTG-3′; p27, forward
5′-GTCAAACGTAAACAGCTCGAAT-3′, reverse 5′- TGCATAATGCTACATCCAACG-3′;
p53, forward 5′- GAGGTTGGCTCTGACTGTACC-3′, reverse 5′-
TCCGTCCCAGTAGATTACCAC-3′; cyclin D1, forward
5′-GTGCTGCGAAGTGGAAACC-3′, reverse 5′-ATCCAGGTGGCGACGATCT-3′;
cyclin E, forward 5′-GTTATAAGGGAGACGGGGAGC-3′, reverse
5′-TGCTCTGCTTCTTACCGCTC-3′; and GAPDH, forward
5′-CGACCACTTTGTCAAGCTCA-3′; and reverse 5′-AGGGGTCTACATGGCAACTG-3′.
PCR was performed at 94°C for 5 min, followed by 40 cycles at 94°C
for 30 sec and 56°C for 30 sec. Relative quantification of gene
expression was performed using the threshold cycle difference
2−ΔΔCq method (17), and
the geometric mean of GAPDH levels was used as an internal control
to normalize the variability in expression level.
Western blot analysis
Cells were washed with cold PBS and lysed with lysis
buffer (1X PBS, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40)
supplemented with freshly added 1 mM phenylmethylsulfonyl fluoride,
1X Roche complete Mini protease inhibitor cocktail (Roche
Diagnostics, Shanghai, China), 10 mM glycerophosphate, 10 mM NaF,
and 1 mM sodium orthovanadate. DNA contained in the lysate was
sheared by sonication with 10 1-sec bursts with a 3 sec interval at
medium power. Following quantification using the bicinchoninic acid
method, proteins (60 µg) were separated by 10–15% SDS-PAGE.
Thereafter, proteins were transferred to polyvinylidene difluoride
membrane and blocked with 5% non-fat milk in PBS-Tween (PBST) at
37°C for 1 h. Antibodies against poly (ADP-ribose) polymerase
(PARP) (cat. no. 9532), caspase-3 (cat. no. 9665), p21 (cat. no.
2947), Erk1/2 (cat. no. 4695), phosphorylated (p)-Erk1/2 (cat. no.
4370), Akt (cat. no. 4691) and p-Akt (cat. no. 4060) were purchased
from CST Biological Reagents Co., Ltd. (Shanghai, China).
Horseradish peroxidase-conjugated goat antibodies against mouse and
rabbit (cat. no. 31430, cat. no. 31460) were purchased from Thermo
Fisher Scientific Inc. Antibodies was prepared in PBST with
dilutions of 1:500 (p-Erk1/2 and p-Akt), 1:8,000 (β-actin) and the
rest of the antibodies were 1:1,000. Primary antibody was added to
the membrane and incubated at 4°C overnight. Following washing in
triplicate with PBST, the secondary antibody was added (1:5,000)
and incubated at room temperature for 1 h, subsequently washed 2
times with PBST and 1 time with PBS. Enhanced chemiluminescence
reagent (Immobilon Western; cat. no. WBKLS0500, EMD Millipore,
Billerica, MA, USA) was added and the x-ray film was exposed.
Protein bands were quantified and normalized to β-actin by using
Image-Pro Plus 6.0 system (Media Cybernetics, Inc., Rockville, MD,
USA).
Statistical analysis
Each experiment was performed at least three times.
GraphPad 5.0 Software (GraphPad Software, Inc., San Diego, CA, USA)
was used for statistical analysis. Data are expressed as the means
± standard deviation, and differences between groups were assessed
by one-way analysis of variance with post-hoc intergroup
comparisons using Turkey test. P<0.05 was considered to indicate
a statistically significant difference.
Results
Pristimerin inhibits OSCC cell
proliferation
To investigate the possible effects of pristimerin
on OSCC cells, CAL-27 and SCC-25 cells were treated with increasing
concentrations of pristimerin for 68 h, prior to determining cell
viability with the MTS assay. Results demonstrated that pristimerin
effectively inhibited the growth of CAL-27 and SCC-25 cells in a
dose-dependent manner with IC50 values of 0.70 and 0.73
µM, respectively (Fig. 1B). The toxic
effects of pristimerin on CAL-27 and SCC-25 viability were compared
to the ones observed after 72 h treatment with the commonly used
antitumor drugs cisplatin and 5-fluorouracil. The results
demonstrated that the IC50 values of cisplatin and 5-
fluorouracil in CAL-27 cells were 7.69 and 11.98 µM, respectively.
The IC50 values of cisplatin and 5-fluorouracil in
SCC-25 cells were 15.96 and 11.45 µM, respectively (Fig. 1C). Compared with cisplatin and
5-fluorouracil, the IC50 values of pristimerin in oral
cancer cells were lowest (~0.7 µM). Because clonogenicity is
believed to better reflect the malignant behavior of tumor cells,
colony formation was determined after CAL-27 and SCC-25 cells were
treated either with increasing concentrations of pristimerin for 24
h, or with 1 µM pristimerin for various durations. The results
demonstrated that pristimerin potently inhibited the colony
formation o CAL-27 and SCC-25 cells in a dose- and time-dependent
manner (Fig. 1D).
Pristimerin induces
G0/G1 phase arrest and modulates the
expression levels of cell cycle-associated molecules
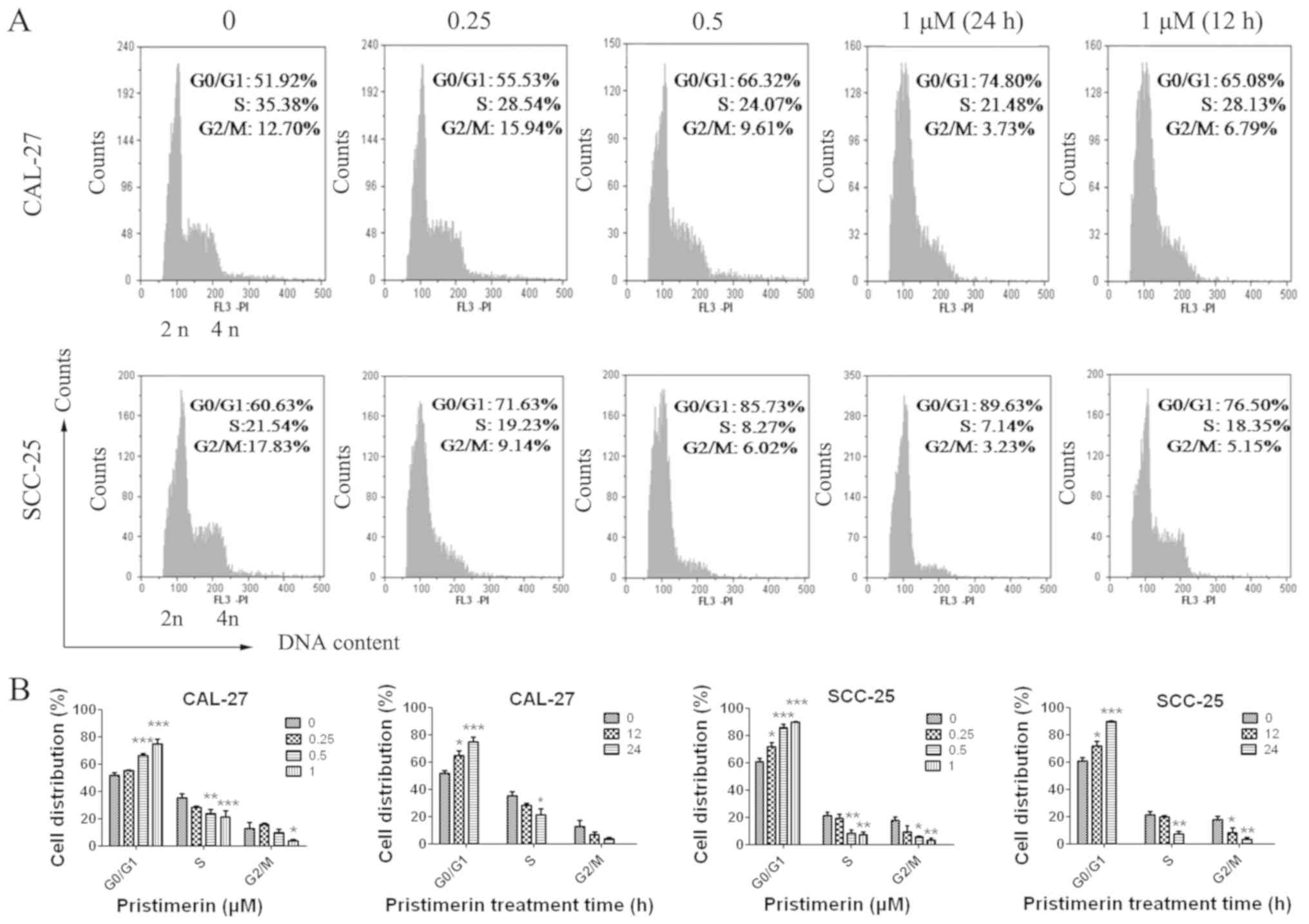
To explore the underlying mechanism of
pristimerin-induced inhibition of cell proliferation, the effects
of pristimerin on cell cycle distribution were examined by flow
cytometry (Fig. 2A). The results
revealed that the proportion of G0/G1 phase
CAL-27 cells treated with 0, 0.25, 0.5 and 1 µM pristimerin for 24
h were 51.92, 55.53, 66.32 and 74.80%, respectively. Similarly, in
SCC-25 cells, G0/G1 phase distributions were
60.63, 71.63, 85.73 and 89.63% following treatment with 0, 0.25,
0.5 and 1 µM pristimerin, respectively, for 24 h. In addition, the
proportion of G0/G1 phase CAL-27 cells was
65.08% and of G0/G1 phase SCC-25 cells was
76.50% following treatment with 1 µM pristimerin for 12 h (Fig. 2A). In parallel, the S and
G2/M phase distributions were reduced. These data
revealed that pristimerin significantly increased the proportion of
G0/G1 phase CAL-27 and SCC-25 cells, thus
suggesting that G0/G1 phase arrest may be
induced following pristimerin treatment. Furthermore, the
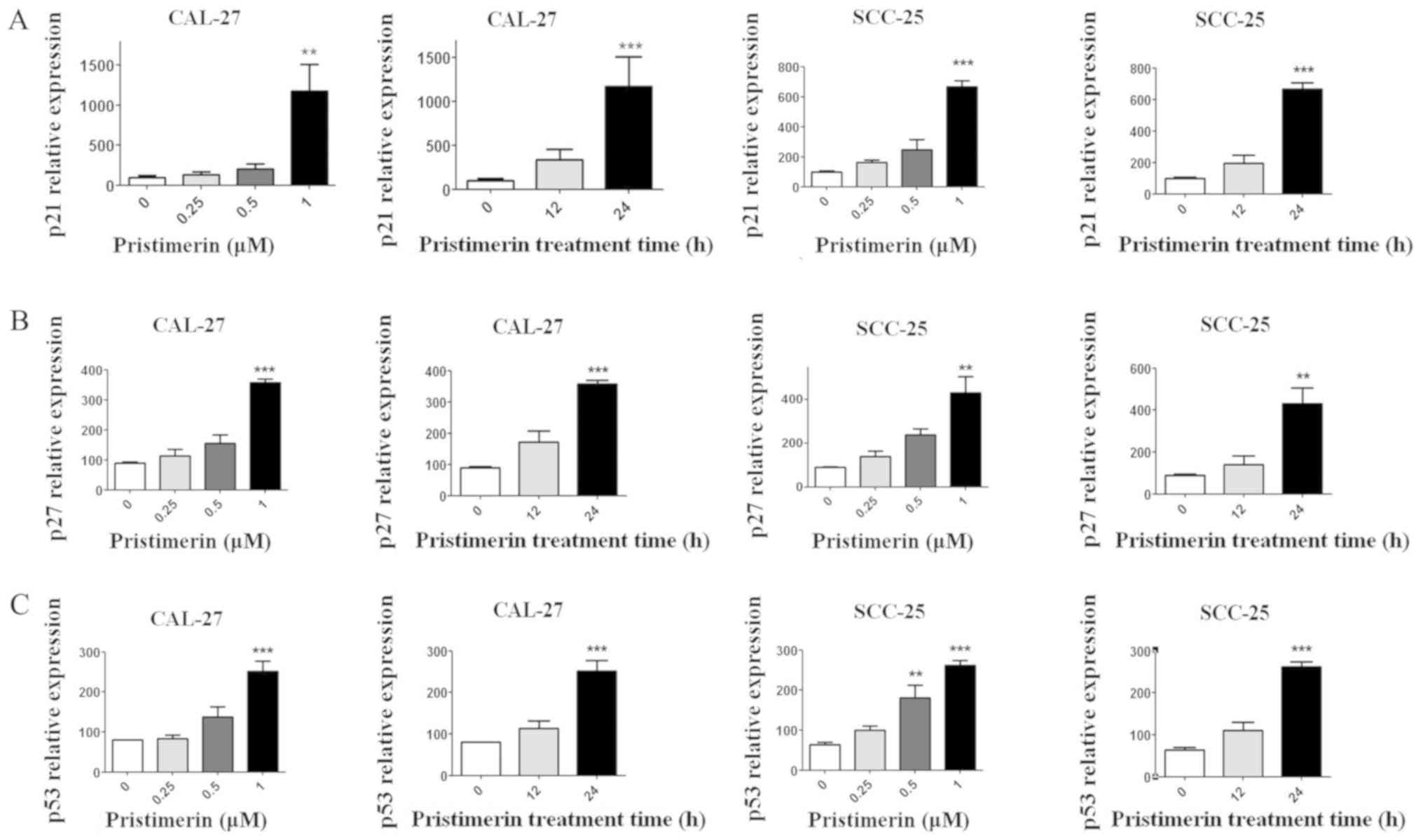
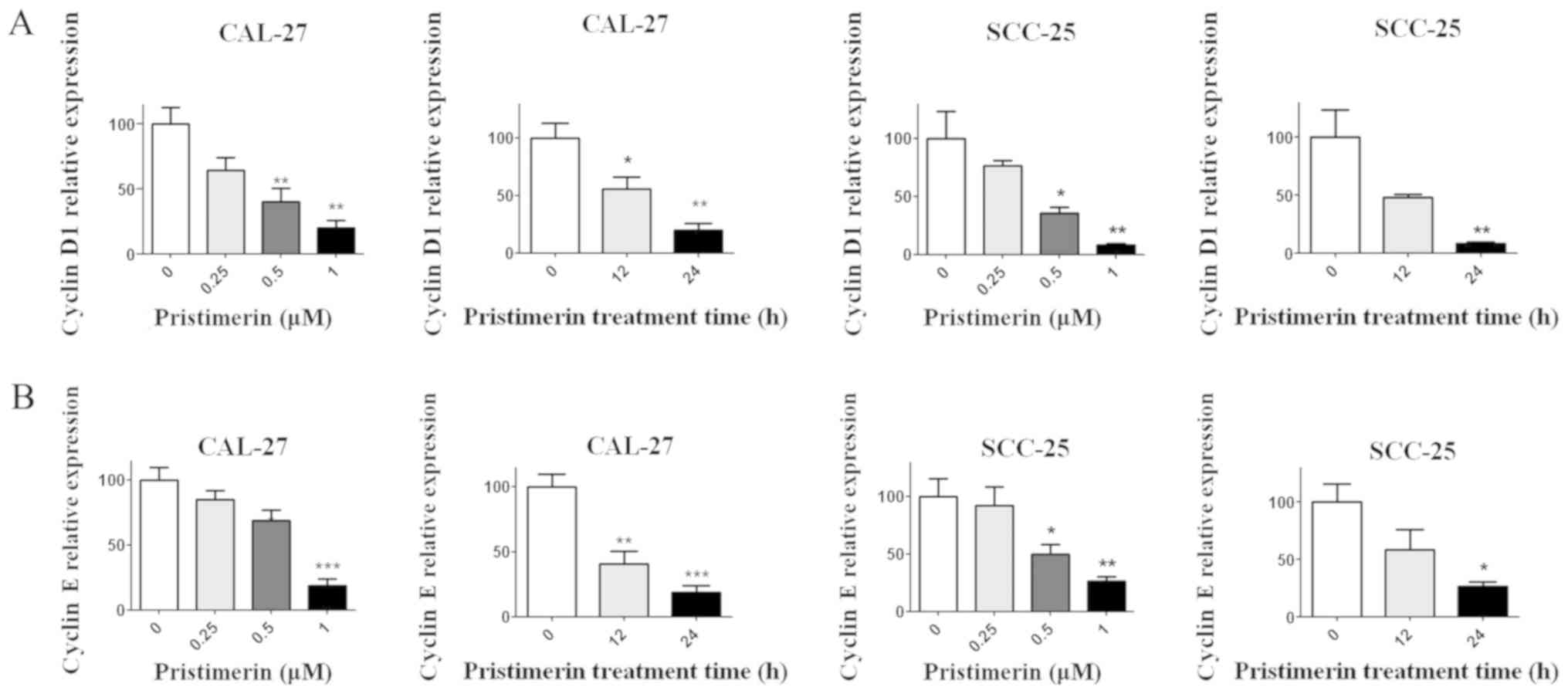
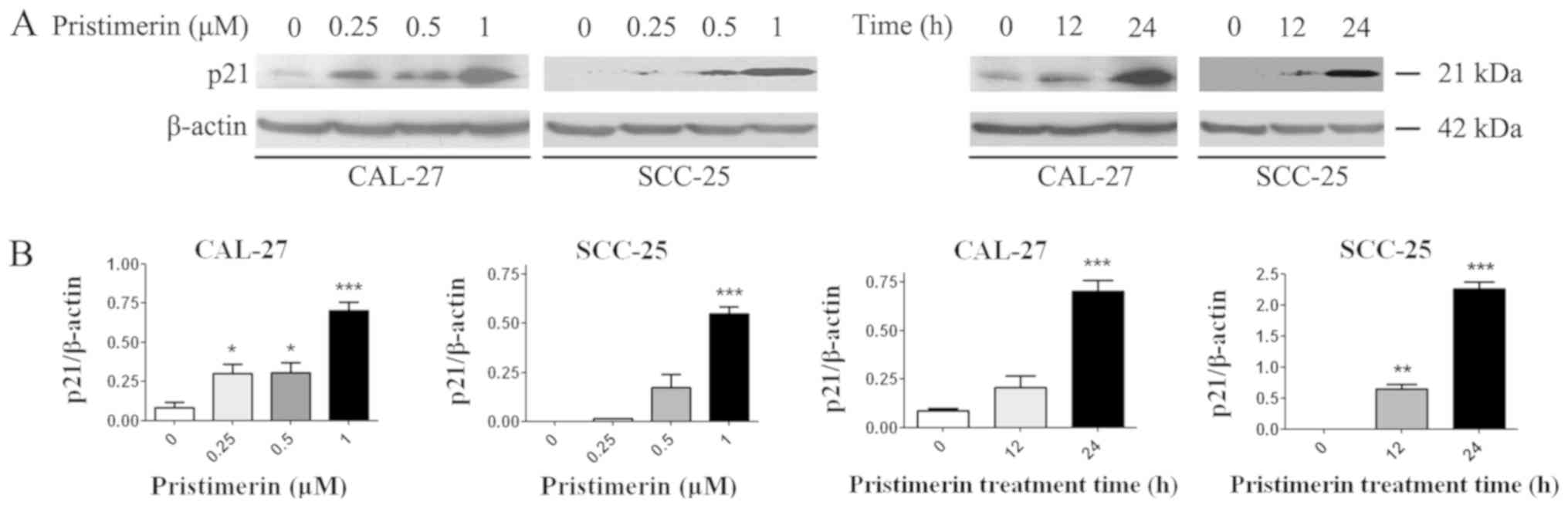
expression levels of molecules involved in the cell cycle process,
including cyclin D1, cyclin E, cyclin-dependent kinase (CDK)
inhibitors (CDKIs) p21 and p27, and p53, were assessed by RT-qPCR.
Results demonstrated that p21, p27 and p53 expression levels were
significantly increased following pristimerin treatment (Fig. 3A-C), whereas cyclin D1 and cyclin E
expression levels were significantly decreased under the same
conditions (Fig. 4A and B). These
modulating effects of pristimerin were observed in a dose- and
time-dependent manner in both cell lines. Moreover, western blot
analysis exhibited increased levels of p21 protein in a dose- and
time-dependent manner in both cell lines (Fig. 5A and B). These results were consistent
with the ones obtained from RT-qPCR analysis of p21 expression
(Fig. 3).
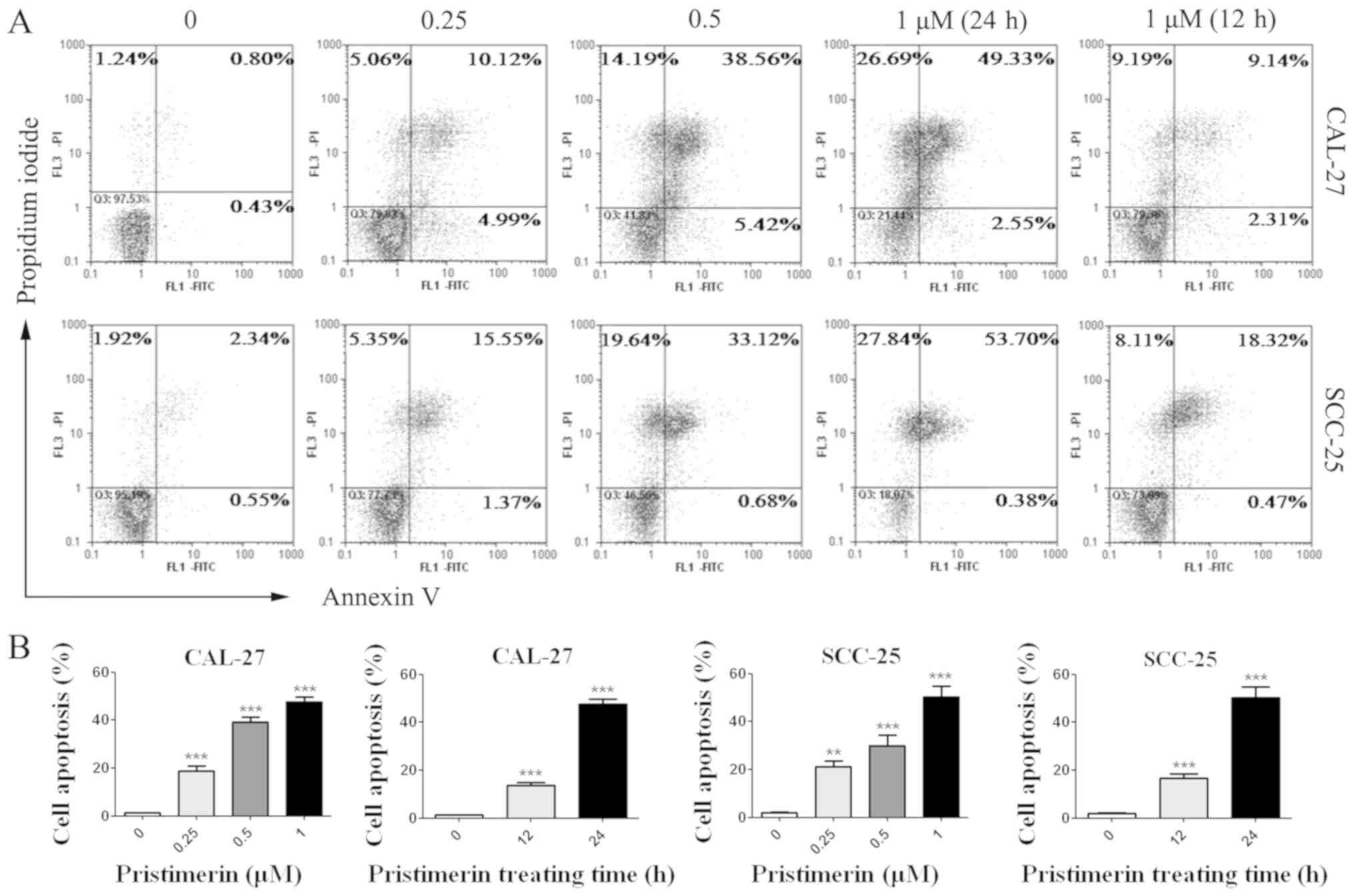
Pristimerin induces apoptosis of
CAL-27 and SCC-25 cells
The effects of pristimerin on cancer cell apoptosis
were assessed by flow cytometry. CAL-27 and SCC-25 cells were
treated with various concentrations of pristimerin for various
durations (Fig. 6), and were
double-stained with Annexin V-FITC/PI. Results demonstrated that
apoptotic cells were significantly increased by pristimerin in a
dose- and time-dependent manner (Fig. 6A
and B). The mean apoptotic cell rates from three independent
experiments were 1.29, 18.81, 39.1 and 47.7% in CAL-27 cells, and
were 2.1, 21.1, 30.05 and 50.23% in SCC-25 cells following
treatment with pristimerin at 0, 0.25, 0.5 and 1 µM, respectively
for 24 h. Following cell treatment with 1 µM pristimerin for 12 h,
the mean apoptotic cell rates were 13.61% in CAL-27 cell and 16.67%
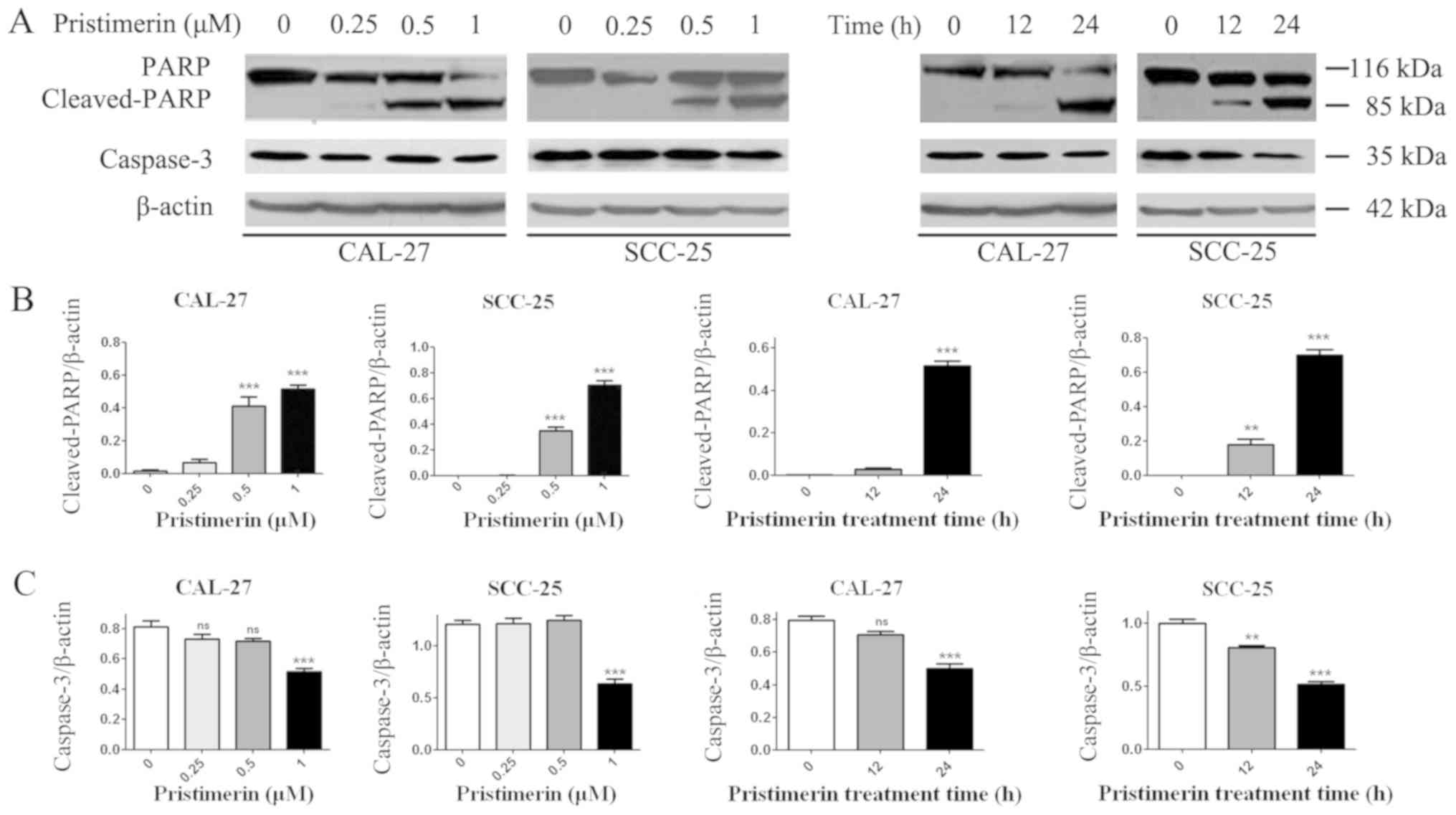
in SCC-25 cells. Additionally, PARP and caspase-3, the hallmark
proteins in apoptosis, were examined by western blot analysis. As
presented in Fig. 7, caspase-3
expression levels were significantly reduced in the two cell lines
in a dose- and time-dependent manner following pristimerin
treatment. In addition, cleaved-PARP (85 kDa) levels were
substantially increased in a dose- and time-dependent manner
following pristimerin treatment in CAL-27 and SCC-25 cells
(Fig. 7). The upregulated
cleaved-PARP level and downregulated caspase-3 level further
suggested that apoptosis was induced following pristimerin
treatment in OSCC cells (Fig. 7).
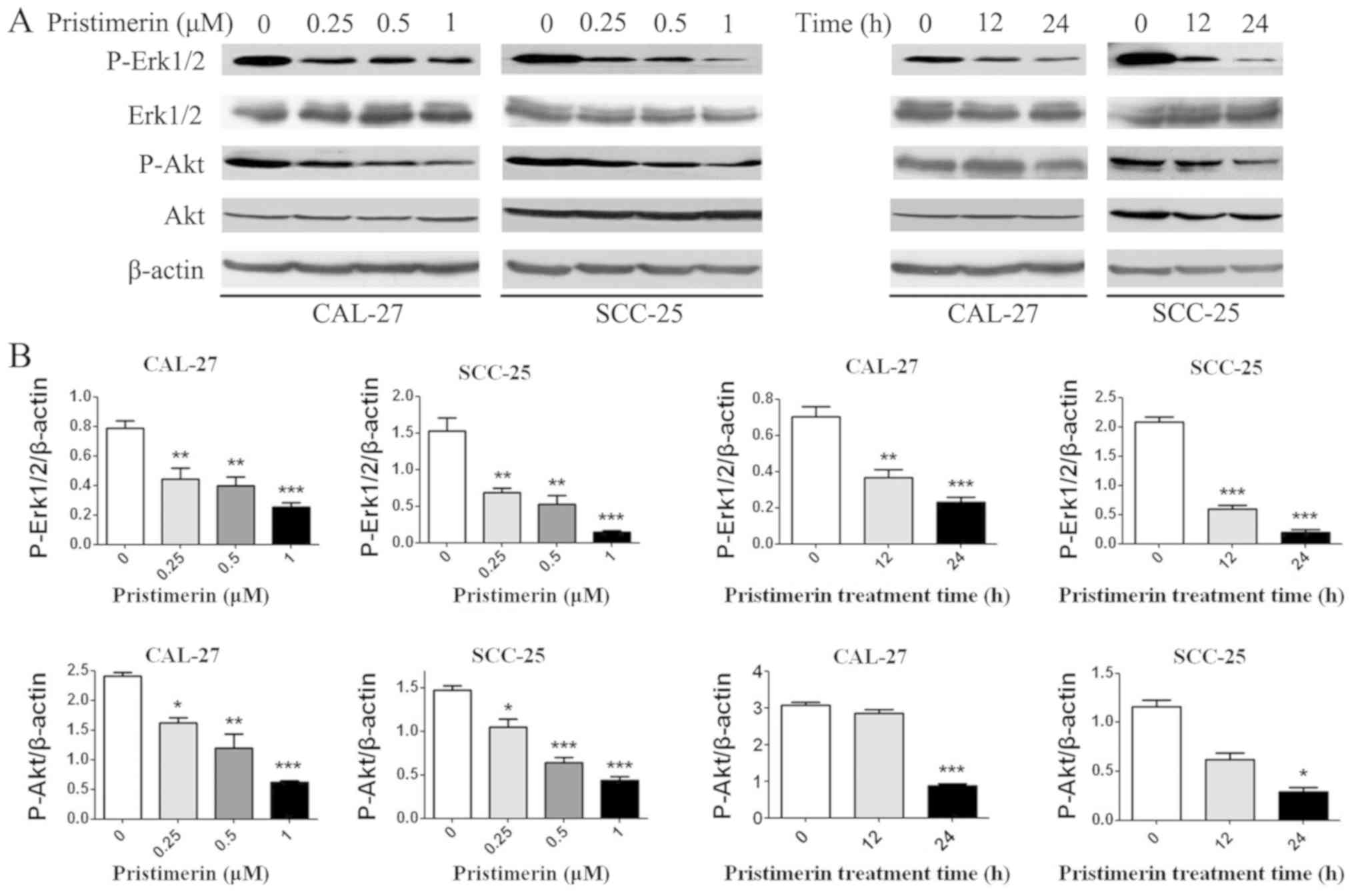
Pristimerin inhibits the MAPK/Erk1/2
and phosphoinositide 3-kinase (PI3K)/Akt signaling pathways
To determine whether the MAPK/Erk1/2 and PI3K/Akt
pathways were involved in the anticancer effects of pristimerin in
OSCC cells, p-Akt, Akt, p-Erk1/2 and Erk1/2 levels were detected by
western blotting (Fig. 8). Results
demonstrated that p-Akt and p-Erk1/2 expression levels were
decreased in a dose- and time-dependent manner; however, there were
no significant differences in the expression levels of total Akt
and Erk1/2.
Discussion
There has been growing interest in the last 30 years
in natural products derived from plants for the development of
novel anticancer therapies (6).
Pristimerin is a quinine methyltriterpenoid extracted from Chinese
plants; it has been reported to possess potential anticancer
effects in various types of cancer (9). To the best of our knowledge, the present
study is the first to determine the cytotoxic potency of
pristimerin against two OSCC cell lines, CAL-27 and SCC-25, in a
time- and dose-dependent manner. Furthermore, the effects of
pristimerin on cell proliferation were greater than those observed
by the conventional drugs cisplatin and 5-fluorouracil. In the
present study, the antitumor activities of pristimerin were only
detected in CAL-27 and SCC-25 cells; our future studies aim to test
the antitumor activities of pristimerin in other OSCC cells.
The disruption of cell cycle regulation is a main
cause for the proliferation of tumor cells. Therefore, modulation
of cell cycle progression in cancer cells is considered a target
for the treatment of human malignancies (18–21).
Cyclins, CDKs and CDKIs are three important modulators of cell
cycle progression. Abnormal expression of these molecules leads to
aberrant cell proliferation that can stimulate tumor development.
Cyclins bind to CDKs and promote cell cycle progression, whereas
CDKIs inhibit CDK activity by binding to cyclin-CDKs, cyclins or
CDKs, ultimately blocking cell proliferation (22–25). In
this study, the upregulation of p21 and p27, and downregulation of
cyclin D1 and cyclin E expression levels were observed and
responsible for the pristimerin-induced G0/G1
phase arrest, eventually leading to cell proliferation suppression
in CAL-27 and SCC-25 cells. Previous studies reported likewise,
that pristimerin can induce G1 phase arrest, which is
mediated by the upregulation of p21 and downregulation of cyclin D1
(10,26,27).
Additionally, the tumor suppressor protein p53, which is known as a
cell cycle regulator and a guardian of genetic integrity, was
increased following pristimerin treatment in CAL-27 and SCC-25
cells.
Most anticancer drugs mediate their effects via cell
apoptosis induction, which is the major mechanism involved in the
treatment of cancer, including oral cancer (28). Apoptosis can be initiated either by
the mitochondria (the intrinsic pathway) or through cell death
receptors (the extrinsic pathway), leading to the activation of
caspase cascades and resulting in apoptosis. Previous studies have
reported that pristimerin may induce cell apoptosis via both the
mitochondria and the death receptor-mediated extrinsic pathways in
U87 glioma cells (29) and cervical
cancer cells (30). Pristimerin
likewise modulates the levels of B-cell lymphoma 2 (Bcl-2) family
proteins, which are known to be involved in mitochondria-mediated
apoptosis, in pancreatic cancer cells (31). In the present study, pristimerin
induced significant apoptosis of CAL-27 and SCC-25 cells, which was
mediated by PARP specific cleavage and caspase-3 downregulation.
Lee et al (26) and Yousef
et al (27) also reported that
pristimerin induces PARP cleavage and apoptosis in breast and
colorectal cancer cells, respectively. Specific cleavage of PARP
indicates cell apoptosis. Caspase-3 acts as a junction between the
exogenous apoptotic pathway and the endogenous apoptotic pathway,
and its activation by cleavage itself ultimately induces cell
apoptosis (32,33). The effects of pristimerin against
apoptosis-associated Bcl-2 family proteins, and whether the
intrinsic and/or extrinsic pathway(s) are involved in the apoptotic
process in CAL-27 and SCC-25 cells requires further
investigation.
The MAPK/Erk1/2 and PI3K/Akt pathways are important
signaling pathways associated with the regulation of cell
proliferation, differentiation, apoptosis and tumor pathogenesis
(34–36). The downregulation of MAPK/Erk1/2 and
PI3K/Akt signaling demonstrated in this study may account for the
proliferation-suppressing and apoptosis-inducing effects of
pristimerin in CAL-27 and SCC-25 cells. Furthermore, previous
studies have reported that pristimerin inhibits MAPK/Erk1/2 and
PI3K/Akt signaling in breast (26,37),
colorectal (27) and pancreatic
cancer cells (13). However, the
complete regulatory mechanisms of pristimerin on MAPK/Erk1/2 and
PI3K/Akt signaling pathways in OSCC cells requires further
elucidation.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Fund of China (grant no. 81460032) and the Natural Science
Fund of Kunming University of Science and Technology (grant no.
KKSY201460068).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Author's contributions
HW and LL conceived and designed the study. HW, LL,
ZA and JY performed the experiments. HW and LL wrote the
manuscript. LC reviewed and edited the manuscript and was also
involved in the conception of the study. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guha N, Warnakulasuriya S, Vlaanderen J
and Straif K: Betel quid chewing and the risk of oral and
oropharyngeal cancers: A meta-analysis with implications for cancer
control. Int J Cancer. 135:1433–1443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pednekar MS, Gupta PC, Yeole BB and Hebert
JR: Association of tobacco habits, including bidi smoking, with
overall and site-specific cancer incidence: Results from the Mumbai
cohort study. Cancer Causes Control. 22:859–868. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Radoï L and Luce D: A review of risk
factors for oral cavity cancer: The importance of a standardized
case definition. Community Dent Oral Epidemiol. 41:97–109,
e178-e191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wikner J, Gröbe A, Pantel K and Riethdorf
S: Squamous cell carcinoma of the oral cavity and circulating
tumour cells. World J Clin Oncol. 5:114–124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Newman DJ, Cragg GM and Snader KM: Natural
products as sources of new drugs over the period 1981–2002. J Nat
Prod. 66:1022–1037. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brinker AM, Ma J, Lipsky PE and Raskin I:
Medicinal chemistry and pharmacology of genus Tripterygium
(Celastraceae). Phytochemistry. 68:732–766. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao JM, Wu WJ, Zhang JW and Konishi Y: The
dihydro-beta-agarofuran sesquiterpenoids. Nat Prod Rep.
24:1153–1189. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salminen A, Lehtonen M, Suuronen T,
Kaarniranta K and Huuskonen J: Terpenoids: Natural inhibitors of
NF-kappaB signaling with anti-inflammatory and anticancer
potential. Cell Mol Life Sci. 65:2979–2999. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tiedemann RE, Schmidt J, Keats JJ, Shi CX,
Zhu YX, Palmer SE, Mao X, Schimmer AD and Stewart AK:
Identification of a potent natural triterpenoid inhibitor of
proteosome chymotrypsin-like activity and NF-kappaB with
antimyeloma activity in vitro and in vivo. Blood. 113:4027–4037.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR
and Wu YC: Pristimerin induces caspase-dependent apoptosis in
MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer
Ther. 4:1277–1285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo Y, Zhang W, Yan YY, Ma CG, Wang X,
Wang C and Zhao JL: Triterpenoid pristimerin induced HepG2 cells
apoptosis through ROS-mediated mitochondrial dysfunction. J BUON.
18:477–485. 2013.PubMed/NCBI
|
|
13
|
Deeb D, Gao X, Liu YB, Pindolia K and
Gautam SC: Pristimerin, a quinonemethide triterpenoid, induces
apoptosis in pancreatic cancer cells through the inhibition of
pro-survival Akt/NF-kappaB/mTOR signaling proteins and
anti-apoptotic Bcl-2. Int J Oncol. 44:1707–1715. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu YB, Gao X, Deeb D, Arbab AS and Gautam
SC: Pristimerin induces apoptosis in prostate cancer cells by
down-regulating Bcl-2 through ROS-dependent Ubiquitin-proteasomal
degradation pathway. J Carcinog Mutagen Suppl. 6:0052013.
|
|
15
|
Byun JY, Kim MJ, Eum DY, Yoon CH, Seo WD,
Park KH, Hyun JW, Lee YS, Lee JS, Yoon MY and Lee SJ: Reactive
oxygen species-dependent activation of Bax and poly(ADP-ribose)
polymerase-1 is required for mitochondrial cell death induced by
triterpenoid pristimerin in human cervical cancer cells. Mol
Pharmacol. 76:734–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu Z, Jin Y, Chen C, Li J, Cao Q and Pan
J: Pristimerin induces apoptosis in imatinib-resistant chronic
myelogenous leukemia cells harboring T315I mutation by blocking
NF-kappaB signaling and depleting Bcr-Abl. Mol Cancer. 9:1122010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Call JA, Eckhardt SG and Camidge DR:
Targeted manipulation of apoptosis in cancer treatment. Lancet.
Oncol. 9:1002–1011. 2008.
|
|
19
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Molinari M: Cell cycle checkpoints and
their inactivation in human cancer. Cell Prolif. 33:261–274. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakanishi M, Shimada M and Niida H:
Genetic instability in cancer cells by impaired cell cycle
checkpoints. Cancer Sci. 97:984–989. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ball KL: p21: Structure and functions
associated with cyclin-CDK binding. Prog Cell Cycle Res. 3:125–134.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Castedo M, Perfettini JL, Roumier T and
Kroemer G: Cyclin-dependent kinase-1: Linking apoptosis to cell
cycle and mitotic catastrophe. Cell Death Differ. 9:1287–1293.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo H, Ray RM and Johnson LR: RhoA
stimulates IEC-6 cell proliferation by increasing
polyamine-dependent Cdk2 activity. Am J Physiol Gastrointest Liver
Physiol. 285:G704–G713. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sherr CJ and Roberts JM: Living with or
without cyclins and cyclin-dependent kinases. Genes Dev.
18:2699–2711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee JS, Yoon IS, Lee MS, Cha EY, Thuong
PT, Diep TT and Kim JR: Anticancer activity of pristimerin in
epidermal growth factor receptor 2-positive SKBR3 human breast
cancer cells. Biol Pharm Bull. 36:316–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yousef BA, Guerram M, Hassan HM, Hamdi AM,
Zhang LY and Jiang ZZ: Pristimerin demonstrates anticancer
potential in colorectal cancer cells by inducing G1 phase arrest
and apoptosis and suppressing various pro-survival signaling
proteins. Oncol Rep. 35:1091–1100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang WH, Hsuan KY, Chu LY, Lee CY, Tyan
YC, Chen ZS and Tsai WC: Anticancer effects of Salvia miltiorrhiza
alcohol extract on oral squamous carcinoma cells. Evid Based
Complement Alternat Med. 2017:53640102017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan YY, Bai JP, Xie Y, Yu JZ and Ma CG:
The triterpenoid pristimerin induces U87 glioma cell apoptosis
through reactive oxygen species-mediated mitochondrial dysfunction.
Oncol Lett. 5:242–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eum DY, Byun JY, Yoon CH, Seo WD, Park KH,
Lee JH, Chung HY, An S, Suh Y, Kim MJ and Lee SJ: Triterpenoid
pristimerin synergizes with taxol to induce cervical cancer cell
death through reactive oxygen species-mediated mitochondrial
dysfunction. Anticancer Drugs. 22:763–773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Zhou Y, Zhou H, Jia G, Liu J, Han
B, Cheng Z, Jiang H, Pan S and Sun B: Pristimerin causes G1 arrest,
induces apoptosis, and enhances the chemosensitivity to gemcitabine
in pancreatic cancer cells. PLoS One. 7:e438262012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ba XH, Cai LP and Han W: Effect of
cilostazol pretreatment on the PARP/AIF-mediated apoptotic pathway
in rat cerebral ischemia-reperfusion models. Exp Ther Med.
7:1209–1214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wan Y, Xin Y, Zhang C, Wu D, Ding D, Tang
L, Owusu L, Bai J and Li W: Fermentation supernatants of
Lactobacillus delbrueckii inhibit growth of human colon cancer
cells and induce apoptosis through a caspase 3-dependent pathway.
Oncol Lett. 7:1738–1742. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cantrell DA: Phosphoinositide 3-kinase
signalling pathways. J Cell Sci. 114:1439–1445. 2001.PubMed/NCBI
|
|
35
|
Liu Z, Zhu G, Getzenberg RH and Veltri RW:
The upregulation of PI3K/Akt and MAP kinase pathways is associated
with resistance of microtubule-targeting drugs in prostate cancer.
J Cell Biochem. 116:1341–1349. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xie G, Yu X, Liang H, Chen J, Tang X, Wu S
and Liao C: Pristimerin overcomes adriamycin resistance in breast
cancer cells through suppressing Akt signaling. Oncol Lett.
11:3111–3116. 2016. View Article : Google Scholar : PubMed/NCBI
|