Introduction
Esophageal cancer was the ninth most common cancer
type and the sixth most common cause of cancer-associated mortality
globally in 2012 (1). Esophageal
squamous cell carcinoma (ESCC) accounts for ~90% of all cases of
esophageal cancer worldwide (2). The
incidence rate of ESCC is particularly high in the so-called
‘esophageal cancer belt’, which stretches from northern China,
where the annual incidence rate is 1/100,000, through central Asia
to northern Iran (1,2). Despite significant advancements in
treatment options, including surgery and chemotherapy, the overall
survival rate has not significantly improved (3). Due to a lack of symptoms during early
stages of ESCC and a lack of non-invasive detection strategies, the
majority of cases are diagnosed during advanced stages of the
disease. Platinum-based therapeutic regimens are currently employed
for the clinical management of esophageal cancer and are associated
with high rates of clinical responses (4). However, a large number of human
malignancies are intrinsic or become insensitive to the cytostatic
effects of platinum, which causes a prominent challenge for the use
of platinum. Therefore, there is a requirement to develop new
therapeutic strategies that improve chemosensitivity.
B lymphoma Mo-MLV insertion region 1 homolog (BMI1)
is a member of the polycomb repressive complex 1 (PRC1). BMI1
functions as an epigenetic regulator and represses gene
transcription via its participation in histone modification and DNA
methylation (5). BMI1 expression is
increased in numerous types of human cancer; therefore, it can be
used as a predictive biomarker of progression and poor prognosis
(6). A number of studies have
demonstrated that aberrant overexpression of BMI1 is associated
with advanced stages, aggressive clinicopathological behaviors,
therapeutic resistance and poor prognosis in melanoma, glioma and
other types of tumor (6,7). BMI1 also serves a crucial role in
cisplatin chemoresistance and inhibition of BMI1 can reverse
cisplatin insensitivity (6,8). A recent study demonstrated that
targeting BMI1-positive cancer stem cells effectively inhibited
head and neck squamous cell carcinoma (HNSCC) growth and eliminated
chemoresistance (9). The small
molecule complex PTC-209 targeting BMI1 can inhibit tumor growth
and enhance chemosensitivity in colorectal cancer and HNSCC
(10,11). ESCC shares numerous biological
characteristics with HNSCC (12);
however, to the best of our knowledge, understanding of BMI1
regarding chemotherapy sensitivity of esophageal cancer remains
absent. Therefore, the current study investigated the association
between BMI1 and cisplatin chemosensitivity in ESCC.
Melanoma nuclear protein 18 (Mel18), a homologue of
BMI1, contains a zinc finger structure and is involved in histone
methylation, ubiquitination, SUMOylation and chromatin remodeling
(13,14). Mel18 is implicated in the regulation
of cell proliferation, differentiation, tumorigenesis, senescence,
apoptosis, cancer stem cell activity, angiogenesis and invasion in
a number of cancer types (15–17).
Although the Mel18 gene product is structurally highly similar to
the BMI1 protein, the role of Mel18 in cancer remains
controversial. Our previous study demonstrated that BMI1 expression
is significantly upregulated in ESCC tissues compared with adjacent
noncancerous tissues, and a strong negative association was
identified between Mel18 and BMI1 expression in ESCC (18). Previous studies have also revealed
that Mel18 acts as a tumor suppressor and is downregulated in
certain types of human cancer, including breast, gastric, prostate
and colorectal cancer (19–22). By contrast, Mel18 may act as an
oncogene as it is highly expressed in several types of tumor,
including Hodgkin's lymphoma, medulloblastoma, salivary gland
adenoid cystic carcinoma and salivary gland myoepithelial tumor
(23–26). Jung et al (27) demonstrated that silencing Mel18
inhibits endothelial cell migration and tube formation. In
addition, Park et al (28)
identified that Mel18 inhibition promotes tube formation in human
umbilical endothelial cells. A number of studies have suggested
that Mel18 downregulates BMI1 in several types of human tumor
(29–31). However, certain studies have
indicated that BMI1 and Mel18 exhibit synergistic roles in the
regulation of homeobox (HOX) genes, skeletal patterning, H3K27
trimethylation and colitis-associated cancer development (15,32,33).
These observations indicate that the biological functions of BMI1
and Mel18 may be different or redundant in different cancer
microenvironments. BMI1 is upregulated in ESCC tissues and cells,
and the expression of Mel18 is negatively associated with BMI1 in
gastric cancer and ESCC (18,31,34).
To the best of our knowledge, the interaction and involvement of
Mel18 and BMI1 in the chemoresistance of ESCC has not been
evaluated. We hypothesized that Mel18 and BMI1 cooperate to
regulate the intrinsic chemosensitivity of ESCC.
Our pilot study suggested that inhibition of BMI1
significantly effects cisplatin-induced proliferation and clonal
growth of ESCC cells. This effect may be strengthened by
co-inhibition of BMI1 and Mel18 in ESCC cells. The present study
investigated the combinational effects of Mel18 and BMI1 on
apoptosis and key molecules of apoptosis. To do so, it was
hypothesized that combined inhibition of BMI1 and Mel18 could
enhance the effects of BMI1-induced cell proliferation inhibition
by regulating apoptosis and associated proteins.
Materials and methods
Cell culture and treatment with
cisplatin
Human ESCC cell lines (EC109 and TE1) were obtained
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). ESCC cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin mix
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells were
cultured at 37°C in a humidified atmosphere containing 5%
CO2. Cisplatin (MedChemExpress, Monmouth Junction, NJ,
USA) was dissolved in dimethyl sulfoxide (DMSO; Beyotime Institute
of Biotechnology, Shanghai, China) at 50 mM and stored at −80°C
until use.
Using a 96-well plate, 1×104 stably
transfected cells in 100 µl of complete growth medium were seeded 1
day prior to treatment. Cells were treated with cisplatin (1, 2, 4,
8, 16 and 32 µM) dissolved in complete growth medium containing
<1% DMSO and control cells were treated with complete growth
medium containing the same concentration of DMSO.
Plasmids construct and
transfection
BMI1 short hairpin RNA (shRNA) was designed and
cloned into the pcDNA3.1-EGFP vector with the neomycin resistant
gene. The BMI1 shRNA target sequence was as follows:
5′-GGTCATCAGCAACTTCTTCT-3′. Mel18 shRNA was designed and cloned
into the psi-LVRU6GP vector with the puromycin resistant gene. The
Mel18 shRNA target sequence was as follows:
5′-GGCTCTGAGTGATGATGAGAT-3′. Human full-length Mel18 (reference
sequence no. NM_007144.2) was isolated from the human complementary
DNA library and connected to the pEZ-M13 vector with the neomycin
resistant gene. All plasmids, including Negative control (NC)
shRNA, were purchased from GenePharma (Shanghai, China).
Transfections of all vectors were performed using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The mass/concentration of
all plasmid transfected was 2 mg/ml. Stably transfected TE1 and
EC109 cells were selected and maintained in DMEM containing 700
µg/ml G418 (Beyotime Institute of Biotechnology) or 1 µg/ml
puromycin (Beyotime Institute of Biotechnology). After 2 weeks,
stable transfected cells were used for subsequent experiments.
Transfection efficiency was evaluated by western blot analysis.
Measurement of cytotoxicity
The cytotoxic effects of cisplatin in ESCC cells
were measured using Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan). Stably transfected cells were
plated in 96-well plates at a density of 1×104
cells/well. Following incubation at 37°C for 24 h, cells were
treated with various concentrations of cisplatin (1, 2, 4, 8, 16
and 32 µM) at 37°C for 24 h. Following 24 h, the complete growth
medium was replaced with serum-free medium, 10 µl CCK-8 was added
to each well and the cell mixtures were incubated at 37°C for 2 h.
Using a fine needle the bubbles were punctured and the absorbance
was then measured at 450 nm using a microplate reader. The half
maximal inhibitory concentration (IC50) was defined as
the concentration resulting in a 50% reduction in growth compared
with the growth of the control cells. Cell viability was calculated
according to the following equation: Cell viability =
[A450(drug)-A450(blank)]/[A450(control)-A450(blank)]. Six replicate
wells were set up in each group and three independent experiments
were performed. The IC50 dose-response curves were
plotted with GraphPad Prism Version 7.0 (GraphPad Software, Inc.,
La Jolla, CA, USA).
Colony formation assay
Following stable transfection of TE1 and EC109
cells, exponentially growing cells were harvested and placed into
60 mm plates (1×103 cells/well) and cultured with 5 µM
cisplatin at 37°C. The medium was changed every 3 days. After 2–3
weeks, cells were washed in PBS three times, fixed in 4% formalin
(Beyotime Institute of Biotechnology) at room temperature for 15
min and stained with 0.1% crystal violet (Beyotime Institute of
Biotechnology) at room temperature for 10 min. The number of
colonies was counted and analyzed using ImageJ software version 1.0
(National Institutes of Health, Bethesda, MD, USA).
Apoptosis detection
Apoptosis was determined with the Annexin
V-APC/7-AAD apoptosis kit (MultiSciences, Hangzhou, Zhejiang,
China). ESCC cells were seeded in 6-well plates and incubated with
5 µM cisplatin and serum-free medium at 37°C for 24 h. The cells
were digested with 0.25% trypsin without EDTA (Thermo Fisher
Scientific, Inc.) and resuspended in binding buffer to a density of
1×106 cells/ml. Annexin V-APC (5 µl) and 7-AAD (10 µl)
were mixed prior to incubation in the dark at room temperature for
10 min. Apoptotic cells were detected using a flow cytometer
(FACSAriaII; Becton, Dickinson and Company, Franklin Lakes, NJ,
USA). FlowJo software (version 10; Becton, Dickinson and Company)
was used for data analysis.
Western blot analysis
Both ESCC cells were seeded in 6-well plates at a
density of 2×105 cells/well and treated with cisplatin
(5 µM) at 37°C for 24 h. Cells were lysed in
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) supplemented with phenylmethylsulfonyl fluoride
(Beyotime Institute of Biotechnology) for 30 min at 4°C and
centrifuged at 10,000 × g for 15 min in 4°C. The supernatant was
then collected. Tumor tissues were cut into 100 mg pieces,
incubated with 500 µl lysis buffer, homogenized completely with a
tissue homogenizer for 5 min and lysed for 30 min on ice. Samples
were then centrifuged at 10,000 × g for 15 min in 4°C, and
supernatant was collected. Protein concentration was determined
using bicinchoninic acid assay (Beyotime Institute of
Biotechnology). Proteins (30 µg) were separated by 10% SDS-PAGE and
transferred onto 0.22 µm polyvinylidene fluoride membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 5%
skimmed milk for 2 h at 37°C and incubated with the appropriate
primary antibodies at 4°C overnight. The following antibodies were
used: Anti-Mel18 (cat. no. ab5267, Abcam, Cambridge, UK), anti-BMI1
(cat. no. 6964; Cell Signaling Technology, Danvers, MA, USA),
anti-B-cell lymphoma-2 (Bcl-2) (cat. no. 4223; Cell Signaling
Technology), anti-Bcl-2-associated X protein (BAX) (cat. no. 5023;
Cell Signaling Technology), anti-caspase3 (cat. no. 9662; Cell
Signaling Technology), anti-nuclear factor-κB (NF-κB) (cat. no.
8242; Cell Signaling Technology), anti-c-Myc (cat. no. 5605; Cell
Signaling Technology), anti-Akt (cat. no. 4685; Cell Signaling
Technology), anti-phosphorylated-Akt (cat. no. 4060; Cell Signaling
Technology) and anti-GAPDH (cat. no. 5174; Cell Signaling
Technology). Subsequently, membranes were incubated with
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:5,000; cat. no. ab97200; Abcam) at room temperature for
1 h. Bands were detected using enhanced chemiluminescence substrate
(EMD Millipore) and Bio-Rad ChemiDoc MP High-end imaging system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Relative
expression level of all proteins was normalized to endogenous
control GAPDH using ImageJ version 1.0 (National Institutes of
Health, Bethesda, MD, USA).
Tumor xenograft model
All animal procedures were approved by the Ethics
Committee of Qianfoshan Hospital (Jinan, China). Mice were housed
(five mice per cage) under specific pathogen-free conditions, at a
constant room temperature of 22–24°C, with a 12-h light/dark cycle
and had unlimited access to food and water. The significance of
BMI1 and Mel18 inhibition in the sensitization of cisplatin in ESCC
in vivo was studied by subcutaneous injection of cancer
cells into nude mice. A total of 20 male BALB/c nude mice (3-week
old) were purchased from Vital River Laboratories Co., Ltd.
(Beijing, China). All 20 nude mice were randomly divided into four
groups. Exponentially growing transfected EC109 cells, including
Mel18 shRNA and BMI1 shRNA-transfected cells, Mel18 and BMI1
shRNA-transfected cells, BMI1 shRNA-transfected cells, and NC
shRNA-transfected cells, were harvested and resuspended in sterile
PBS. Equal cell numbers (5×106) of each group were
injected subcutaneously in the right flank of BALB/c nude mice.
Tumor volume was calculated using the following formula: Tumor
volume (mm3) = length × width2 ×0.5. When the
tumor volume reached approximately 200 mm3, cisplatin
was intraperitoneally injected every 3 days with a dose of 5 mg/kg,
according to the manufacturer's protocol and a previously published
study (35). After 3 weeks of
treatment, mice were sacrificed in a chamber with increasing
concentrations of carbon dioxide. The tumor volume was measured by
a caliper and calculated using the aforementioned formula.
Statistical analysis
All statistical analysis was performed using SPSS
software (version 20; IBM Corp., Armonk, NY, USA). Unless otherwise
indicated, data were presented as the means ± standard deviation.
An unpaired Student's t-test of two independent samples was used
for statistical comparison between two groups. Analysis of variance
was performed to compare the mean among multiple groups and
Student-Newman-Keuls method was used for pairwise comparison
between different treatment groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
BMI1 knockdown increases the
sensitivity of cells to cisplatin
To study the biological role of BMI1 on cell
survival in ESCC with the treatment of cisplatin, two BMI1 shRNA
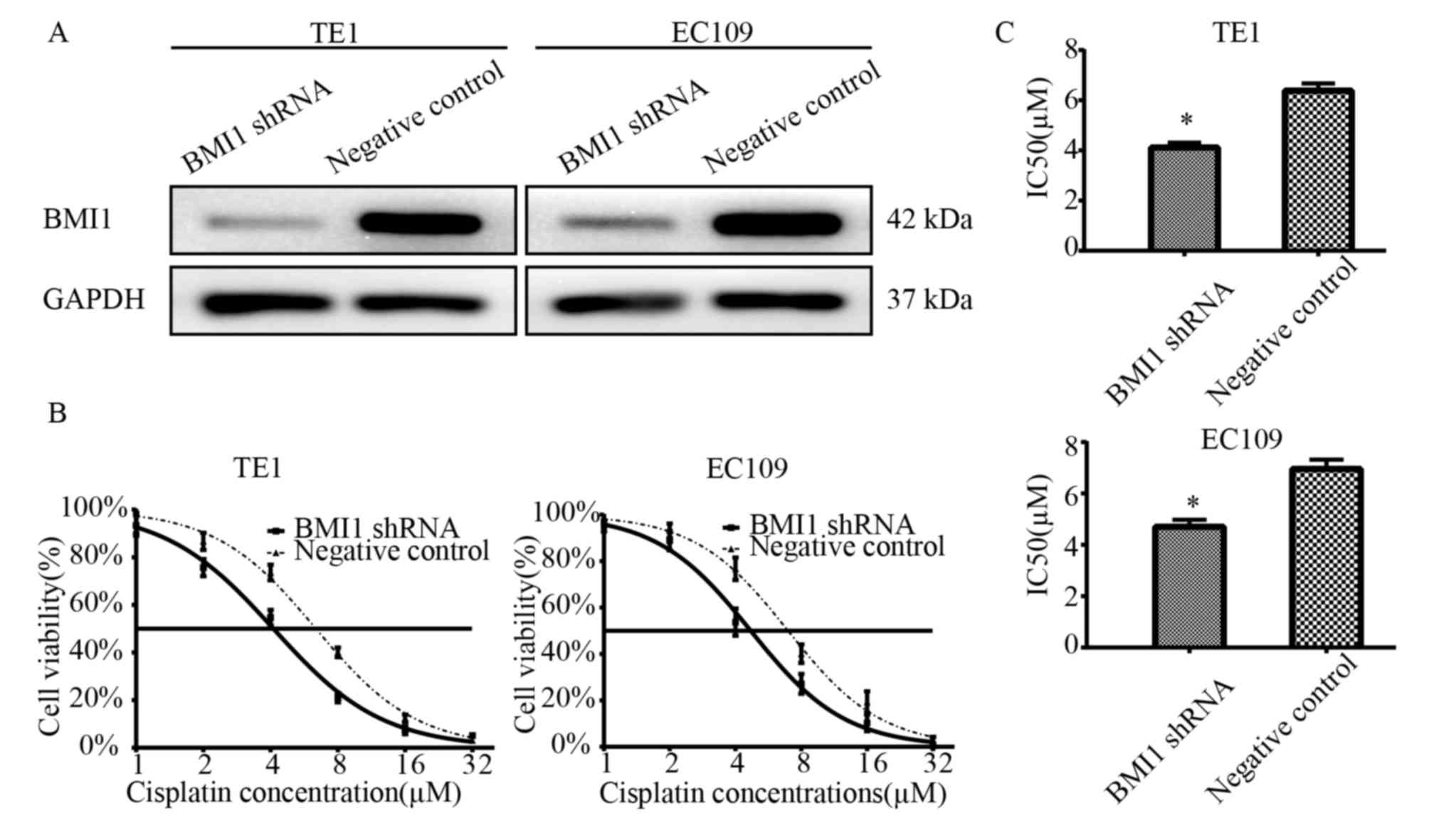
stably transfected cell lines were established. As demonstrated in
Fig. 1A, BMI1 expression was
markedly lower in the transfected EC109 and TE1 cells compared with
the negative control cells. To examine the effects of cisplatin on
the survival of BMI1 knockdown cells, a CCK-8 assay was performed
following treatment of the cells with cisplatin for 24 h. Following
treatment with various concentrations of cisplatin, it was
identified that the BMI1 shRNA-transfected cells demonstrated lower
cell viabilities compared with the NC shRNA-transfected cells
(Fig. 1B). The IC50
values of cisplatin in the BMI shRNA-transfected and the
NC-transfected EC109 cells were 4.695±0.287 and 6.953±0.369 µM,
respectively (P<0.05). The IC50 values of cisplatin
in the BMI shRNA-transfected and the NC-transfected TE1 cells were
4.117±0.192 and 6.376±0.294 µM, respectively (P<0.05; Fig. 1C). These results indicate that BMI1
knockdown increases sensitivity to cisplatin.
Co-inhibition of Mel18 and BMI1
further sensitizes ESCC cells to cisplatin compared with inhibition
of BMI1 alone
To determine the effects of Mel18 and BMI1 on the
chemosensitivity of ESCC cells, the present study established four
stably transfected ESCC cell lines for each TE1 and EC109 cells,
including Mel18 shRNA and BMI1 shRNA-transfected cells, Mel18 and
BMI1 shRNA-transfected cells, BMI1 shRNA-transfected cells, and NC
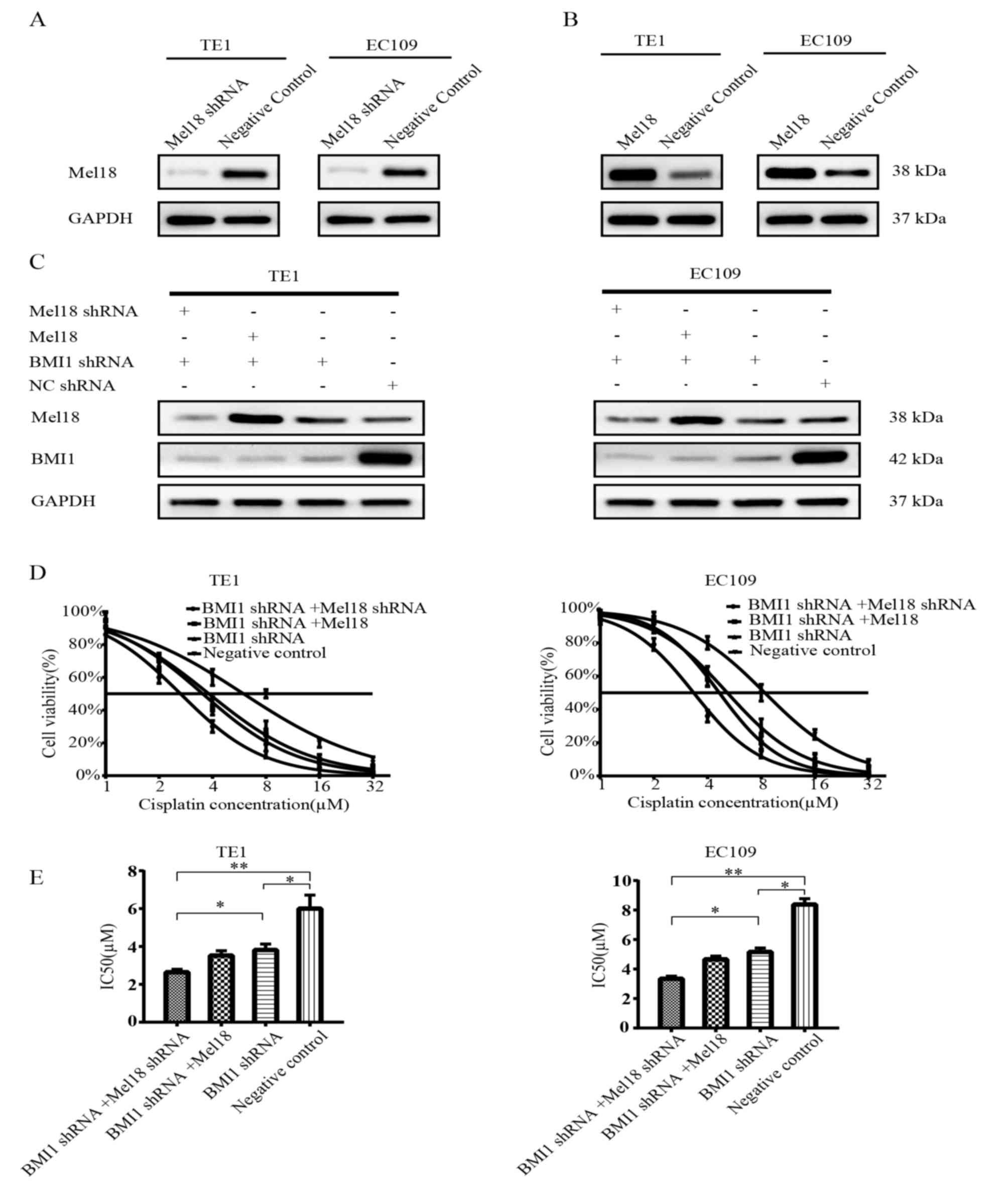
shRNA-transfected cells. As presented in Fig. 2A-C, the transfection efficiency was
verified by western blot analysis.
Subsequently, a survival assay was performed with
the transfected cells treated with different concentrations of
cisplatin. As expected, the cell viability was significantly lower
for the BMI1 shRNA-transfected cells compared with the NC cells
following treatment with cisplatin in EC109 cells (P<0.05;
Fig. 2D and E; right panel). A
combination of Mel18 and BMI inhibition significantly enhanced the
sensitivity to cisplatin compared with BMI inhibition alone in
EC109 cells (P<0.05, Fig. 2D and
E; right panel). Overexpression of Mel18 combined with BMI1
inhibition did not enhance the sensitivity to cisplatin compared
with BMI inhibition alone in EC109 cells (Fig. 2D and E; right panel). Similar results
were observed in TE1 cells (Fig. 2D and
E; left panel). In comparison with the NC group, the three
remaining groups demonstrated significantly lower IC50
values (all P<0.05; Fig. 2E). The
EC109 cells with the most significant reduction in IC50
were those in which both Mel18 and BMI1 had been inhibited
(P<0.01; Fig. 2E). Similar
results were observed in TE1 cells (Fig.
2E).
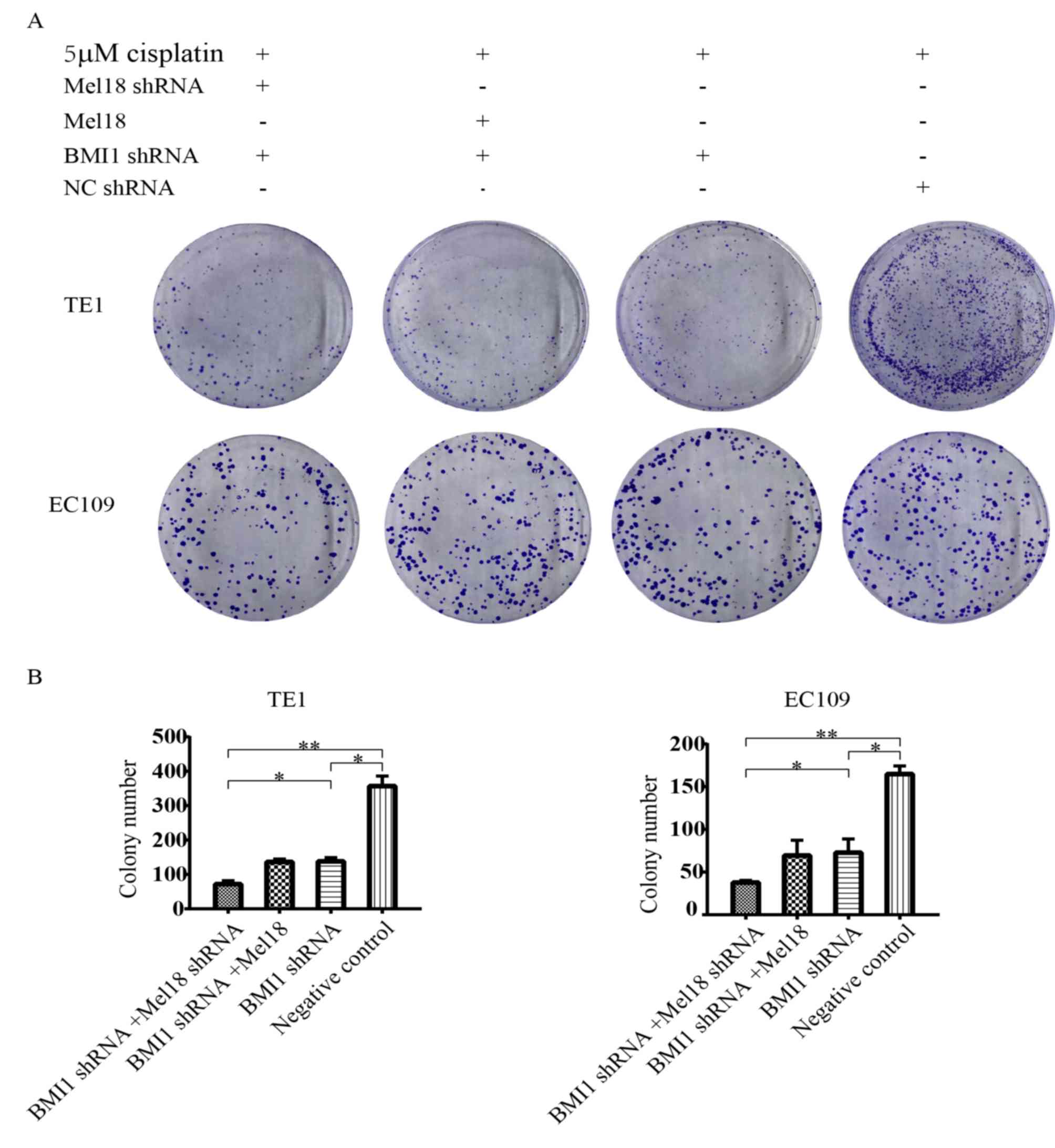
To further determine the effects of Mel18 and BMI1
inhibition on cisplatin sensitization, the current study
investigated the long-term effects of BMI1 and Mel18 inhibition by
colony formation assay. It was revealed that depletion of BMI1
increased sensitivity to cisplatin (Fig.
3A and B). Furthermore, depletion of Mel18 and BMI1 in the two
ESCC cell lines further increased sensitivity to cisplatin compared
with individual depletion of BMI1 (Fig.
3A and B). In summary, the current results indicate that
combined inhibition of Mel18 and BMI1 sensitizes ESCC cells to
cisplatin. By contrast, overexpression of Mel18 combined with BMI1
inhibition did not markedly reduce cell survival compared with
individual knockdown of BMI1.
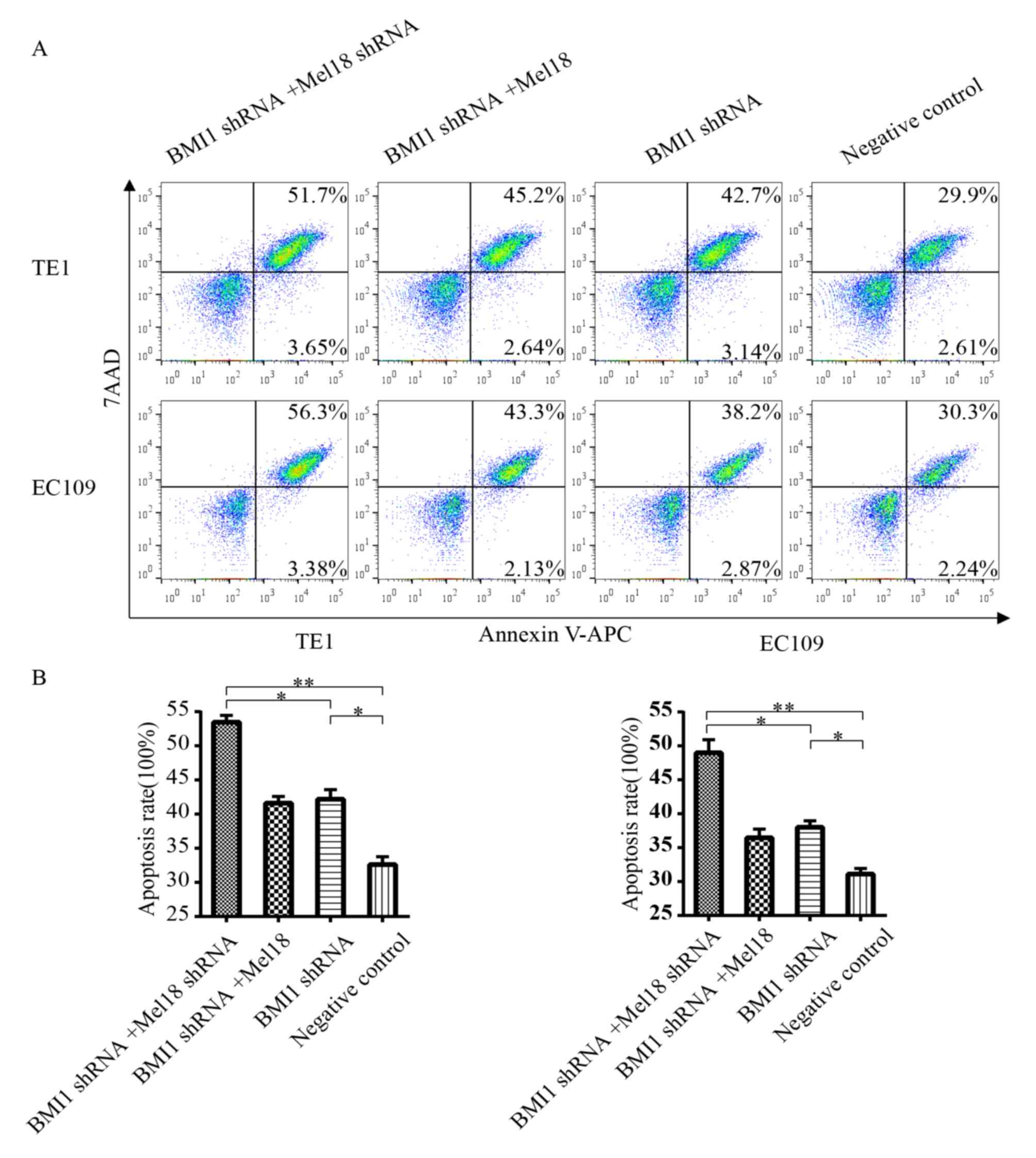
Inhibition of BMI1 and Mel18 enhances
cisplatin-induced apoptosis
Cisplatin-induced cytotoxic effects predominantly
occur via apoptosis (36). To
evaluate the effect of BMI1 and Mel18 knockdown on
cisplatin-induced apoptosis, the apoptotic rates of stably
transfected ESCC cells were examined following treatment with 5 µM
of cisplatin for 24 h. Consistent with the cytotoxic effects
observed with the cell viability assay, combined inhibition of BMI1
and Mel18 increased the rate of apoptosis compared with the BMI1
inhibition group (P<0.05) or the NC group (P<0.01), as
presented in Fig. 4A and B. Similar
results were revealed in the EC109 and TE1 cells. In summary, Mel18
and BMI1 inhibition can prompt an increase in the rate of apoptosis
with BMI1 inhibition alone in ESCC cells.
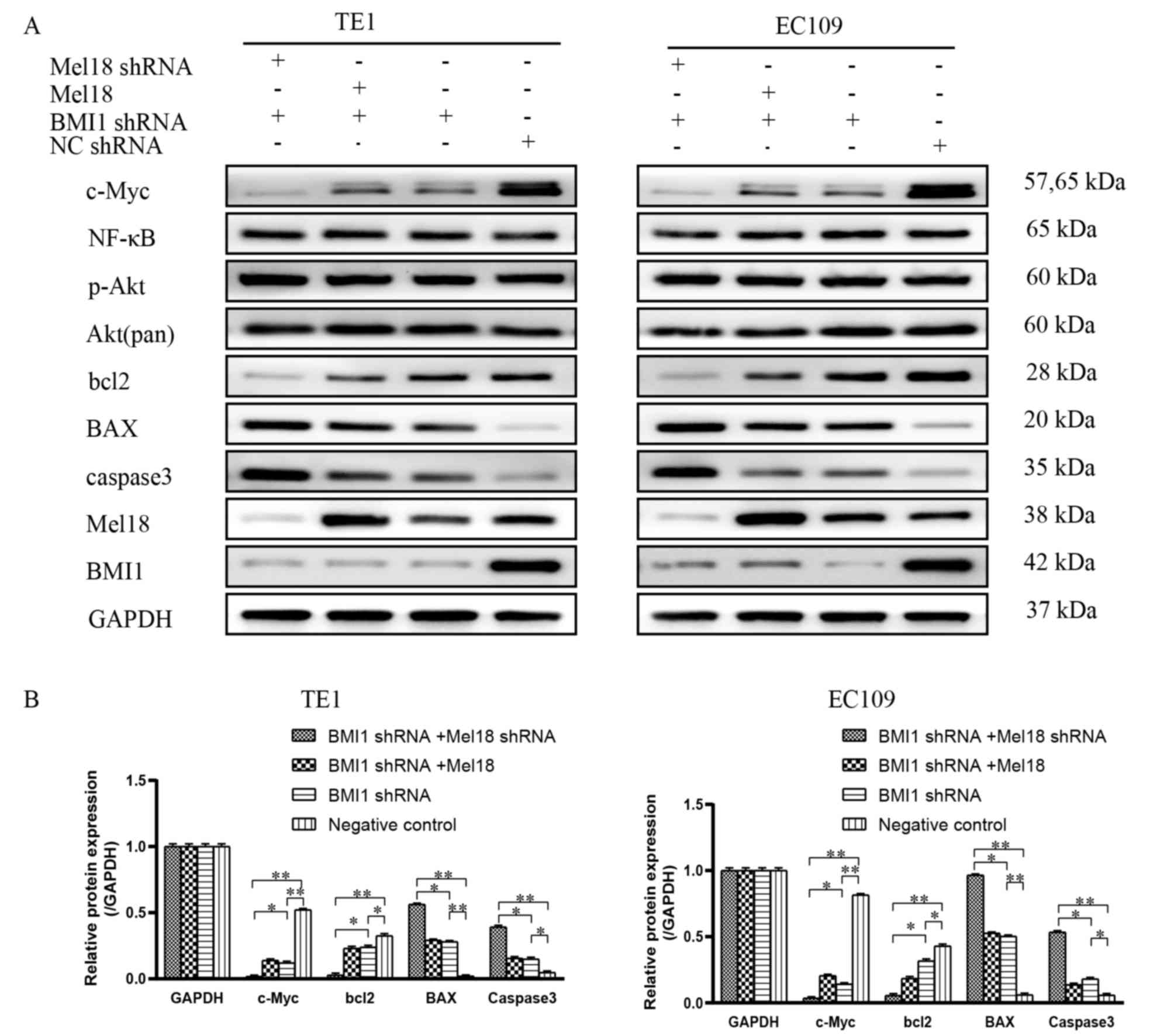
c-Myc may be essential for BMI-1 and
Mel18-induced sensitization to cisplatin
Cisplatin-induced apoptosis predominantly occurs via
phosphoinositide 3-kinase/Akt, NF-κB and c-Myc dysregulation
(36). Mel18 and BMI1 can regulate
proliferation, apoptosis, angiogenesis, tumorigenesis and
development via these pathways (28,37,38). To
further investigate the mechanism underlying the enhancement of
cisplatin-induced apoptosis by the silencing of BMI1 and Mel18, the
present study examined the expression levels of proteins associated
with these pathways, including total NF-κB, total-Akt,
phosphorylated-Akt, c-Myc, caspase-3, BAX and Bcl-2, in the stably
transfected cells. The protein expression levels were evaluated by
western blot analysis. As presented in Fig. 5, the expression levels of caspase-3,
BAX markedly increased and Bcl-2 markedly decreased in cells
transfected with Mel18 shRNA and BMI1 shRNA compared with cells
transfected with NC or BMI shRNA (all P<0.05), which indicated
that BMII1 and Mel18-induced apoptosis may be closely associated
with the mitochondrial apoptotic pathway. Inhibition of BMI1 was
identified to reduce the expression levels of proteins associated
with these signaling pathways to different extents, which is
consistent with previous studies (6,7,9). Compared with BMI1 inhibition or NC
group, the effect of inhibiting BMI1 and Mel18 on c-Myc was more
notable (P<0.05), while the effect on NF-κB and Akt was
limited.
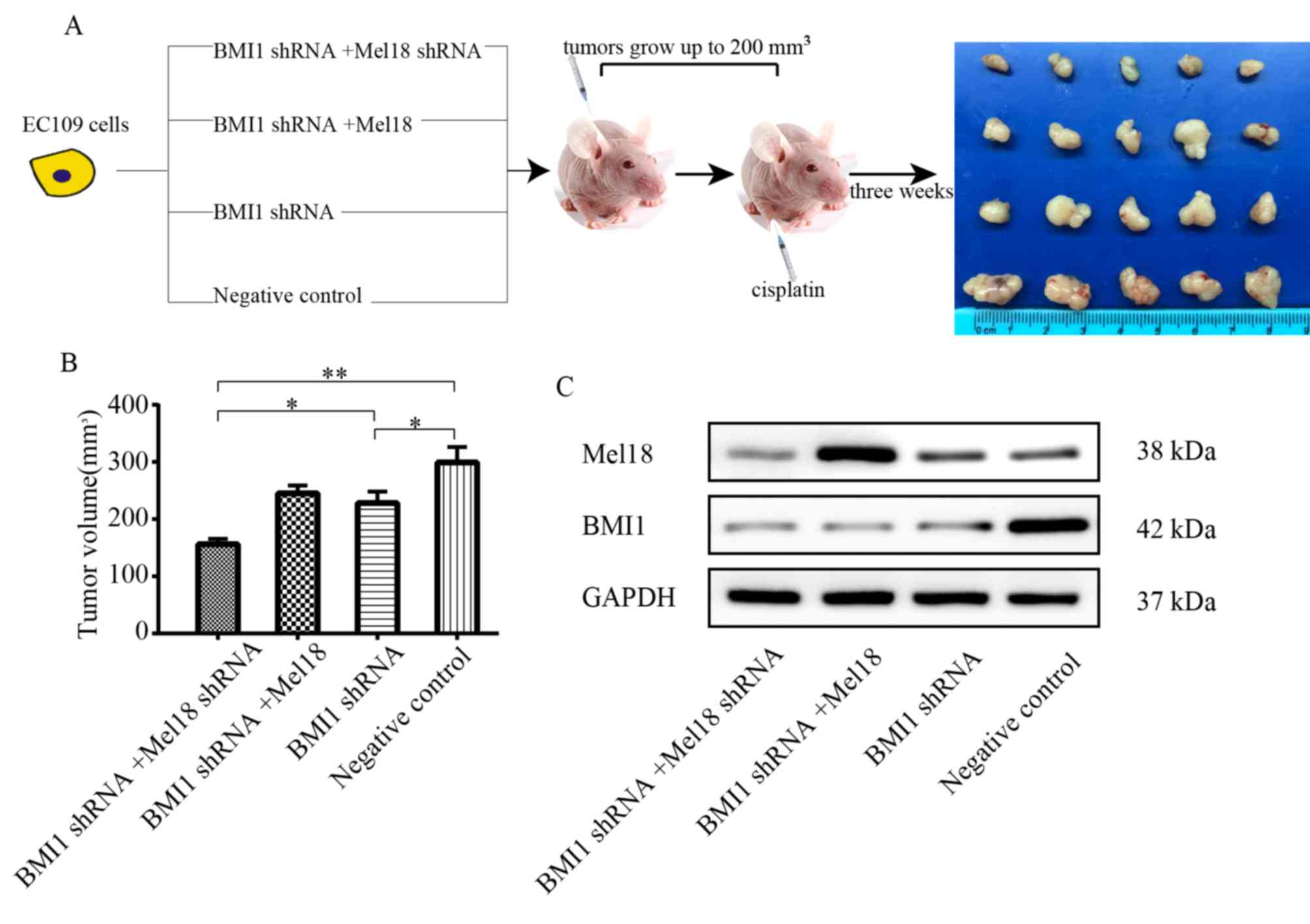
BMI1 and Mel18 inhibition sensitize
esophageal xenograft tumors to cisplatin
The effect of cisplatin treatment in combination
with BMI1 and Mel18 inhibition on the growth of esophageal tumors
was further determined in vivo. An ESCC xenograft model was
established by subcutaneous injection of ESCC cells into the right
flank of nude mice. The treatments were initiated once tumor
volumes reached ~200 mm3 (Fig. 6A). With the same does of cisplatin,
the size of EC109 ×enograft tumors in the BMI1 shRNA group were
significantly smaller compared with the NC group (P<0.05;
Fig. 6B). In addition, the tumor
volume was markedly lower in the BMI1 and Mel18 shRNA group
compared with the BMI1 inhibition alone group (P<0.05; Fig. 6B). The expression levels of Mel18 and
BMI1 in xenograft tumors were verified by western blot analysis
(Fig. 6C). These results suggest
that Mel18 inhibition can cooperate with BMI1 inhibition to enhance
chemosensitivity of EC109 cells in vivo.
Discussion
Platinum-based chemoradiotherapy is the standard
treatment for advanced esophageal cancer and postoperative
recurrent esophageal cancer (4,39). A
limitation of this therapy is the frequent occurrence of resistance
to platinum. BMI1 is an important epigenetic regulator and stem
cell marker (7,10). Previous studies have demonstrated
that inhibition of BMI1 can regulate the expression of MDR1, affect
platinum transport and hydration, and promote the chemotherapy
sensitivity of ovarian, breast and pancreatic cancer (40–42). As
a homologue of BMI1, the function of Mel18, and the interaction of
BMI1 and Mel18 have been widely studied. However, the role of Mel18
remains controversial. The present study demonstrated that Mel18
inhibition, combined with BMI1 knockdown, can promote the
chemosensitivity of esophageal cancer cells via cisplatin-induced
apoptosis.
The current study established BMI1 knockdown EC109
and TE1 cells by transfection with BMI1 shRNA, followed by
treatment with an increasing gradient of cisplatin. Compared with
the negative control group, the cell survival rate and the
IC50 value of the BMI1 inhibition group was
significantly lower, which indicates that BMI1-inhibited esophageal
cancer cells are sensitive to cisplatin. Therefore, there is a
requirement to further investigate the association between BMI1 and
cisplatin chemosensitivity.
Mel18 is a homologue of BMI1 and a component of
PRC1. The structures of BMI1 and Mel18 are similar, and their roles
are similar in embryonic development; however, the association
between Mel18 and BMI1 remains unknown (32). Previous studies have suggested that
Mel18 can inhibit BMI1, which subsequently inhibits tumorigenesis,
angiogenesis and tumor progression (20,31,43). By
contrast, Liu et al (15)
confirmed that BMI1 and Mel18 contribute to the development of
colorectal cancer by promoting proliferation and reducing apoptosis
via suppression of Reg3b expression. The interaction between BMI1
and Mel18, and the effects on tumor sensitivity to chemotherapy
remain unknown and require further investigation.
In the present study, Mel18 was knocked down or
overexpressed in esophageal cancer cell lines by stable
transfection with BMI1 shRNA. Compared with the BMI1-inhibited
group and the negative control group, inhibition of Mel18 and BMI1
markedly enhanced the short-term and long-term sensitivity of
esophageal cancer cells to cisplatin, and these effects were
consistent with the impacts on apoptosis and changes of protein
levels associated with the mitochondrial apoptotic pathway. During
an investigation of the core proteins associated with the
mitochondrial apoptosis pathway, the expression level of c-Myc was
identified to exhibit the most notable change following Mel18 and
BMI1 inhibition. A number of studies have demonstrated that PRC1s,
including BMI1 and Mel18, can regulate c-Myc to affect neoplastic
cell proliferation and apoptosis (5,44). BMI1
has been identified to cooperate with c-Myc within the cell nucleus
and BMI1 overexpression can inhibit c-Myc-induced apoptosis via
negative regulation of the Ink4a-Arf pathway (5). Mel18 can regulate the cell cycle via a
c-Myc/Cdc25 cascade (44). Combined
with these previous studies, the results of the present study
indicate that the regulation of chemotherapy sensitivity of
esophageal cancer by BMI1 and Mel18 may be achieved via c-Myc
regulation of the mitochondrial apoptosis pathway.
In summary, the present study confirmed the role of
BMI1 in the regulation of tumor chemosensitivity and revealed a
combined effect of Mel18 and BMI1 on tumor chemosensitivity, and
confirmed this effect in vivo. To the best of our knowledge,
the current study was the first to demonstrate that inhibition of
BMI1 can increase the chemosensitivity of ESCC to platinum-based
chemotherapy. In addition, it was demonstrated that Mel18
inhibition can enhance the effects of BMI1 inhibition. These
effects were identified to be achieved via apoptosis-associated
pathways. However, the current study also had numerous limitations
and complete understanding of the associated mechanisms remains
unknown. Liu et al (15)
confirmed that BMI1 and Mel18 synergistically promote the
development of colon cancer; however, the present study did not
verify that the combined effects of the two molecules were achieved
through synergy. In future studies it may be beneficial to study
whether there is a direct interaction between the two molecules;
this could be achieved by investigating whether the combined
effects of the two molecules are achieved via synergistic or
additional effects. Although further clinical studies are required,
Mel18 and BMI1 may serve as prominent therapeutic targets for ESCC
chemotherapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shandong
Science and Technology Department Science and Technology
Development Foundation (grant no. 2013GSF11836) and the Natural
Science Foundation of Shandong Province (grant no. ZR2016HQ50).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
JW and HJ designed the study and performed all in
vitro experiments. QZ, JD and XY performed the animal
experiments. ZJ designed the experiments and performed the
statistical analysis.
Ethics approval and consent to
participate
All animal procedures were approved by Qianfoshan
Hospital Ethics Committee (Jinan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Global Burden of Disease Cancer
Collaboration, ; Fitzmaurice C, Dicker D, Pain A, Hamavid H,
Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, et
al: The global burden of cancer 2013. JAMA Oncol. 1:505–527. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Waddell T, Chau I, Cunningham D, Gonzalez
D, Okines AF, Okines C, Wotherspoon A, Saffery C, Middleton G,
Wadsley J, et al: Epirubicin, oxaliplatin, and capecitabine with or
without panitumumab for patients with previously untreated advanced
oesophagogastric cancer (REAL3): A randomised, open-label phase 3
trial. Lancet Oncol. 14:481–489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hingorani M, Crosby T, Maraveyas A, Dixit
S, Bateman A and Roy R: Neoadjuvant chemoradiotherapy for
resectable oesophageal and gastro-oesophageal junction cancer-do we
need another randomised trial. Clin Oncol (R Coll Radiol).
23:696–705. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Benetatos L, Vartholomatos G and
Hatzimichael E: Polycomb group proteins and MYC: The cancer
connection. Cell Mol Life Sci. 71:257–269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ferretti R, Bhutkar A, McNamara MC and
Lees JA: BMI1 induces an invasive signature in melanoma that
promotes metastasis and chemoresistance. Genes Dev. 30:18–33. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jin X, Kim LJY, Wu Q, Wallace LC, Prager
BC, Sanvoranart T, Gimple RC, Wang X, Mack SC, Miller TE, et al:
Targeting glioma stem cells through combined BMI1 and EZH2
inhibition. Nat Med. 23:1352–1361. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ren H, Du P, Ge Z, Jin Y, Ding D, Liu X
and Zou Q: TWIST1 and BMI1 in cancer metastasis and
chemoresistance. J Cancer. 7:1074–1080. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen D, Wu M, Li Y, Chang I, Yuan Q,
Ekimyan-Salvo M, Deng P, Yu B, Yu Y, Dong J, et al: Targeting
BMI1(+) cancer stem cells overcomes chemoresistance and inhibits
metastases in squamous cell carcinoma. Cell Stem Cell. 20:621–634.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kreso A, van Galen P, Pedley NM,
Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W,
Sydorenko N, et al: Self-renewal as a therapeutic target in human
colorectal cancer. Nat Med. 20:29–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Q, Li Z, Wu Y, Huang R, Zhu Y, Zhang
W, Wang Y and Cheng J: Pharmacological inhibition of Bmi1 by
PTC-209 impaired tumor growth in head neck squamous cell carcinoma.
Cancer Cell Int. 17:1072017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cancer Genome Atlas Research N, Analysis
Working Group, ; Asan U, Agency BCC, et al: Integrated genomic
characterization of oesophageal carcinoma. Nature. 541:169–175.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jo S, Lee YL, Kim S, Lee H and Chung H:
PCGF2 negatively regulates arsenic trioxide-induced PML-RARA
protein degradation via UBE2I inhibition in NB4 cells. Biochim
Biophys Acta. 1863:1499–1509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang CY, Kuo CH, Pai PY, Ho TJ, Lin YM,
Chen RJ, Tsai FJ, Vijaya Padma V, Kuo WW and Huang CY: Inhibition
of HSF2 SUMOylation via MEL18 upregulates IGF-IIR and leads to
hypertension-induced cardiac hypertrophy. Int J Cardiol.
257:283–290. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Wei W, Li X, Shen P, Ju D, Wang Z,
Zhang R, Yang F, Chen C, Cao K, et al: BMI1 and MEL18 promote
colitis-associated cancer in Mice via REG3B and STAT3.
Gastroenterology. 153:1607–1620. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Won HY, Lee JY, Shin DH, Park JH, Nam JS,
Kim HC and Kong G: Loss of Mel-18 enhances breast cancer stem cell
activity and tumorigenicity through activating notch signaling
mediated by the Wnt/TCF pathway. FASEB J. 26:5002–5013. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jo S, Lee H, Kim S, Hwang EM, Park JY,
Kang SS and Chung H: Inhibition of PCGF2 enhances granulocytic
differentiation of acute promyelocytic leukemia cell line HL-60 via
induction of HOXA7. Biochem Biophys Res Commun. 416:86–91. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ji H, Cao M, Ren K, Sun N, Wang W, Zhu Q,
Zang Q and Jiang Z: Expression and clinicopathological significance
of Mel-18 and Bmi-1 in esophageal squamous cell carcinoma. Technol
Cancer Res Treat. 1533034617705055. 2017. View Article : Google Scholar
|
|
19
|
Guo BH, Zhang X, Zhang HZ, Lin HL, Feng Y,
Shao JY, Huang WL, Kung HF and Zeng MS: Low expression of Mel-18
predicts poor prognosis in patients with breast cancer. Ann Oncol.
21:2361–2369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu YW, Li J and Guo WJ: Expression and
clinicopathological significance of Mel-18 and Bmi-1 mRNA in
gastric carcinoma. J Exp Clin Cancer Res. 29:1432010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang W, Lin T, Huang J, Hu W, Xu K and Liu
J: Analysis of Mel-18 expression in prostate cancer tissues and
correlation with clinicopathologic features. Urol Oncol.
29:244–251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tao J, Liu YL, Zhang G, Ma YY, Cui BB and
Yang YM: Expression and clinicopathological significance of Mel-18
mRNA in colorectal cancer. Tumour Biol. 35:9619–9625. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dukers DF, van Galen JC, Giroth C, Jansen
P, Sewalt RG, Otte AP, Kluin-Nelemans HC, Meijer CJ and Raaphorst
FM: Unique polycomb gene expression pattern in Hodgkin's lymphoma
and Hodgkin's lymphoma-derived cell lines. Am J Pathol.
164:873–881. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zakrzewska M, Zakrzewski K, Gresner SM,
Piaskowski S, Zalewska-Szewczyk B and Liberski PP: Polycomb genes
expression as a predictor of poor clinical outcome in children with
medulloblastoma. Child's Nerv Syst. 27:79–86. 2011. View Article : Google Scholar
|
|
25
|
Vekony H, Raaphorst FM, Otte AP, van
Lohuizen M, Leemans CR, van der Waal I and Bloemena E: High
expression of Polycomb group protein EZH2 predicts poor survival in
salivary gland adenoid cystic carcinoma. J Clin Pathol. 61:744–749.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vekony H, Roser K, Loning T, Raaphorst FM,
Leemans CR, Van der Waal I and Bloemena E: Deregulated expression
of p16INK4a and p53 pathway members in benign and malignant
myoepithelial tumours of the salivary glands. Histopathology.
53:658–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jung JH, Choi HJ, Maeng YS, Choi JY, Kim
M, Kwon JY, Park YW, Kim YM, Hwang D and Kwon YG: Mel-18, a
mammalian polycomb gene, regulates angiogenic gene expression of
endothelial cells. Biochem Biophys Res Commun. 400:523–530. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park JH, Lee JY, Shin DH, Jang KS, Kim HJ
and Kong G: Loss of Mel-18 induces tumor angiogenesis through
enhancing the activity and expression of HIF-1alpha mediated by the
PTEN/PI3K/Akt pathway. Oncogene. 30:4578–4589. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo WJ, Datta S, Band V and Dimri GP:
Mel-18, a polycomb group protein, regulates cell proliferation and
senescence via transcriptional repression of Bmi-1 and c-Myc
oncoproteins. Mol Biol Cell. 18:536–546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo WJ, Zeng MS, Yadav A, Song LB, Guo BH,
Band V and Dimri GP: Mel-18 acts as a tumor suppressor by
repressing Bmi-1 expression and down-regulating Akt activity in
breast cancer cells. Cancer Res. 67:5083–5089. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang XW, Sheng YP, Li Q, Qin W, Lu YW,
Cheng YF, Liu BY, Zhang FC, Li J, Dimri GP and Guo WJ: BMI1 and
Mel-18 oppositely regulate carcinogenesis and progression of
gastric cancer. Mol Cancer. 9:402010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akasaka T, van Lohuizen M, van der Lugt N,
Mizutani-Koseki Y, Kanno M, Taniguchi M, Vidal M, Alkema M, Berns A
and Koseki H: Mice doubly deficient for the polycomb group genes
Mel18 and Bmi1 reveal synergy and requirement for maintenance but
not initiation of Hox gene expression. Development. 128:1587–1597.
2001.PubMed/NCBI
|
|
33
|
Gao Z, Zhang J, Bonasio R, Strino F, Sawai
A, Parisi F, Kluger Y and Reinberg D: PCGF homologs, CBX proteins,
and RYBP define functionally distinct PRC1 family complexes. Mol
Cell. 45:344–356. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu WL, Guo XZ, Zhang LJ, Wang JY, Zhang
G, Guan S, Chen YM, Kong QL, Xu LH, Li MZ, et al: Prognostic
relevance of Bmi-1 expression and autoantibodies in esophageal
squamous cell carcinoma. BMC Cancer. 10:4672010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi Q, Shen LY, Dong B, Fu H, Kang XZ,
Yang YB, Dai L, Yan WP, Xiong HC, Liang Z and Chen KN: The
identification of the ATR inhibitor VE-822 as a therapeutic
strategy for enhancing cisplatin chemosensitivity in esophageal
squamous cell carcinoma. Cancer Lett. 432:56–68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sanchez-Beato M, Sanchez E, Garcia JF,
Pérez-Rosado A, Montoya MC, Fraga M, Artiga MJ, Navarrete M,
Abraira V, Morente M, et al: Abnormal PcG protein expression in
Hodgkin's lymphoma. Relation with E2F6 and NFkappaB transcription
factors. J Pathol. 204:528–537. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Silva J, Garcia JM, Peña C, García V,
Domínguez G, Suárez D, Camacho FI, Espinosa R, Provencio M, España
P and Bonilla F: Implication of polycomb members Bmi-1, Mel-18, and
Hpc-2 in the regulation of p16INK4a, p14ARF, h-TERT, and c-Myc
expression in primary breast carcinomas. Clin Cancer Res.
12:6929–6936. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Merkow RP, Bilimoria KY, McCarter MD, Chow
WB, Ko CY and Bentrem DJ: Use of multimodality neoadjuvant therapy
for esophageal cancer in the United States: Assessment of 987
hospitals. Ann Surg Oncol. 19:357–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Banerjee Mustafi S, Chakraborty PK, Naz S,
Dwivedi SK, Street M, Basak R, Yang D, Ding K, Mukherjee P and
Bhattacharya R: MDR1 mediated chemoresistance: BMI1 and TIP60 in
action. Biochim Biophys Acta. 1859:983–993. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao Q, Qian Q, Cao D, Yang J, Gui T and
Shen K: Role of BMI1 in epithelial ovarian cancer: Investigated via
the CRISPR/Cas9 system and RNA sequencing. J Ovarian Res.
11:312018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
M JR and S V: BMI1 and PTEN are key
determinants of breast cancer therapy: A plausible therapeutic
target in breast cancer. Gene. 678:302–311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Riis ML, Luders T, Nesbakken AJ, Vollan
HS, Kristensen V and Bukholm IR: Expression of BMI-1 and Mel-18 in
breast tissue-a diagnostic marker in patients with breast cancer.
BMC Cancer. 10:6862010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tetsu O, Ishihara H, Kanno R, Kamiyasu M,
Inoue H, Tokuhisa T, Taniguchi M and Kanno M: mel-18 negatively
regulates cell cycle progression upon B cell antigen receptor
stimulation through a cascade leading to c-myc/cdc25. Immunity.
9:439–448. 1998. View Article : Google Scholar : PubMed/NCBI
|