Introduction
Multiple myeloma (MM) is the second most frequent
hematological neoplasm in the USA in 2018 (1) and is characterized by the infiltration
of clonal plasma cells in the bone marrow, secretion of monoclonal
immunoglobulins and end organ damage (2). Over the last decade, the introduction
of proteasome inhibitors [bortezomib (BTZ) and carfilzomib] and
immunomodulatory drugs (thalidomide and lenalidomide), combined
with autologous stem cell transplantation, have significantly
improved the prognosis of patients with MM. The 5-year overall
survival (OS) rate of patients diagnosed between 2005 and 2011 was
nearly double the rate of patients diagnosed between 1975 and 1977
(49 and 25%, respectively) (3).
However, MM remains an incurable disease, and the majority of
patients will eventually relapse and become chemorefractory to
currently available drugs. Therefore, further development of novel
therapies is required.
Histone deacetylase (HDAC) inhibitors (HDACIs), are
believed to be promising therapeutic drugs in the treatment of
cancer. To date, only 4 HDACIs (panobinostat, belinostat,
romidepsin and vorinostat) have been approved by the US Food and
Drug Administration (FDA) for the treatment of several types of
cancer. Deregulation of histone acetylation has been recognized to
serve a critical role in the pathogenesis of MM (4). HDACs are overexpressed in plasma cells
derived from patients with MM, compared with those from healthy
controls. High expression levels of HDAC1 protein are associated
with decreased overall survival time (OS) in patients with MM
(4). In addition, HDACI treatment
induces the upregulation of 21 genes, which were reported to be
associated with improved OS in MM (5). Therefore, HDACs have been considered
promising targets for MM therapies. Several HDACI-based regimens
are in clinical trials for MM, with overall response rates of
42–61% (6–8). The phase 3 study PANORAMA-1
demonstrated that the chemotherapy regimen including panobinostat
(a pan-HDACI) resulted in a longer progression-free survival, when
compared with that in the placebo group (7). As a result, panobinostat was approved
by the FDA for the treatment of relapsed/refractory MM in 2015.
However, a number of adverse events have been observed with
pan-HDACIs, including diarrhea, anorexia, nausea, fatigue, thrombus
and thrombocytopenia (7). To reduce
the adverse events of pan-HDACIs, selective HDACIs targeting class
I isoforms with improved efficacy and lower toxicity may be
promising drugs for the treatment of MM (9).
Chidamide (CM), a novel benzamide type HDACI,
selectively suppresses the activity of class I HDACs, including
HDAC1, HDAC2, HDAC3 and HDAC10 (10,11).
Previous studies have revealed that CM induces cell proliferation
inhibition and apoptosis in several hematological malignancies,
including myelodysplastic syndromes (12), leukemia (13–15) and
natural killer (NK)/T-cell lymphoma (16), as well as non-hematological
malignancies, including lung cancer (17), hepatocellular carcinoma (18), colon cancer (19) and pancreatic cancer (20). CM was approved by the China FDA for
the treatment of peripheral T-cell lymphoma (PTCL) in 2014
(21). A study in 2017 revealed that
the overall response rate to CM alone or in combination was 39.06
and 51.18%, respectively, for relapsed/refractory PTCL (22). At present, clinical trials of CM are
being conducted in China and the US for the treatment of solid
tumors. However, to the best of our knowledge, its biological
effects on MM and relevant mechanisms have not been
investigated.
In the present study, the cytotoxic effect of CM on
MM cells was evaluated and the possible mechanisms involved were
investigated. The aim of the present study was to provide evidence
for the clinical application of chidamide in multiple myeloma.
Materials and methods
Cell lines and cell culture
The human MM RPMI8226 and U266 cell lines were
obtained from the Hematology Institute of Zhejiang University
(Hangzhou, China). All cells were cultured in a humidified
atmosphere containing 5% CO2 at 37°C. All cells were
maintained in RPMI-1640 medium with 10% fetal bovine serum,
penicillin (100 U/ml) and streptomycin (100 µg/ml) (all Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Bone marrow was
collected from 4 newly diagnosed patients with MM admitted to the
Second Affiliated Hospital, Zhejiang University School of Medicine
(Hangzhou, China) between January 2018 and March 2018. Patient 1
was a 59-year-old male with IgG/κ subtype, patient 2 was a
58-year-old male with IgG/λ subtype, patient 3 was a 72-year-old
male with IgG/λ subtype and patient 4 was a 77-year-old female with
IgG/λ subtype. Once informed consent was obtained, an additional 3
ml of bone marrow was extracted during the diagnostic bone marrow
puncture. Subsequently, primary myeloma cells were isolated by
Ficoll-Hypaque density gradient centrifugation (2,000 × g, 15 min
at room temperature). All experiments were performed according to
the protocol approved by the Ethics Committee, The Second
Affiliated Hospital, Zhejiang University School of Medicine
(approval no. 2019022; Hangzhou, China).
Reagents and antibodies
CM was obtained from Shenzhen Chipscreen Biosciences
Co., Ltd. (Shenzhen, China). CM was prepared in dimethylsulfoxide
(DMSO) and stored at −20°C at 20 mmol/l. The final concentration of
DMSO in the culture medium was <0.1%. The molecular weight of CM
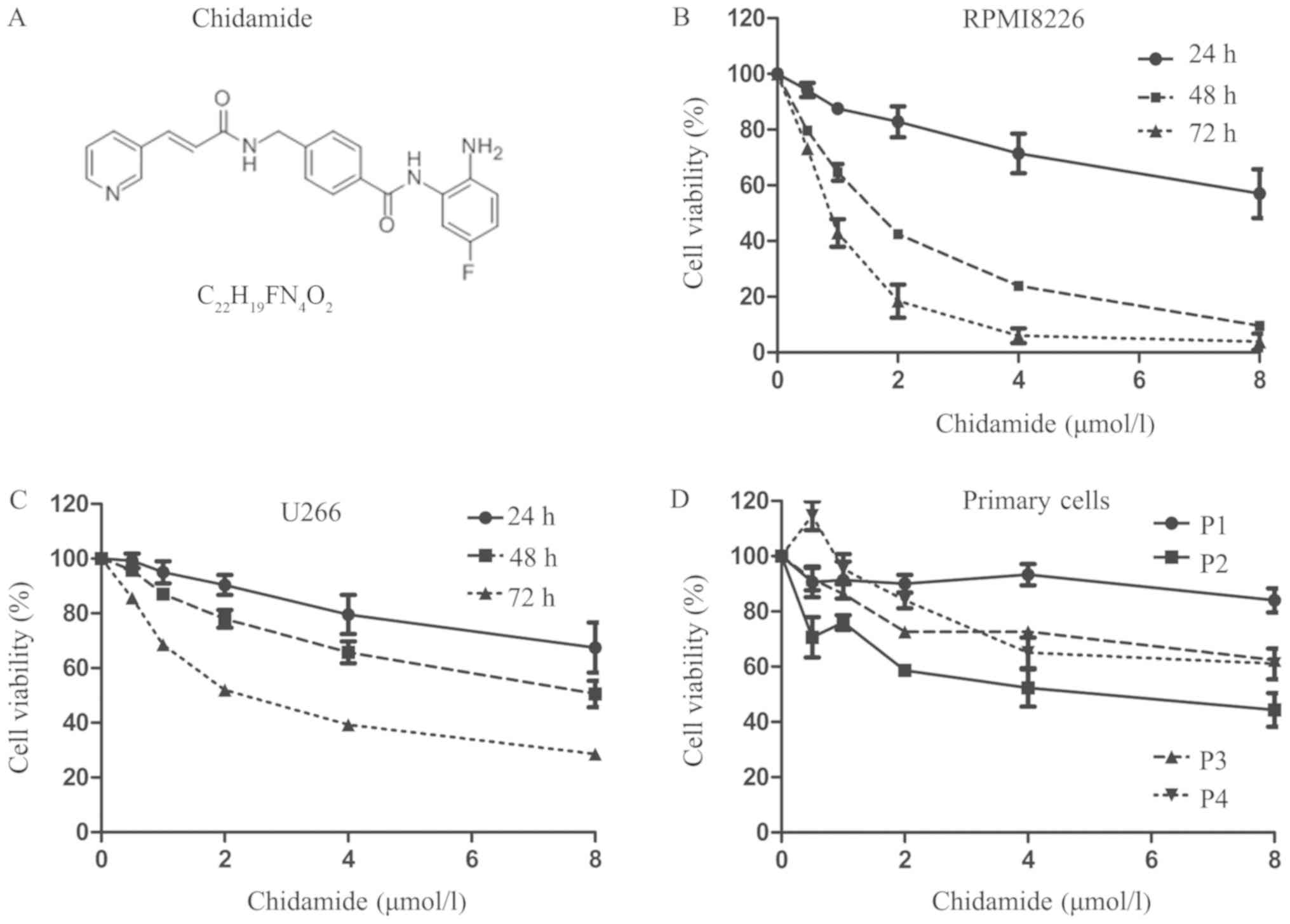
is 747.25 and its formula is provided in Fig. 1A. BTZ was purchased from Xian Janssen
Pharmaceutical Ltd. (Beijing, China). Anti-c-myc (cat. no. 9402),
anti-caspase-3 (cat. no. 9662), anti-caspase-8 (cat. no. 4790),
anti-cleaved-caspase-8 (cat. no. 9496), anti-caspase-9 (cat. no.
9502), anti-Bcl-2 (cat. no. 15071), anti-myeloid cell leukemia 1
(mcl-1; cat. no. 94296), anti-t-p53 (cat. no. 2524), anti-p-p53
(cat. no. 2521), anti-Bax (cat. no. 5023), anti-p21 (cat. no.
2947), anti-cyclin D1 (cat. no. 2922), anti-HDAC1 (cat. no. 5356),
anti-HDAC2 (cat. no. 2540) and anti-HDAC3 (cat. no. 3949)
antibodies were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Anti-GAPDH (cat. no. ab181602), horseradish
peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody
(cat. no. ab6721), HRP-conjugated goat anti-mouse secondary
antibody (cat. no. ab6789), anti-acetyl-histone H3 (acetyl K9 + K14
+ K18 + K23 + K27; cat. no. ab47915) and anti-acetyl-histone H4
(acetyl K5 + K8 + K12 + K16; cat. no. ab177790) antibodies were
obtained from Abcam (Cambridge, MA, USA). The anti-poly ADP ribose
polymerase (PARP) antibody (cat. no. sc53643) was obtained from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). All antibodies
from Cell Signaling Technology and Santa Cruz Biotechnology were
diluted at 1:1,000 and all antibodies from Abcam were diluted at
1:10,000.
Cell viability assay
Cells were seeded in 96-well plates at
2×104 cells/well and incubated in 0.25–8 µmol/l CM for
24, 48 or 72 h. Following treatment, cell viability was measured
using the Cell Counting Kit-8 (CCK-8) viability assay (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan). According to the
manufacturer's instruction, 10 µl CCK-8 solution was added to each
well for 3 h at 37°C. The optical density (OD) was measured at 450
nm using a microplate reader (Biotek, Winooski, VT, USA). To
determine whether CM sensitized MM cells to BTZ, the cells were
cultured with media containing 0.5 µmol/l CM for RPMI8226 cells or
2 µmol/l CM for U266 cells in the absence or presence of BTZ (2.5
and 5 ng/ml) for 24 h, and subsequently cell viability was assessed
using the CCK-8 assay. The cell viability was calculated as [OD
(treatment)-OD (control)]/[OD (control)-OD (blank)] ×100. The
viability inhibition rate (%) was calculated as [100-cell viability
rate (%)]. Time- and dose-response curves were produced using
GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA).
IC50 values were predicted based on the available data
using the SPSS probit model analysis (SPSS v17.0; SPSS, Inc.,
Chicago, IL, USA).
Western blot analysis
Following exposure to CM (0, 0.5, 1, 2 µmol/l for
PRMI8226 cells, and 0, 2, 4, 8 µmol/l for U266 cells) for 48 h, the
cells were harvested and washed with ice-cold PBS. Proteins were
extracted using ice-cold radioimmunoprecipitation assay lysis
buffer (Beyotime Institute of Biotechnology) with
phenylmethylsulfonyl fluoride. Following a brief sonication, the
cell lysates were kept on ice and subsequently centrifuged at
15,984 × g for 10 min. Supernatant was collected and the protein
concentrations were quantified with a bicinchoninic acid assay
(Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Total protein (40 µg/lane) was separated
using 10–12% SDS-PAGE and transferred to polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA). The membrane was
subsequently blocked with TBST (0.05% Tween-20) containing 5%
skimmed milk for 1 h and probed overnight with the corresponding
primary antibodies (diluted in primary antibody dilution buffer
from Beyotime Institute of Biotechnology) at 4°C. Subsequently the
membrane was washed with TBST, exposed to the corresponding
HRP-conjugated secondary antibodies for 1 h at room temperature,
and detected by enhanced chemiluminescence (EMD Millipore).
Flow cytometric analysis
Apoptosis was investigated using an Annexin
V-fluorescein isothiocyanate (FITC) apoptosis kit according to the
manufacturer's protocol (BD Biosciences, San Jose, CA, USA).
Briefly, following CM treatment (0, 0.5, 1, 2 µmol/l for PRMI8226
cells, and 0, 2, 4, 8 µmol/l for U266 cells) for 48 h, the cells
were harvested and washed with ice-cold PBS and adjusted to
1×106 cells/ml with binding buffer. Subsequently, the
cells were incubated with 5 µl FITC-labeled Annexin V and 5 µl
propidium iodide (PI) at room temperature in the dark for 15 min.
Finally, the stained cells were analyzed using a flow cytometer (BD
Biosciences). The storing and processing of data was performed with
FlowJo software v10.0.7 (Tree Star, Inc., Ashland, OR, USA).
Cell cycle analysis was also performed by flow
cytometry. Following CM treatment (0, 0.5, 1, 2 µmol/l for PRMI8226
cells, and 0, 2, 4, 8 µmol/l for U266 cells) for 48 h, the cells
were washed with ice-cold PBS and subsequently fixed in 70%
ice-cold ethanol overnight. Subsequently, the cells were harvested
and incubated in PBS with 100 mg/ml PI and 100 µg/ml RNase A for 30
min (Beyotime Institute of Biotechnology). The PI-stained cells
were subjected to cell cycle profiling analysis using a flow
cytometer (BD Biosciences). Finally, the cell-cycle distribution
was analyzed using ModFit LT software (v3.1, Verity Software House,
Inc., Topsham, ME, USA).
Morphological analysis of
apoptosis
To investigate the effect of CM on cell morphology,
myeloma cells were treated in the presence or absence of CM (2
µmol/l for PRMI8226 cells, and 8 µmol/l for U266 cells) for 48 h.
Cytospin slides were prepared and subsequently stained with
Giemsa-Wright staining for 20 min at room temperature. Cell
morphology was observed under a light microscope (×400
magnification). Characteristics of apoptosis including nuclear
fragmentation, chromatin condensation and apoptotic bodies were
observed.
Statistical analysis
All data are presented as the means ± standard
deviation for ≥3 separate experiments. All analyses were performed
using SPSS. Inter-group comparison was performed using 2-sided
Student's t-test (2 groups) or one-way analysis of variance (≥3
groups), followed by a Student-Newman-Keuls test for subsequent
multiple comparisons between groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
CM suppresses the viability of myeloma
cell lines and primary myeloma cells
The effect of CM on myeloma cell lines (RPMI8226 and
U266) and primary myeloma cells from 4 patients with MM was
investigated using the CCK-8 assay. The 50% inhibitory
concentration (IC50) was measured using different
concentrations (0.25–8 µmol/l for 24–72 h). CM inhibited the
viability of the MM cell lines in a time- and dose-dependent manner
(Fig. 1B and C). The IC50
values at 24, 48 and 72 h were significantly different from each
other in RPMI8226 and U266 cells; the IC50 values in the
RPMI8226 cells were 9.09±0.58, 1.19±0.36 and 0.77±0.21 µmol/l
(P<0.001), respectively, whereas the values for the U266 cells
were 16.16±2.51, 8.06±1.02 and 2.86±0.58 µmol/l (P<0.001),
respectively. The sensitivity of primary myeloma cells to CM was
lower compared with that of the myeloma cell lines, with 48 h
IC50 values between 5.08 and 14.21 µmol/l (patient 1 was
not sensitive to CM and the IC50 could not be
calculated; IC50 for patient 2 was 5.08±0.47 µmol/l;
IC50 for patient 3 was 13.96±0.64 µmol/l;
IC50 for patient 4 was 14.21±1.03 µmol/l) (Fig. 1D).
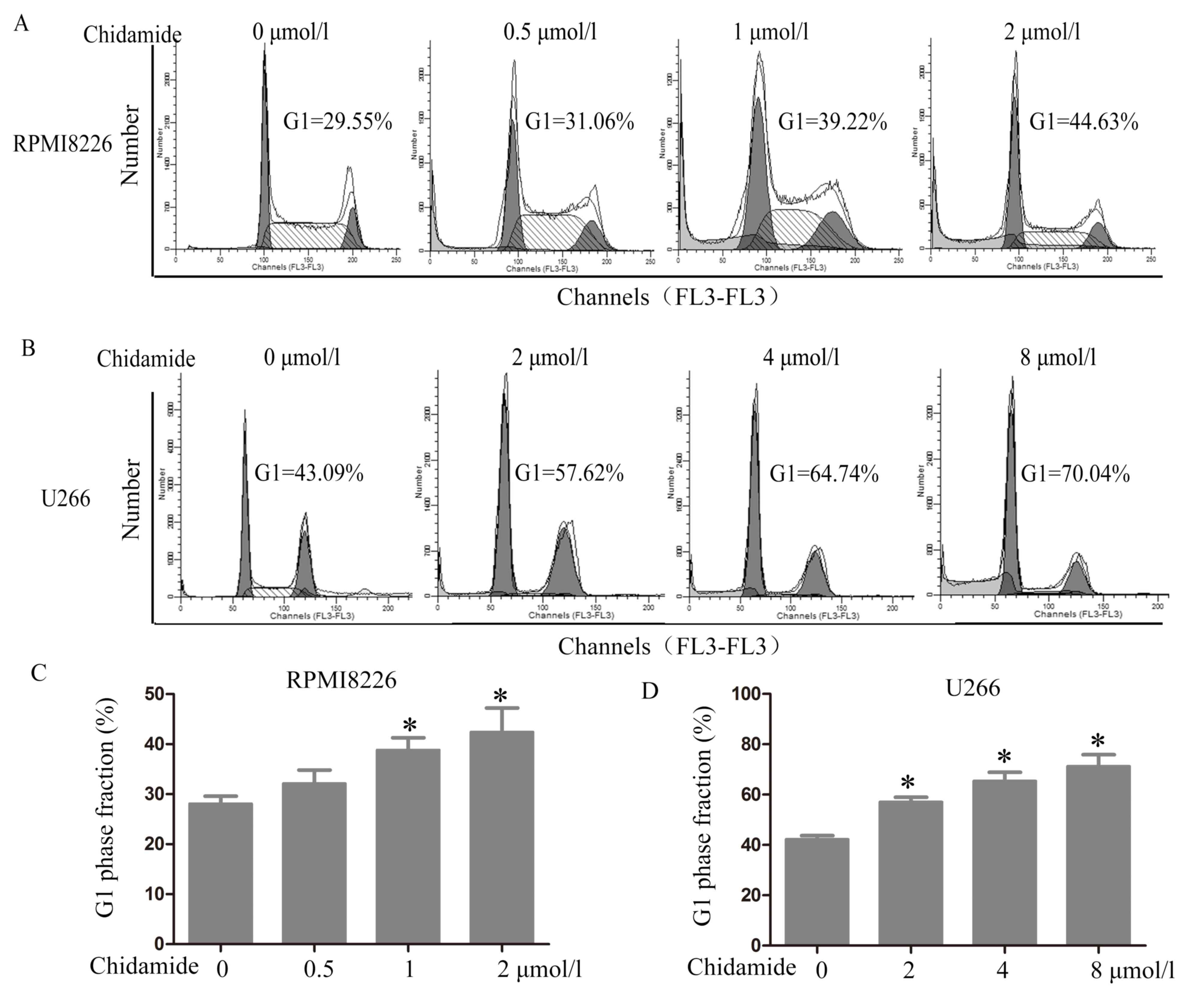
CM induces G0/G1
phase arrest in myeloma cells
To investigate whether CM alters the cell cycle, the
DNA content of the cells was examined (PI staining) using flow
cytometry. The cell cycle analysis demonstrated that CM induced
G0/G1 phase arrest in the RPMI8226 and U266
cells (Fig. 2). The cell number
within the G0/G1 phase increased from
28.12±1.50 to 42.42±4.80% in the RPMI8226 cells following exposure
to 2 µmol/l CM for 48 h compared with no CM treatment (P=0.008).
The number of cells within the G0/G1 phase
increased from 42.31±1.43 to 71.25±4.65% in the U266 cells
following exposure to 8 µmol/l CM for 48 h compared with no CM
treatment (P<0.001).
CM induces apoptosis in MM cell
lines
To demonstrate whether the inhibition of cell
viability was due to the induction of cell apoptosis, flow
cytometry with Annexin V positive staining (the lower right and
upper right quadrants represent cells undergoing apoptosis) was
used. Following treatment for 48 h, CM significantly promoted
apoptosis in a dose-dependent manner in the RPMI8226 and U266
cells. Compared with untreated cells, the proportion of apoptotic
cells increased from 7.12±2.50 to 31.51±4.52% in the RPMI8226 cells
following exposure to 2 µmol/l CM for 48 h (P<0.001; Fig. 3A, C) and from 6.82±2.41 to
18.21±3.51% in the U266 cells following exposure to 8 µmol/l CM for
48 h (P=0.004; Fig. 3B, D).
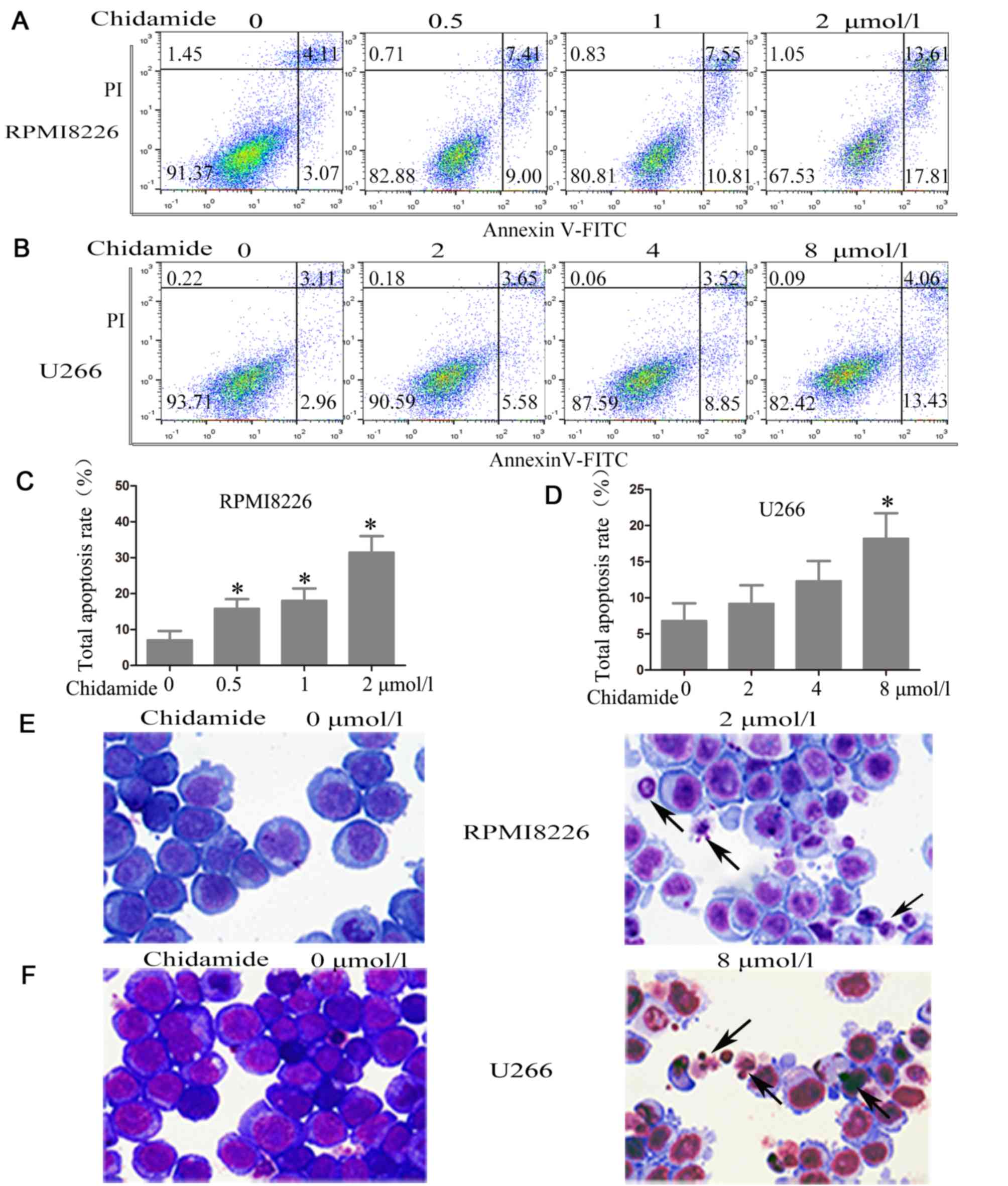 | Figure 3.Chidamide induces apoptosis in
myeloma cells. (A) RPMI8226 cells were treated with (0, 0.5, 1, 2
µmol/l) chidamide for 48 h and (B) U266 cells were treated with (0,
2, 4, 8 µmol/l) chidamide for 48 h, then the percentages of
apoptotic cells were assessed by Annexin V-FITC/PI flow cytometry
(typical results from one experiment are presented). The total
apoptosis rate of (C) RPMI8226 cells and (D) U266 cells following
chidamide treatment for 48 h. The quantitative data were pooled
from three independent experiments. *P<0.05 vs. no treatment.
Giemsa-Wright staining of (E) RPMI8226 and (F) U266 cells was
performed following treatment in the presence or absence of
chidamide (2 µmol/l for RPMI8226 cells and 8 µmol/l for U266 cells)
for 48 h (×400 magnification). The arrows indicate typical
apoptotic cells including nuclear fragmentation, chromatin
condensation and apoptotic bodies. FITC, fluorescein
isothiocyanate; PI, propidium iodide. |
To confirm the apoptotic effect of CM, morphological
observation was performed using Giemsa-Wright staining. The
morphology of myeloma cells treated in the presence or absence of
CM (2 µmol/l for RPMI8226 cells and 8 µmol/l for U266 cells) for 48
h was examined. Morphological changes, including nuclear
fragmentation, chromatin condensation and apoptotic bodies, which
are characteristics of apoptosis, were observed (Fig. 3E and F).
CM decreases the expression of HDACs
and increases the acetylation levels of histones H3 and H4
To investigate the possible mechanisms of CM, the
expression of class I HDACs (HDAC1, HDAC2 and HDAC3) in MM cells
was evaluated by western blot analysis. CM treatment markedly
suppressed the expression of HDAC1, HDAC2 and HDAC3 in a
dose-dependent manner in RPMI8226 (Fig.
4A) and U266 cells (Fig. 4B)
following treatment for 48 h. In addition, the acetylation of
histones H3 and H4 was also markedly increased following exposure
to CM for 48 h for the RPMI8226 and U266 cells. These results
demonstrate that CM can inhibit class I HDAC enzyme activity, and
subsequently induce the acetylation of histones H3 and H4 in
myeloma cells.
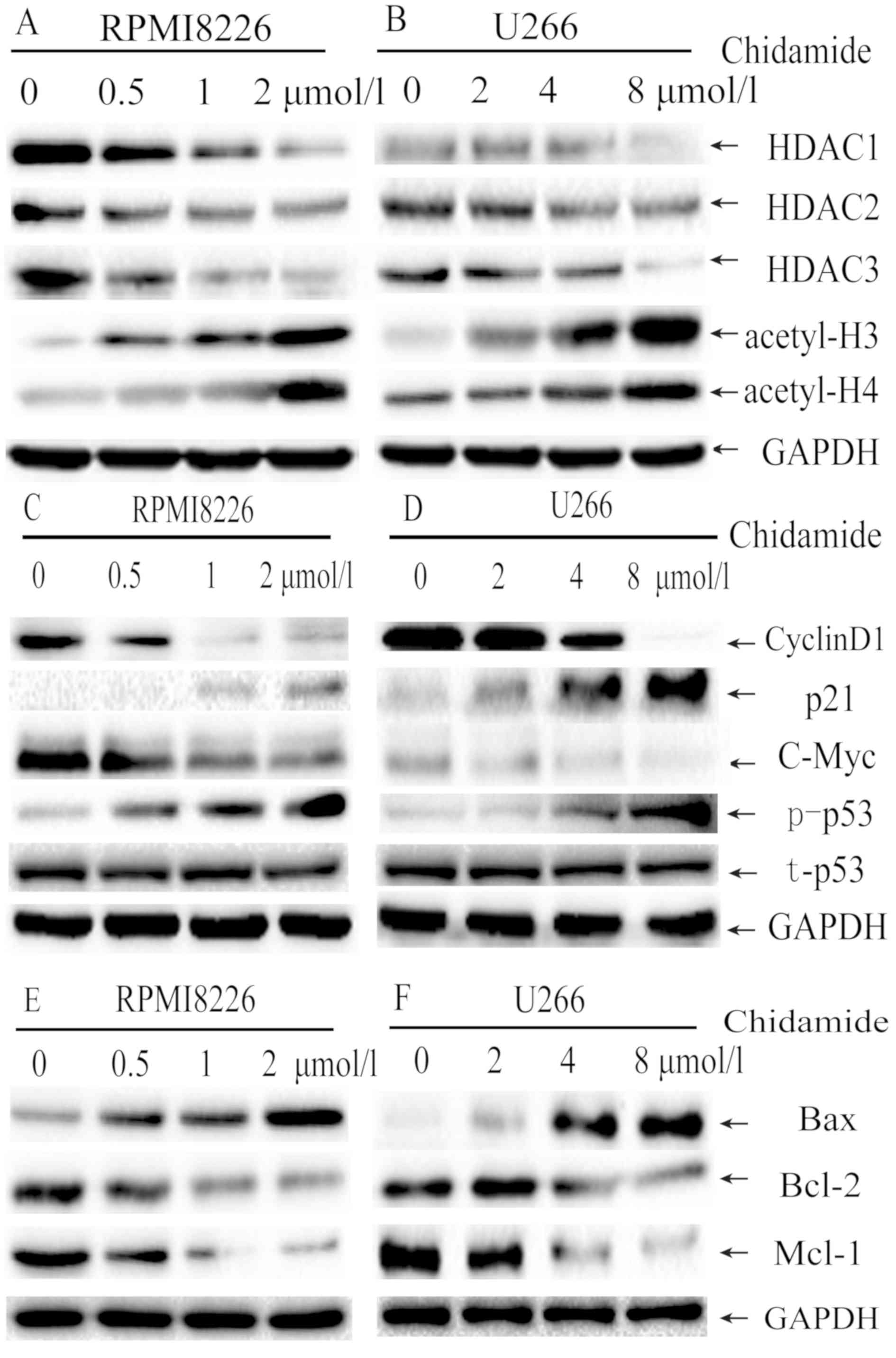 | Figure 4.Molecular mechanisms of
chidamide-mediated cell cycle arrest and apoptosis induction in
myeloma cell lines. (A) RPMI8226 cells were treated with 0, 0.5, 1
or 2 µmol/l chidamide for 48 h and (B) U266 cells were treated with
0, 2, 4 or 8 µmol/l chidamide for 48 h, and HDACs expression was
assessed using western blot analysis. The expression of cyclin D1,
p53, p-p53, c-myc and p21 in (C) RPMI8226 and (D) U266 cells,
whereas Bax, Bcl-2 and mcl-1 in (E) RPMI8226 and (F) U266 cells
following chidamide treatment were assessed using western blot
analysis. GAPDH served as the internal control in all experiments.
HDAC, histone deacetylase; Bcl-2, B-cell lymphoma 2; Bax, apoptosis
regulator Bax; mcl-1, myeloid cell leukemia-1; p-, phosphorylated;
t-, total; p21, cyclin-dependent kinase inhibitor 1; p53, cellular
tumor antigen p53. |
CM downregulates the expression of
Bcl-2, mcl-1, c-myc and cyclin D1, and upregulates the expression
of p-p53, Bax and p21
To understand the molecular mechanisms involved in
CM-induced cell cycle arrest and apoptosis, the protein expression
of Bcl-2, Bax, mcl-1, c-myc, p53, p-p53, cyclin D1 and p21 was
investigated. As expected, CM treatment markedly deceased cyclin D1
and c-myc expression, but increased p21 and p-p53 expression in a
dose-dependent manner, which markedly induced
G0/G1 arrest (Fig.
4C and D). The total p53 expression was not changed. CM
treatment increased the expression of Bax, but decreased the
expression of Bcl-2 and mcl-1 (Fig. 4E
and F) in a dose-dependent manner, which may induce cell
apoptosis by upregulating the Bax/Bcl-2 ratio.
CM promotes apoptosis in a
caspase-dependent manner
The role of members of the caspase family in
CM-induced apoptosis of MM cells was investigated using western
blot analysis. The treatment of RPMI8226 and U266 cells with CM for
48 h induced the cleavage of caspase-8, caspase-9, caspase-3 and
PARP in a dose-dependent manner, which is a hallmark of apoptosis
(Fig. 5). These results indicate
that CM induces apoptosis in a caspase-dependent manner.
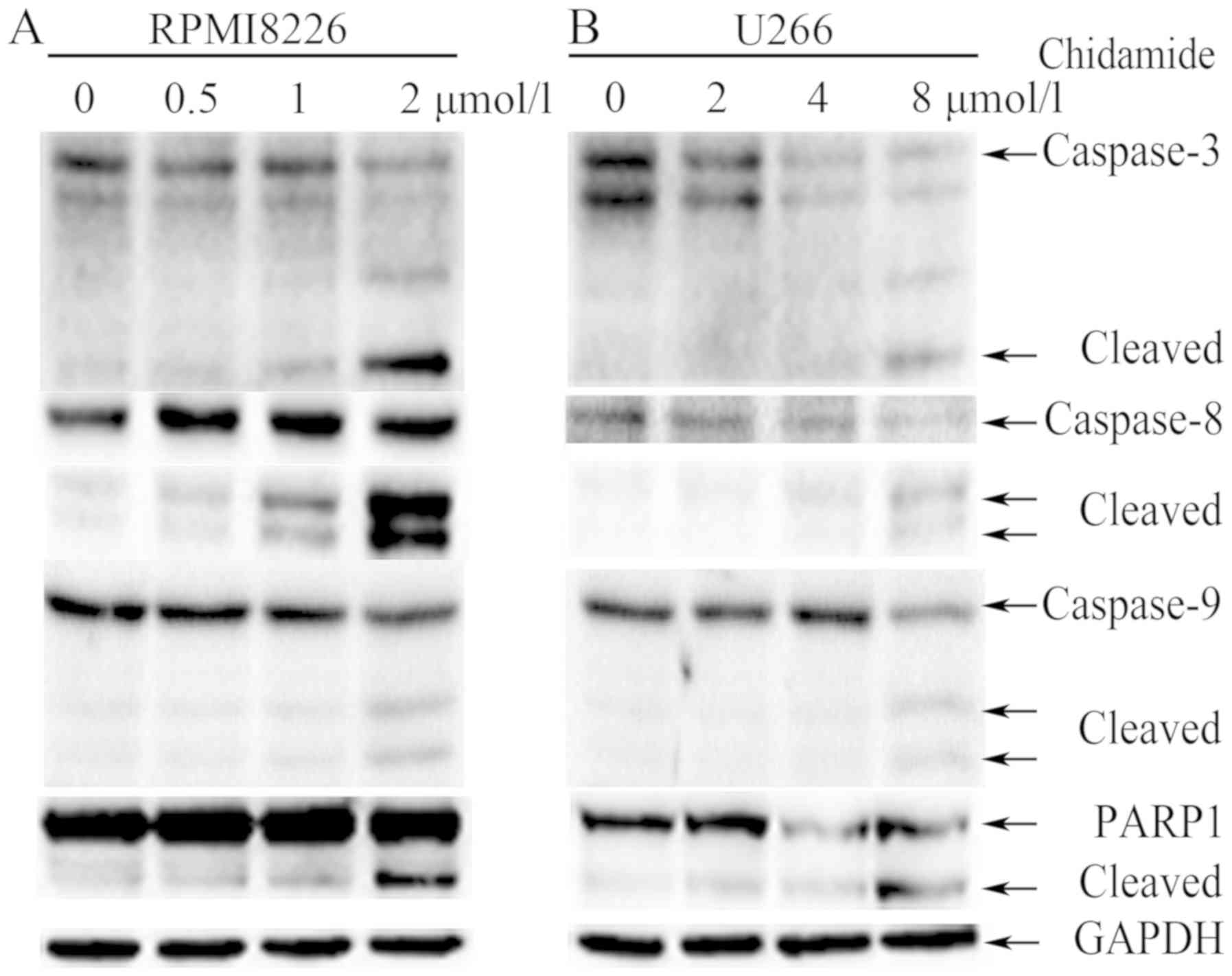 | Figure 5.Chidamide induces apoptosis in a
caspase-dependent manner in myeloma cells. (A) RPMI8226 cells were
treated with (0, 0.5, 1, 2 µmol/l) chidamide for 48 h and (B) U266
cells were treated with (0, 2, 4, 8 µmol/l) chidamide for 48 h, and
then the expression of the cleaved and full-length caspase-8,
caspase-9, caspase-3 and PARP was determined using western blot
analysis. GAPDH served as the internal control. PARP, poly ADP
ribose polymerase. |
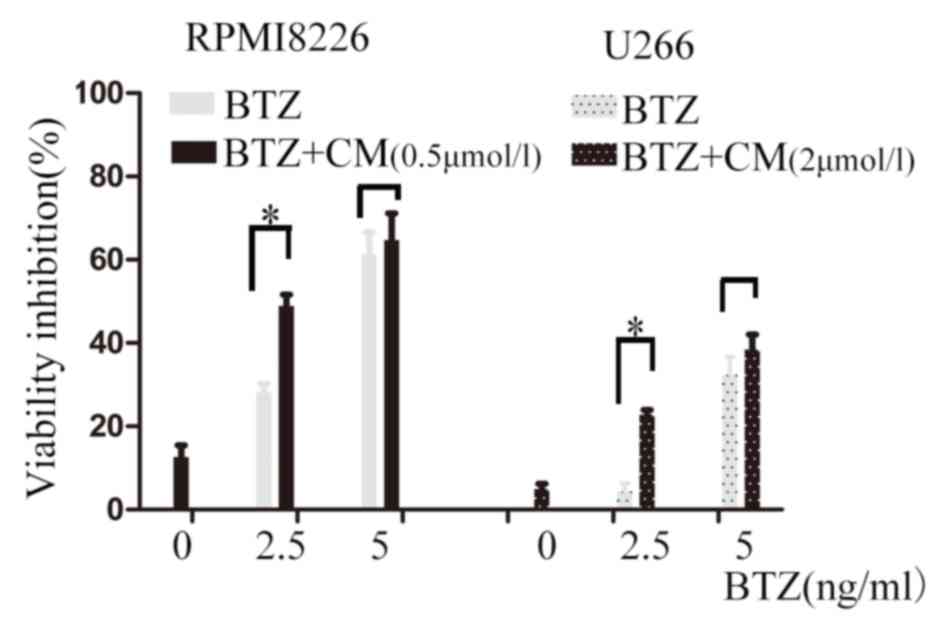
CM sensitizes MM cells to BTZ
CM was investigated further to examine whether it
enhanced the cytotoxic effect of BTZ on MM cells. RPMI8226 and U266
cells were exposed to CM alone (0.5 µmol/l for RPMI8226 cells and 2
µmol/l for U266 cells) and in combination with BTZ (2.5 or 5 ng/ml)
for 24 h. A low dose of CM slightly suppressed cell viability when
used alone; the viability inhibition rate was 12.60±2.82% in the
RPMI8226 cells and 4.92±1.28% in U266. In RPMI8226 cells, compared
with 2.5 ng/ml BTZ treatment alone, the inhibition rate of BTZ
co-treatment with 0.5 µmol/l CM for 24 h increased from 28.11±2.12
to 48.84±2.82% (P<0.001). In U266 cells, compared with treatment
with 2.5 ng/ml BTZ alone, the inhibition rate of BTZ co-treatment
with 2 µmol/l CM for 24 h increased from 4.35±1.94 to 22.83±1.10%
(P<0.001; Fig. 6). The
combination index (CI) values were calculated using the
Chou-Talalay equation (23). The CIs
of the low-dose combination were 0.947 and 0.385 in the RPMI8226
and U266 cells, respectively. All of the CIs were <1, which
suggests a synergistic combination of CM and BTZ at a low dose.
Discussion
In the present study, CM induced an anti-myeloma
effect by decreasing the expression of class I HDACs and increasing
the acetylation levels of histones H3 and H4. In addition, CM
treatment induced G0/G1 arrest by decreasing
the expression of cyclin D1 and c-myc, and increasing the
expression of p-p53 and p21. Furthermore, it promoted cell
apoptosis by upregulating the Bax/Bcl-2 ratio in a
caspase-dependent manner.
The therapeutic potential of HDACIs in cancer
treatment has attracted increased attention and interest. Some well
known mechanisms of HDACIs include the regulation of the cell
cycle, apoptosis, metastasis, DNA-damage responses (DDRs),
autophagy, angiogenesis and other cellular processes (24). CM induces anti-tumor effects through
various mechanisms, depending on the type of cancer and its dose.
These mechanisms include: i) Cell cycle arrest, CM arrests tumor
cells at the G0/G1 phase (12,16,25); ii)
apoptosis induction, CM induces apoptosis by regulating the balance
of pro- and anti-apoptotic genes, activating intrinsic apoptotic
pathways (25); iii) suppression of
cellular signaling pathways, CM inhibits the Janus kinase/STAT,
PI3K/AKT and mitogen-activated protein kinase/JNK signaling
pathways (16,19,26,27); iv)
reactive oxygen species generation and induction of DNA damage
(10,25); v) energy metabolism modulation, CM
suppresses mitochondrial aerobic respiration by downregulation of
mcl-1 (20); vi) activation of
cellular antitumor immunity mediated by NK cells and
antigen-specific cluster of differentiation 8-positive cytotoxic T
lymphocytes (11); vii) reversion of
transforming growth factor-β-induced epithelial-mesenchymal
transition in tumor cells (28); and
viii) upregulation of the tumor suppressor genes Spi-1
proto-oncogene and Krüppel-like factor-4 (29). In the present study, it was
demonstrated that CM serves a role in suppressing the viability of
MM cells. The results suggested that CM was able to inhibit the
expression of class I HDACs and further upregulated the acetylation
of histones H3 and H4, which confirmed the selective HDACI role of
CM in MM. In the present study, the molecular mechanisms underlying
cell cycle arrest and apoptosis induction were investigated.
Aberrant cell cycle is considered a hallmark of a
number of cancer types. The G1/S checkpoint, which
controls the cell cycle transition from G0/G1
to S-phase, is frequently lost in cancer cells. The 2 cell cycle
kinases, cyclin D-cyclin-dependent kinase (CDK)4/6 and cyclin
E-CDK2 complexes, are critical in controlling this checkpoint
(30). Cyclin D1 is a key cell cycle
protein belonging to the G1 phase family. Following
activation by extrinsic or intrinsic mitotic signals, cyclin D1
cooperates with CDK4/6 to drive the cell cycle progression from
G0/G1 to S phase by phosphorylating
retinoblastoma (RB) and releasing E2 factor (E2F) (31). In addition, cyclin D1 overexpression
is significantly associated with tumor invasiveness and metastasis
(32). p21 protein, a CDK inhibitor,
can regulate the cell cycle transition from
G0/G1 to S-phase in response to a number of
stimuli (33). p21 binds to and
inhibits the activity of cyclin E/CDK2, which decreases the
CDK2-dependent phosphorylation of RB and E2F1-dependent gene
transcription (34). p21 is
primarily regulated by the DDR kinases ataxia-telangiectasia
mutated (ATM), p53 and checkpoint kinase 2 (Chk2) (35). At this checkpoint, p53 can be
activated by Chk2 and ATM, thus causing inhibition of cyclin
E/CDK2. The c-myc protein, a transcription factor, can drive cell
proliferation by the upregulation of CDKs, cyclins and E2F
transcription factors, as well as the downregulation of p15, p16,
p21 and p27 (36). Previous studies
revealed that almost all HDACIs, including TF2357, valproate,
LBH589, NVP-LAQ824, sodium butyrate and suberoylanilide hydroxamic
acid (SAHA), induce G0/G1 arrest due to the
upregulation of CDK inhibitor by p53-dependent and -independent
mechanisms in MM cells (37–42). Similarly, it has been reported that
treatment with CM induces G0/G1 cell cycle
arrest in a number of cancer types. In NK/T-cell lymphoma cell
lines, CM decreases cyclin E expression, increases p21 expression
and activates the DDR cell cycle checkpoint pathway
(ATM-Chk2-p53-p21 pathway) (16). In
colon cancer cells, CM increases the level of p53, p-p53 and p21,
but suppresses CDK4 (43). In
myelodysplastic syndromes, CM downregulates CDK2 and upregulates
p-p53 and p21 protein expression (12). The data from the present study
revealed that CM promotes G0/G1 cell cycle
arrest in MM cells. p53 is mutated in the myeloma cell lines
RPMI8226 and U266 (44). Although
p53 expression was not changed following CM treatment, the
expression of p-p53 and its downstream target p21 were increased,
whereas the expression of c-myc and cyclin D1 were decreased,
suggesting that activation of the DDR cell cycle checkpoint pathway
may contribute, at least in part, to CM-induced
G0/G1 arrest in MM.
Induction of apoptosis has been revealed to be a
promising strategy for the development of novel anticancer agents.
HDACIs can induce the intrinsic and extrinsic apoptotic pathways
(45). HDACIs upregulate the Bcl-2
family proteins (Bax, Noxa, BH3-interacting domain death agonist,
BCL-2-like protein 11, Bcl-2 homologous antagonist killer and
Bcl-2-binding component 3), but downregulate anti-apoptotic Bcl-2
family proteins (mcl-1, Bcl-2 and Bcl-xl), thus activating the
intrinsic pathway. Previous studies investigating the treatment of
HDACIs (KD5170, depsipeptide, ITF2357, SAHA and LBH589) in MM cells
found increased mitochondrial permeability and cytosolic release of
cytochrome c and second mitochondria-derived activator of
caspases following activation of the intrinsic pathway (37,39,42,46,47). On
the other hand, HDACIs increase the expression of tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL) receptors and
their susceptibility to TRAIL-induced extrinsic apoptosis, as seen
in MM cells following LBH589, valproate and SAHA treatment
(39,42,48).
Similarly, it has been reported that treatment with CM induces the
intrinsic pathway in a number of cancer types. In the NK/T-cell
lymphoma cell lines, CM downregulates Bcl-2 and induces the
cleavage of PARP, suggesting a mitochondria-mediated
caspase-dependent apoptotic pathway (16). In pancreatic cancer, CM upregulates
the Bax/Bcl-2 ratio, thus suppressing cellular proliferation by
promoting mitochondrial pathway-dependent cell apoptosis (25). In leukemia cell lines, CM increases
Bcl-2 family protein expression and promotes the generation of
reactive oxygen species, mitochondrial dysfunction and cytochrome
c release, inducing caspase-dependent apoptosis (13,27,49). The
data from the present study revealed that CM induces apoptosis in
MM cells in a time- and dose-dependent manner. CM activates
caspase-3, caspase-8, caspase-9 and PARP, and increases the
Bax/Bcl-2 expression ratio, promoting mitochondrial
pathway-dependent cell apoptosis in MM cells.
The present study has several limitations. First,
the most common types of inhibitors of apoptosis include the Bcl-2
family and inhibitor of apoptosis proteins (IAP) family. As
apoptosis was induced by CM, only the effect of CM on Bcl-2 family
(downregulation of mcl-1 and Bcl-2) was investigated, but whether
CM can decrease IAP expression will be explored in future
experiments. Secondly, it was revealed that CM treatment increased
the sensitivity against BTZ in myeloma cells, however, the possible
mechanisms involved were not investigated, requiring further study.
Thirdly, the anti-myeloma effect of CM was examined only in
vitro, therefore the effect should also be investigated in
vivo.
In conclusion, it was demonstrated that CM induces
an anti-myeloma effect. CM promotes G0/G1
arrest and apoptosis in myeloma cells in a caspase-dependent
manner. Future studies will focus on the in vivo efficacy of
this treatment and define the optimal combination regimens. The
present study provides evidences for the clinical administration of
CM in MM.
Acknowledgements
Not applicable.
Funding
This study was supported by the Zhejiang Provincial
Key Innovation Team (grant no. 2011R50015), the National Natural
Science Foundation of China (grant no. 81572920), the National
Basic Research Program of China (grant no. 2013CB911303) and the
Natural Science Foundation of Zhejiang Province of China (grant no.
LY15H160038).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XGY performed the research and wrote the manuscript.
YRH, TY and HWJ performed the research. YX performed the
statistical analysis. XYZ designed and supervised the research
project. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study has been approved by the Ethics Committee
of The Second Affiliated Hospital, Zhejiang University School of
Medicine (Hangzhou, China), and written informed consent was
obtained from all participants.
Patient consent for publication
The study participants provided consent for the data
to be published.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mithraprabhu S, Kalff A, Chow A, Khong T
and Spencer A: Dysregulated Class I histone deacetylases are
indicators of poor prognosis in multiple myeloma. Epigenetics.
9:1511–1520. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moreaux J, Reme T, Leonard W, Veyrune JL,
Requirand G, Goldschmidt H, Hose D and Klein B: Gene
expression-based prediction of myeloma cell sensitivity to histone
deacetylase inhibitors. Br J Cancer. 109:676–685. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dimopoulos M, Siegel DS, Lonial S, Qi J,
Hajek R, Facon T, Rosinol L, Williams C, Blacklock H, Goldschmidt
H, et al: Vorinostat or placebo in combination with bortezomib in
patients with multiple myeloma (VANTAGE 088): A multicentre,
randomised, double-blind study. Lancet Oncol. 14:1129–1140. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
San-Miguel JF, Hungria VT, Yoon SS, Beksac
M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Günther A, Nakorn
TN, Siritanaratkul N, et al: Panobinostat plus bortezomib and
dexamethasone versus placebo plus bortezomib and dexamethasone in
patients with relapsed or relapsed and refractory multiple myeloma:
A multicentre, randomised, double-blind phase 3 trial. Lancet
Oncol. 15:1195–1206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Raje NS, Bensinger W, Cole CE, Lonial S,
Jagannath S, Arce-Lara CE, Valent J Rosko AE, Harb WA, Sandhu PG,
et al: Ricolinostat (ACY-1215), the first selective HDAC6
inhibitor, combines safely with pomalidomide and dexamethasone and
shows promosing early results in relapsed- and- refractory myeloma
(ACE-MM-102 Study). Blood. 126:42282015.
|
|
9
|
Bradner JE, West N, Grachan ML, Greenberg
EF, Haggarty SJ, Warnow T and Mazitschek R: Chemical phylogenetics
of histone deacetylases. Nat Chem Biol. 6:238–243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pan DS, Yang QJ, Fu X, Shan S, Zhu JZ,
Zhang K, Li ZB, Ning Q and Lu XP: Discovery of an orally active
subtype-selective HDAC inhibitor, chidamide, as an epigenetic
modulator for cancer treatment. Med Chem Commun. 5:1789–1796. 2014.
View Article : Google Scholar
|
|
11
|
Ning ZQ, Li ZB, Newman MJ, Shan S, Wang
XH, Pan DS, Zhang J, Dong M, Du X and Lu XP: Chidamide
(CS055/HBI-8000): A new histone deacetylase inhibitor of the
benzamide class with antitumor activity and the ability to enhance
immune cell-mediated tumor cell cytotoxicity. Cancer Chemoth Pharm.
69:901–909. 2012. View Article : Google Scholar
|
|
12
|
Liu Z, Ding K, Li L, Liu H, Wang Y, Liu C
and Fu R: A novel histone deacetylase inhibitor Chidamide induces
G0/G1 arrest and apoptosis in myelodysplastic syndromes. Biomed
Pharmacother. 83:1032–1037. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong K, Xie J, Yi H and Li W: CS055
(Chidamide/HBI-8000), a novel histone deacetylase inhibitor,
induces G1 arrest, ROS-dependent apoptosis and differentiation in
human leukaemia cells. Biochem J. 443:735–746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Yan X, Guo W, Huang X, Huang J, Yu
M, Ma Z, Xu Y, Huang S, Li C, et al: Chidamide in FLT3-ITD positive
acute myeloid leukemia and the synergistic effect in combination
with cytarabine. Biomed Pharmacother. 90:699–704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi P, Zhang L, Chen K, Jiang Z, Deng M,
Zha J, Guo X, Li P and Xu B: Low-dose decitabine enhances
chidamide-induced apoptosis in adult acute lymphoblast leukemia,
especially for p16-deleted patients through DNA damage.
Pharmacogenomics. 18:1259–1270. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou J, Zhang C, Sui X, Cao S, Tang F, Sun
S, Wang S and Chen B: Histone deacetylase inhibitor chidamide
induces growth inhibition and apoptosis in NK/T lymphoma cells
through ATM-Chk2-p53-p21 signalling pathway. Invest New Drugs.
36:571–580. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou Y, Pan DS, Shan S, Zhu JZ, Zhang K,
Yue XP, Nie LP, Wan J, Lu XP, Zhang W and Ning ZQ: Non-toxic dose
chidamide synergistically enhances platinum-induced DNA damage
responses and apoptosis in non-small-cell lung cancer cells. Biomed
Pharmacother. 68:483–491. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang H, Guo Y, Fu M, Liang X, Zhang X,
Wang R, Lin C and Qian H: Antitumor activity of Chidamide in
hepatocellular carcinoma cell lines. Mol Med Rep. 5:1503–1508.
2012.PubMed/NCBI
|
|
19
|
Liu L, Chen B, Qin S, Li S, He X, Qiu S,
Zhao W and Zhao H: A novel histone deacetylase inhibitor Chidamide
induces apoptosis of human colon cancer cells. Biochem Biophys Res
Commun. 392:190–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He M, Qiao Z, Wang Y, Kuai Q, Li C, Wang
Y, Jiang X, Wang X, Li W, He M, et al: Chidamide inhibits aerobic
metabolism to induce pancreatic cancer cell growth arrest by
promoting Mcl-1 degradation. PLoS One. 11:e01668962016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gu R, Liu T, Zhu X, Gan H, Wu Z, Li J,
Zheng Y, Dou G and Meng Z: Development and validation of a
sensitive HPLC-MS/MS method for determination of chidamide
(epidaza), a new benzamide class of selective histone deacetylase
inhibitor, in human plasma and its clinical application. J
Chromatogr B Analyt Technol Biomed Life Sci. 1000:181–186. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi Y, Jia B, Xu W, Li W, Liu T, Liu P,
Zhao W, Zhang H, Sun X, Yang H, et al: Chidamide in relapsed or
refractory peripheral T cell lymphoma: A multicenter real-world
study in China. J Hematol Oncol. 10:692017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bose P, Dai Y and Grant S: Histone
deacetylase inhibitor (HDACI) mechanisms of action: Emerging
insights. Pharmacol Ther. 143:323–336. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao B and He T: Chidamide, a histone
deacetylase inhibitor, functions as a tumor inhibitor by modulating
the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol Rep.
33:304–310. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Chen K, Zhou Y, Xiao Y, Deng M,
Jiang Z, Ye W, Wang X, Wei X, Li J, et al: A new strategy to target
acute myeloid leukemia stem and progenitor cells using chidamide, a
histone deacetylase inhibitor. Curr Cancer Drug Targets.
15:493–503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao S, Guo J, Zhao Y, Fei C, Zheng Q, Li
X and Chang C: Chidamide, a novel histone deacetylase inhibitor,
inhibits the viability of MDS and AML cells by suppressing
JAK2/STAT3 signaling. Am J Transl Res. 8:3169–3178. 2016.PubMed/NCBI
|
|
28
|
Lin SH, Wang BY, Lin CH, Chien PJ, Wu YF,
Ko JL and Chen JJ: Chidamide alleviates TGF-β-induced
epithelial-mesenchymal transition in lung cancer cell lines. Mol
Biol Rep. 43:687–695. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang T, Wang F, Hu L, Cheng X, Zheng Y,
Liu T and Jia Y: Chidamide and decitabine can synergistically
induce apoptosis of Hodgkin lymphoma cells by up-regulating the
expression of PU.1 and KLF4. Oncotarget. 8:77586–77594. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fu M, Wang C, Li Z, Sakamaki T and Pestell
RG: Minireview: Cyclin D1: Normal and abnormal functions.
Endocrinology. 145:5439–5447. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Imoto M, Doki Y, Jiang W, Han EK and
Weinstein IB: Effects of cyclin D1 overexpression on G1
progression-related events. Exp Cell Res. 236:173–180. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hirao A, Kong YY, Matsuoka S, Wakeham A,
Ruland J, Yoshida H, Liu D, Elledge SJ and Mak TW: DNA damage-
induced activation of p53 by the checkpoint kinase Chk2. Science.
287:1824–1827. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bretones G, Delgado MD and León J: Myc and
cell cycle control. Biochim Biophys Acta. 1849:506–516. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Golay J, Cuppini L, Leoni F, Micò C,
Barbui V, Domenghini M, Lombardi L, Neri A, Barbui AM, Salvi A, et
al: The histone deacetylase inhibitor ITF2357 has anti-leukemic
activity in vitro and in vivo and inhibits IL-6 and VEGF production
by stromal cells. Leukemia. 21:1892–1900. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kaiser M, Zavrski I, Sterz J, Jakob C,
Fleissner C, Kloetzel PM, Sezer O and Heider U: The effects of the
histone deacetylase inhibitor valproic acid on cell cycle, growth
suppression and apoptosis in multiple myeloma. Haematologica.
91:248–251. 2006.PubMed/NCBI
|
|
39
|
Maiso P, Carvajal-Vergara X, Ocio EM,
López-Pérez R, Mateo G, Gutiérrez N, Atadja P, Pandiella A and San
Miguel JF: The histone deacetylase inhibitor LBH589 is a potent
antimyeloma agent that overcomes drug resistance. Cancer Res.
66:5781–5789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Catley L, Weisberg E, Tai YT, Atadja P,
Remiszewski S, Hideshima T, Mitsiades N, Shringarpure R, LeBlanc R,
Chauhan D, et al: NVP-LAQ824 is a potent novel histone deacetylase
inhibitor with significant activity against multiple myeloma.
Blood. 102:2615–2622. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lavelle D, Chen YH, Hankewych M and
DeSimone J: Histone deacetylase inhibitors increase p21(WAF1) and
induce apoptosis of human myeloma cell lines independent of
decreased IL-6 receptor expression. Am J Hematol. 68:170–178. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mitsiades N, Mitsiades CS, Richardson PG,
McMullan C, Poulaki V, Fanourakis G, Schlossman R, Chauhan D,
Munshi NC, Hideshima T, et al: Molecular sequelae of histone
deacetylase inhibition in human malignant B cells. Blood.
101:4055–4062. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu L, Qiu S, Liu Y, Liu Z, Zheng Y, Su X,
Chen B and Chen H: Chidamide and 5-flurouracil show a synergistic
antitumor effect on human colon cancer xenografts in nude mice.
Neoplasma. 63:193–200. 2016.PubMed/NCBI
|
|
44
|
Mazars GR, Portier M, Zhang XG, Jourdan M,
Bataille R, Theillet C and Klein B: Mutations of the p53 gene in
human myeloma cell lines. Oncogene. 7:1015–1018. 1992.PubMed/NCBI
|
|
45
|
Miller CP, Singh MM, Rivera-Del Valle N,
Manton CA and Chandra J: Therapeutic strategies to enhance the
anticancer efficacy of histone deacetylase inhibitors. J Biomed
Biotechnol. 2011:5142612011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Feng R, Ma H, Hassig CA, Payne JE, Smith
ND, Mapara MY, Hager JH and Lentzsch S: KD5170, a novel
mercaptoketone-based histone deacetylase inhibitor, exerts
antimyeloma effects by DNA damage and mitochondrial signaling. Mol
Cancer Ther. 7:1494–1505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Khan SB, Maududi T, Barton K, Ayers J and
Alkan S: Analysis of histone deacetylase inhibitor, depsipeptide
(FR901228), effect on multiple myeloma. Br J Haematol. 125:156–161.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schwartz C, Palissot V, Aouali N, Wack S,
Brons NH, Leners B, Bosseler M and Berchem G: Valproic acid induces
non-apoptotic cell death mechanisms in multiple myeloma cell lines.
Int J Oncol. 30:573–582. 2007.PubMed/NCBI
|
|
49
|
Li Y, Chen K, Zhou Y, Xiao Y, Deng M,
Jiang Z, Ye W, Wang X, Wei X, Li J, et al: A new strategy to target
acute myeloid leukemia stem and progenitor cells using chidamide, a
histone deacetylase inhibitor. Curr Cancer Drug Targets.
15:493–503. 2015. View Article : Google Scholar : PubMed/NCBI
|