Introduction
Intrahepatic cholangiocarcinoma (ICC) is a form of
liver cancer typically diagnosed at advanced stages and with poor
prognosis; in the United States its incidence has increased to
3,000 cases annually (1). Despite
the fact that surgery is the preferred treatment for early ICC,
only ~35% of patients have early stage disease that may be treated
by surgical resection (2,3). For patients with advanced stage or
unresectable cholangiocarcinoma, the available systemic therapies
are of limited effectiveness. The median overall survival time with
the current standard of care chemotherapy regimen of gemcitabine
and cisplatin is <1 year (3,4).
Therefore, the identification of novel antitumor targets for ICC is
urgently required.
Glucagon-like peptide-1 (GLP-1) is an incretin
hormone secreted by intestinal L cells in the distal intestine in
response to nutrient ingestion (5,6). Upon
binding to the GLP-1 receptor (GLP-1R), GLP-1 affects blood glucose
levels by stimulating insulin secretion, inhibiting glucagon
secretion and gastric emptying, and reducing food intake (7–9). Due to
its ability to regulate blood glucose levels, GLP-1 is now widely
used in clinic for patients with diabetes (10). In addition to its hypoglycemic
effect, GLP-1 alleviates inflammation in intraepithelial
lymphocytes of intestinal mucosal epithelium, confers antioxidative
and neurogenerative effects in the brain, and protects vascular
functions in the cardiovascular system (11). GLP-1 and GLP-1R-associated signaling
also promote tumor progression. Long-term GLP-1R activation was
reported to increase the risk of pancreatic cancer development
(12), and GLP-1R agonists were
reported to promote neoplastic intestinal growth (13). In addition, GLP-1-based therapies
have been used to treat thyroid carcinoma (14). It has been previously reported that
GLP-1R protein expression is upregulated in ICC and that GLP-1R
expression correlated with lymph node metastasis (15), indicating that GLP-1R is involved in
ICC tumor progression. However, Exendin-4 with regards to diabetes,
a GLP-1 analog that has similar functions as GLP-1, enhanced
oxaliplatin-mediated tumor suppression in ICC (16). These conflicting data call for an
in-depth study on the GLP-1R underlying mechanism of action in
ICC.
Forkhead box O1 (FoxO1) is a member of the forkhead
box family of transcription factors with a highly conserved
DNA-binding domain (17). Although
FOXO-mediated signal transduction pathways are evolutionarily
conserved in most species, they seem to have been co-opted by
differentiated tissues for a variety of specialized functions
(18). For example, FoxO1 deficiency
in T cells confers a survival defect and increased apoptosis,
whereas in B cells, enforced expression of FoxO1 results in partial
cell cycle arrest in addition to increased apoptosis (18). FoxO1 act as a tumor repressor while
also maintaining cancer stem cells in some digestive malignancies
including liver cancer, colorectal cancer, and gastric cancer
(19). Some FoxO transcription
factors promote antitumor activity by negatively regulating the
expression of immunosuppressive proteins and by controlling the
antitumor immune response, as well as the homeostasis and
development of immune cells (20).
It has been reported that GLP-1R signals activate FoxO1 in
hepatocellular carcinoma (HCC) cells (21). However, it remains unclear whether
GLP-1R regulates the phosphorylation of FoxO1 in other types of
cancer. Thus, the present study aimed to determine whether GLP-1R
regulates FoxO1 signaling and its function in ICC.
Materials and methods
Patients and specimens
ICC tumor specimens were obtained from patients who
underwent surgical resection without preoperative treatment between
April 2004 and May 2008 in the department of Hepatobiliary Surgery
at the General Hospital of Ningxia Medical University (Yinchuan,
China). All of the methods were approved by the Research Medical
Ethics Committee of Ningxia Medical University (Yinchuan, China)
and were carried out in accordance with the ethical guidelines of
the Helsinki Declaration. Written informed consent was obtained
from all patients.
Cell lines
The human ICC cell lines RBE and HCCC-9810, and the
extrahepatic cholangiocarcinoma (ECC) cell lines QBC939 and SSP-25,
were obtained from the Shanghai Cell Bank of the Chinese Academy of
Sciences (Shanghai, China). Cells were cultured in DMEM
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C with 5% CO2. For oxaliplatin
treatment, cells were cultured in normal culture media with 1 mg/ml
oxaliplatin (Dalian Meilun Biotech Co., Ltd.,) at 37°C for 12
h.
Western blot analysis
Briefly, HCC tissues or RBE, HCCC-9810, QBC939,
SS_-25 cells were homogenized in SDS sample buffer (10% Glycerol,
2% SDS, 0.01% bromophenol blue, 1.25% 2-β-mercaptoethanol, 25 mM
Tris-HCl, pH 6.8) using ULTRA-TURRAX® (IKA-Works, Inc.,
Wilmington, NC, USA) at 4°C. The protein concentration was
determined with the Quick Start™ Bradford protein assay kit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein extracts
(10 µg) were loaded on 10% SDS-PAGE gels, and transferred to 0.45
µm PVDF membranes (EMD Millipore, Billerica, MA, USA) using
electro-blotting apparatus (Bio-Rad Laboratories, Inc.). After
overnight blocking with 3% BSA (Sigma-Aldrich; Merck KGaA) at 4°C,
membranes were incubated with the following primary antibodies:
GLP-1R (dilution, 1:1,000; cat. no. 26196-1-AP; Proteintech Group
Inc., Chicago, IL, USA), FoxO1 (1:1,000; cat no. 18592-1-AP;
Proteintech Group Inc.), phosphorylated (p)-FoxO1 (S256) (dilution,
1:1,000; cat. no. 9461; Cell Signaling Technology, Inc., Danvers,
MA, USA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH;
dilution, 1:3,000; cat. no. 10494-1-AP; Proteintech Group Inc.)
overnight at 4°C, following with HRP-conjugated secondary antibody
(dilution, 1:3,000; cat. no. 715-035-150; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) at 37°C for 2 h. Enhanced
chemiluminescence was used for detection. The protein bands were
quantified by using ImageJ software v1.42 (National Institutes of
Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA of all cell lines was extracted from cells
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocols. The RNA was
subsequently reverse transcribed at 95°C for 10 sec followed by 40
cycles at 95°C for 5 sec and at 60°C for 45 sec. The cDNA was used
as the template for quantitative PCR using a Takara RNA PCR kit and
SYBR Premix Ex Taq (Takara Bio Inc., Otsu, Japan) using Applied
Biosystem's 7500 qPCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocols. GAPDH
was used as an internal control. The following primers were used:
Human GLP-1R, sense 5′-GGTGCAGAAATGGCGAGAATA-3′, anti-sense
5′-CCGGTTGCAGAACAAGTCTGT-3′; human FoxO1, sense
5′-GGATGTGCATTCTATGGTGTACC-3′, anti-sense
5′-TTTCGGGATTGCTTATCTCAGAC-3′; human Cadherin 1 (CDH1), sense
5′-CCTAGATGAACCTTATGAGGCCA-3′, anti-sense
5′-GCTGTAGAGGAGACGAGCATTAT-3′; human GAPDH, sense
5′-GAGTCAACGGATTTGGTCGT-3′, anti-sense 5′-TTGATTTTGGAGGGATCTCG-3′;
human CDH2, sense 5′-TGTATGTGGGCAAGATCCACT-3′, anti-sense
5′-CTCGTCGATCAGGAAGATGGT-3′; human Snail family transcriptional
repressor 1 (SNAI1), sense 5′-TCGGAAGCCTAACTACAGCGA-3′, anti-sense
5′-AGATGAGCATTGGCAGCGAG-3′; human SNAI2, sense
5′-CGAACTGGACACACATACAGTG-3′, anti-sense
5′-CTGAGGATCTCTGGTTGTGGT-3′; human cyclin D2 (CCND2), sense
5′-ACCTTCCGCAGTGCTCCTA-3′, anti-sense 5′-CCCAGCCAAGAAACGGTCC-3′;
human cyclin-dependent kinase inhibitor 1A (CDKN1A), sense
5′-TGTCCGTCAGAACCCATGC-3′, anti-sense 5′-AAAGTCGAAGTTCCATCGCTC-3′;
Bcl2-interacting mediator (BIM), sense 5′-TGGAGCGGACATGATAAGCAT-3′,
anti-sense 5′-AGCACAGGTGTCAACTAAATCC-3′; NADPH oxidase activator
(NOXA), sense 5′-TGCTACACAATGTGGCGTC-3′, anti-sense
5′-ACTTGGACATGGCCTCCCTA-3′; and superoxide dismutase 2 (SOD2),
sense 5′-TTTCAATAAGGAACGGGGACAC-3′, anti-sense
5′-GTGCTCCCACACATCAATCC-3′. The thermocycling conditions were as
follows: 95°C for 2 min, 95°C for 5 sec, 60°C for 30 sec, 72°C for
30 sec, 40 cycles and dissociation curve. All data were calculated
using the 2−ΔΔCq method (22).
Immunohistochemistry (IHC)
IHC was performed on formalin-fixed
paraffin-embedded surgical specimens. In brief, tissue samples were
fixed with 4% formalin overnight at 4°C, followed by dehydration in
70, 80, 95, and 100% ethanol. After paraffin embedding, the
specimen was cut to 5 to 8 µm thick sections. Tissue sections were
dried at 60°C for 6 h, dewaxed in xylene, rehydrated in a gradient
concentration of ethyl alcohol (100, 75, 50 and 25%) and endogenous
peroxidase activity was blocked using 3% hydrogen peroxide at 37°C
for 10 min. Following antigen retrieval with citrate buffer in a
microwave oven, tissue sections were incubated with GLP-1R (1:200
dilution), FoxO1 (1:100 dilution), or p-FoxO1 (1:100 dilution)
primary antibodies at 4°C overnight. The tissue sections were
treated with Primary Antibody Amplifier Quanto and HRP Polymer
Quanto (Thermo Fisher Scientific, Inc.) prior to visualization with
DAB solution (Thermo Fisher Scientific, Inc.) and counterstaining
with hematoxylin (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Sections were visualized using a Nikon
Eclipse Ti-s microscope (Nikon Corporation, Tokyo, Japan) at ×20
magnification and analyzed using ImageJ software v1.42 (National
Institutes of Health, Bethesda, MD, USA). Two independent
pathologists of the Department of Pathology at the General Hospital
of Ningxia Medical University (Yinchuan, China), who were blinded
to the patients' clinicopathological data, provided the IHC
staining scores. The extent of tumor cell staining in the tissues
was graded as follows: 0, <5; 1, 5–25; 2, 26–50; 3, 51–75 and 4
for 76–100% (21). The intensity of
IHC staining was scored as follows: 0, no IHC signal; 1, weak
signal; 2, moderate signal and 3, strong signal. The final score
used in the analysis was calculated by multiplying the extent score
by the intensity score, and the final score ranged between 0–12.
Values ≤6 were considered low expression based on Receiver
Operating Characteristic analysis. The images presented were
selected randomly.
Small interfering RNA (siRNA) and
plasmids
The siRNA (5′-GCGCTTCATCAAGCTGTTTAC-3′) targeting
GLP-1R was purchased from Sigma-Aldrich (Merck KGaA). A scrambled
siRNA precursor (Scr) was used as the negative control. The siRNAs
(10 µg for cells in 10 cm dish, 0.01 µg/µl) were transfected into
cells using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocols.
Lentiviral vectors for the overexpression of GLP-1R were
constructed by cloning target genes into pCDH-CMV-MCS-EF1-Puro
(System Biosciences, Palo Alto, CA, USA; cat. no. CD510B-1) between
the EcoRI and XbaI restriction sites; these vectors
were also used for transfection (10 µg for cells in 10 cm dish,
0.01 µg/µl). The FoxO1 S256D and FoxO1 S256A plasmids were
constructed by site-directed mutagenesis. Point mutations of FOXO1
were generated by PCR-based mutagenesis using the
GeneArt® Site-Directed Mutagenesis System (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocols. Briefly, mutagenesis PCR was performed in the reaction
mixture, including wild-type (WT)-FoxO1, DNA methylase, Pfx DNA
polymerase, and point-mutated primers. The PCR products were
transformed into DH5α-T1 E. coli competent cells (Tiangen
Biotech, Co., Ltd., Beijing, China), and the positive colonies were
analyzed by sequencing. Mutagenesis primer sequences for S256D were
forward, 5′-AGGAGAAGAGCTGCAAGTATGGACAACAACAGT-3′ and reverse,
5′-ACTGTTGTTGTCCATACTTGCAGCTCTTCTCCT-3′. Primer sequences for S256A
were forward, 5′-AGGAGAAGAGCTGCAGCAATGGACAACAACAGT-3′ and reverse,
5′-ACTGTTGTTGTCCATTGCTGCAGCTCTTCTCCT-3′. For all overexpression
experiments, empty pCDH-CMV-MCS-EF1-Puro vector was used as the
control, and transfection efficiency was assessed using qPCR and
western blot analysis. Further experiments were performed 48 h
after transfection.
Transwell assays
Transwell migration and invasion assays were
performed in 12-well Transwell plates (8-µm pore size), according
to the manufacturer's protocols (Corning Incorporated, Corning, NY,
USA). For invasion assays, the bottom of a Transwell chamber was
coated with BD Matrigel Basement Membrane Matrix (BD Biosciences,
San Jose, CA, USA). Cells (1×105) in basic culture
medium without serum were added to the upper chamber, and the lower
chamber was filled with culture medium containing 20% FBS
(Invitrogen; Thermo Fisher Scientific, Inc.) as a chemoattractant.
Cell migration and invasion were determined after 24 and 48 h,
respectively. Cells on the upper side of the chamber were removed
from the surface of the membrane by scrubbing, and cells on the
lower surface of the membrane were fixed with 4% paraformaldehyde
at room temperature for 10 min and stained with 0.1% crystal violet
at room temperature for 10 min. The numbers of cells were counted
in five randomly selected microscopic fields for each filter using
a Nikon Eclipse Ti-s microscope (Nikon Corporation, Tokyo, Japan)
at ×20 magnification.
Statistical analysis
All data were presented as mean ± standard
deviation. All statistical data were based on three separate
repeated trials. Statistical analysis was performed with GraphPad
Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Differences between two groups were examined by a Student's
two-tailed t-test; multiple comparisons between the groups were
performed using Student-Newman-Keuls method following one way
analysis of variance. Correlations between two groups were analyzed
using a nonparametric Spearman's R test. P<0.05 was considered
to indicate a statistically significant difference.
Results
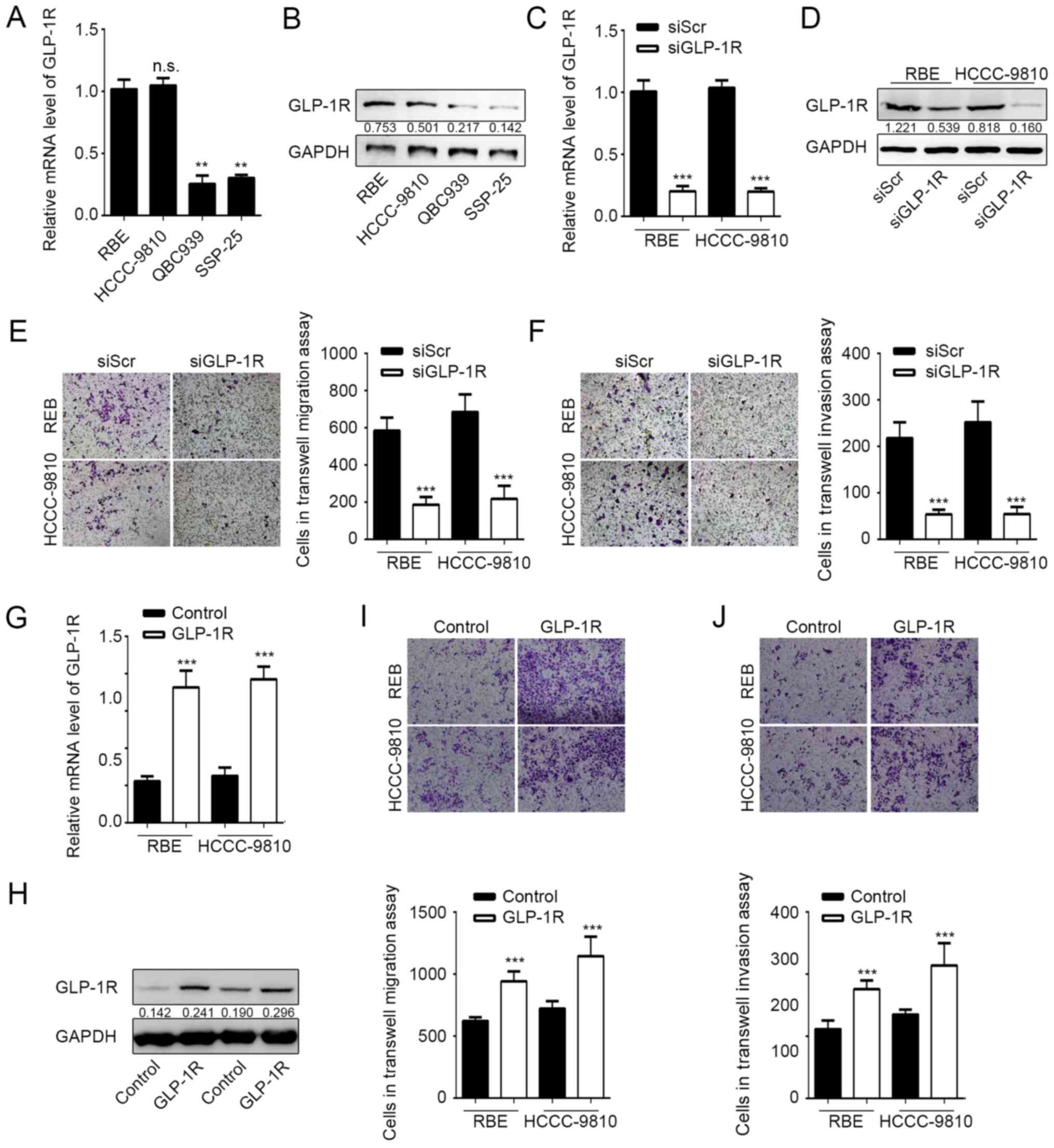
GLP-1R promotes migration and invasion
of ICC cells
It has been indicated previously that GLP-1R is
upregulated in ICC tumor tissues (15). To investigate the role of GLP-1R in
ICC cells, GLP-1R expression was measured in different
cholangiocarcinoma cell lines by RT-qPCR and western blot analysis.
It was indicated that mRNA and protein expression levels of GLP-1R
were significantly higher in the ICC cell lines RBE and HCCC-9810
compared with the ECC lines QBC939 and SSP-25 (Fig. 1A and B). The expression of GLP-1R was
subsequently knocked down in RBE and HCCC-9810 cells by RNA
interference to determine the effects of GLP-1R expression on tumor
cell migration and invasion. Knockdown of GLP-1R expression was
confirmed by RT-qPCR and western blot analysis (Fig. 1C and D), and the Transwell assay
demonstrated that RBE and HCCC-9810 tumor cells exhibited
significantly reduced migration and invasion upon GLP-1R silencing
(Fig. 1E and F). Furthermore,
overexpression of GLP-1R significantly promoted ICC cell migration
and invasion compared with the control (Fig. 1G-J). These data demonstrated that
GLP-1R promotes tumor cell migration and invasion during ICC
progression.
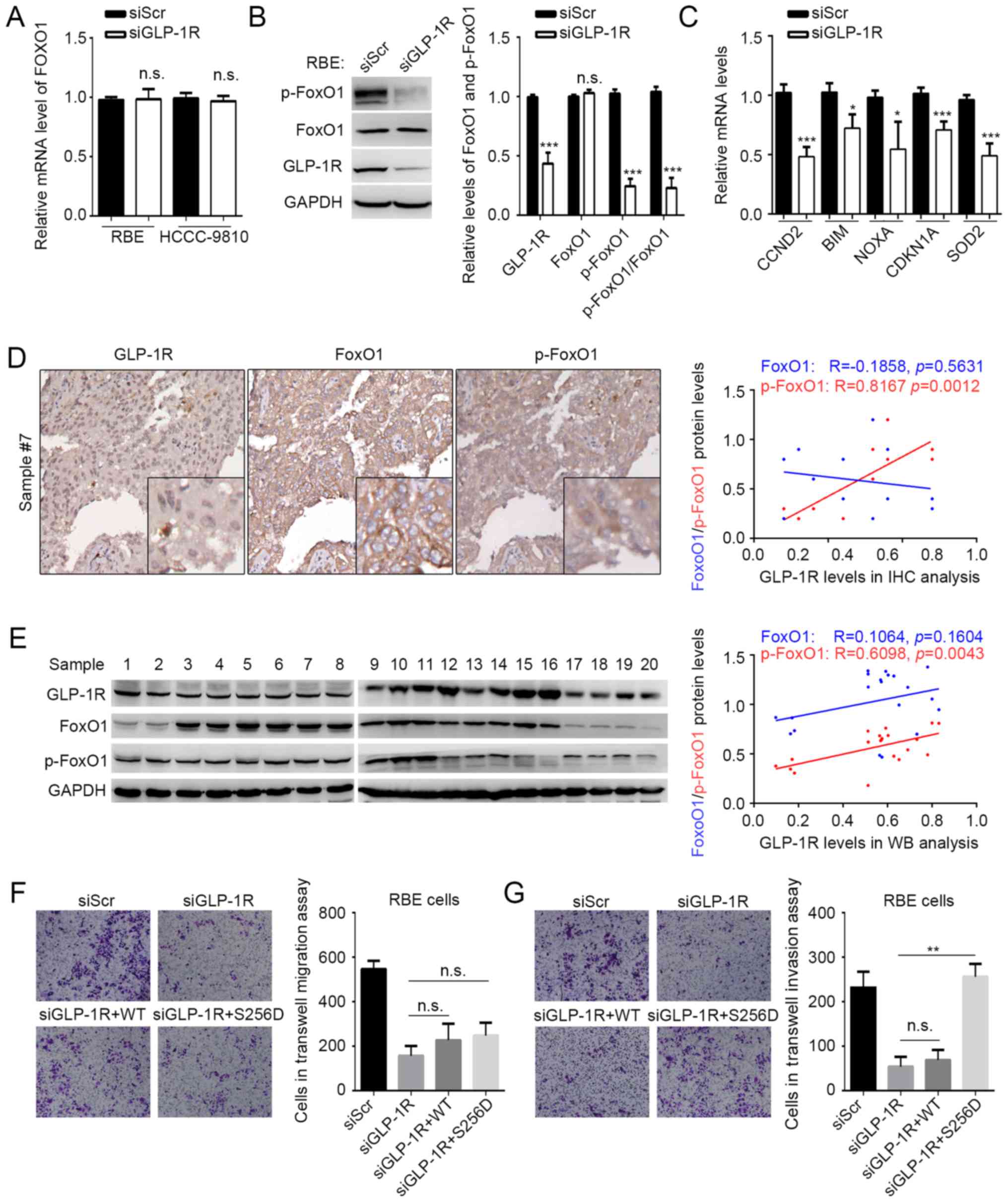
GP-1R functions in ICC by regulating
FoxO1 signaling
GLP-1R has been indicated to activate FoxO1 in obese
mouse (21), and activated FoxO1 has
been reported to be involved in maintaining cancer stem cells
(19), benefiting tumor metastasis.
Considering the effects of GLP-1R on tumor cell migration and
invasion, it was speculated that GLP-1R may also modulate FoxO1 in
ICC. The expression and phosphorylation state of FoxO1 under GLP-1R
knockdown conditions was examined, and it was indicated that
suppressed GLP-1R expression resulted in reduced phosphorylation of
FoxO1 without affecting the FoxO1 mRNA and protein levels in ICC
(Fig. 2A and B). The expression of
some FoxO1-regulating genes, including CCND2, CDKN1A, BIM, NOXA and
SOD2 were also examined using RT-qPCR under GLP-1R knockdown
conditions (18). As expected,
GLP-1R knockdown significantly suppressed the expression of genes
regulated by FoxO1, compared with the control (Fig. 2C). To verify this result, a
correlation analyses was performed to determine the association
between GLP-1R and FoxO1 expression, and phosphorylated FoxO1
expression by IHC of 20 tumor tissues from patients with ICC. IHC
analysis revealed that the levels of GLP-1R protein correlated with
the phosphorylation levels of FoxO1 (R=0.8167; P=0.0012), but not
with FoxO1 expression (R=−0.1858; P=0.5631) (Fig. 2D). However, the immunohistochemical
staining using this antibody was not recommended by the
manufacturer. Thus, to confirm IHC results of the present study,
western blot analysis was performed. As expected by using another
20 tissues for western blot analysis, GLP-1R protein expression
correlated with the levels of phosphorylated FoxO1 (R=0.6098;
P=0.0043), but not with FoxO1 expression (R=0.1064; P=0.1064)
(Fig. 2E). Our previous findings
indicated that upregulation of GLP-1R in ICC correlated with
elevated lymph node metastasis (15); however, we failed to observe any
significant correlation between IHC data and clinicopathological
factors, which may be due to the small sample size (n=20) in the
current study. The aforementioned data suggested that GLP-1R
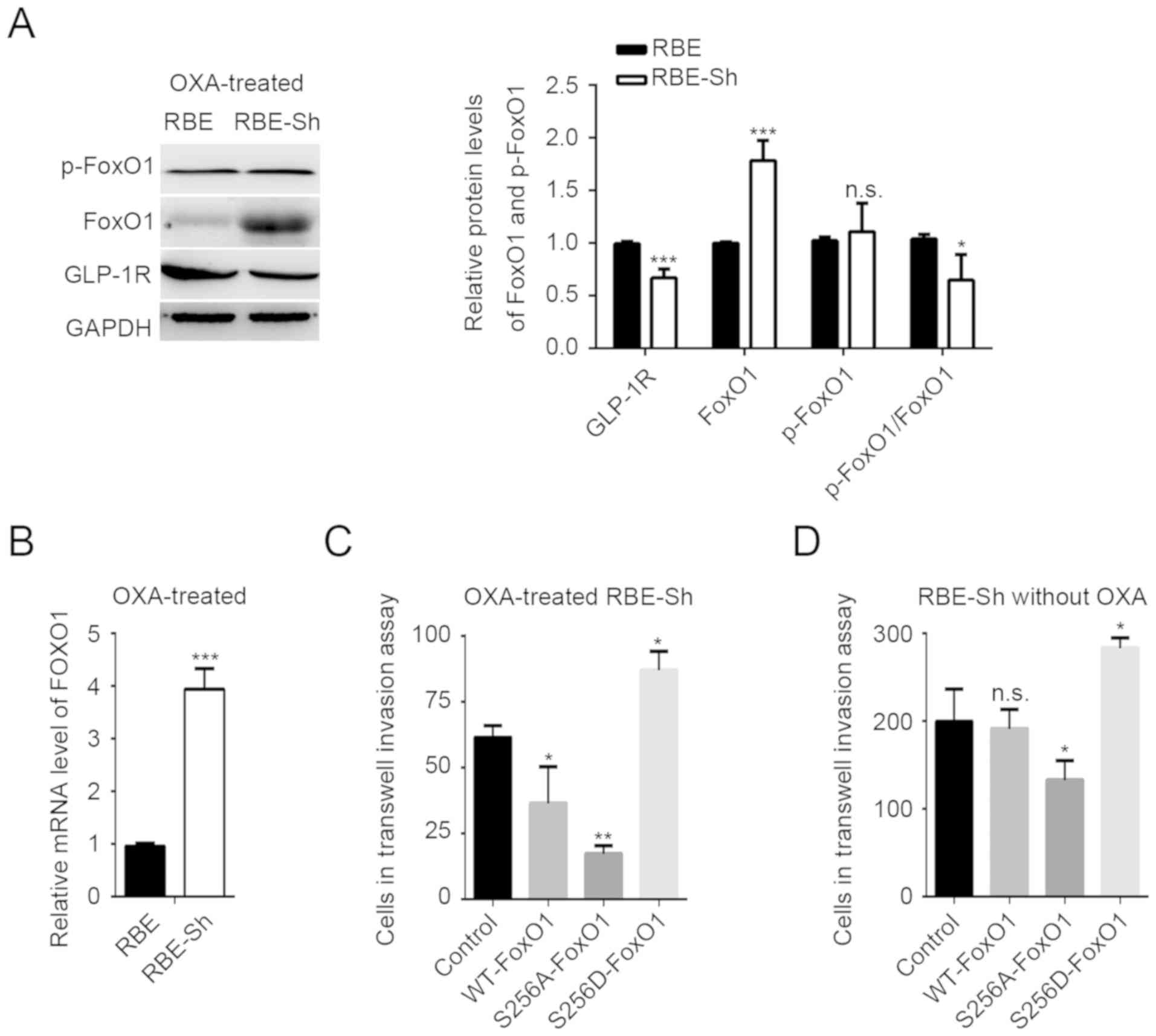
regulates FoxO1 activation. In addition, a mutation (S256D) was
constructed in FoxO1 that affects the phosphorylation of FoxO1. The
mutation significantly increased the number of invasive cells
compared with GLP-1R silencing, but not the abundance of migrating
ICC cells under GLP-1R knockdown conditions (Fig. 2F and G). This indicated that GLP-1R
functions in ICC by modulating the phosphorylation of FoxO1.
 | Figure 2.GLP-1R functions in ICC by regulating
FoxO1 signaling. (A) The mRNA levels of FoxO1 in RBE and HCCC-9810
cells with GLP-1R knockdown were determined by reverse
transcription-quantitative polymerase chain reaction analysis. (B)
The protein level and the phosphorylation state of FoxO1 in RBE
cells were determined by western blot analysis. The p-FoxO1/FoxO1
ratio was calculated as (p-protein/control)/(total
protein/control). (C) Expression of genes targeted by FoxO1 were
measured in RBE cells with GLP-1R knockdown. Correlations between
GLP-1R levels, FoxO1 levels, and FoxO1 phosphorylation states were
evaluated in 20 tumor tissues by (D) immunohistochemical and (E)
western blot analyses. Transwell assays were used to determine the
effects of FoxO1 and FoxO1S256D in the (F) migration and (G)
invasion of RBE cells with GLP-1R knockdown. n.s., not significant;
GLP-1R, glucagon-like peptide-1; FoxO1, forkhead box O1; p,
phosphorylated; si, small interfering RNA. Representative images
for Transwell assays were obtained at ×20 magnification. n.s., not
significant, *P<0.05 vs. siGLP-1R, **P<0.01 vs. siGLP-1R or
siGLP-1R+S256D, ***P<0.001 vs. siGLP-1R. |
Oxaliplatin reverses the effects of
GLP-1R on ICC invasion
Previously, it has been reported that during
oxaliplatin treatment, an agonist of GLP-1R functions in ICC tumor
suppression (16), and it has been
indicated that the functions of tumor suppressors and oncogenic
proteins are reversed during chemotherapy. For example, the effects
of p53-mediated senescence on tumor cells could be reversed under
chemotherapy (23–25). It was speculated that chemotherapy
may also alter the functions of GLP-1R in ICC. In the present
study, oxaliplatin treatment significantly reduced the migration of
tumor cells with or without GLP-1R-silencing. Also, GLP-1R
knockdown did not suppress the inhibitory effects of oxaliplatin
treatment on the migration of RBE and HCCC-9810 cells, indicating
that GLP-1R may not be involved in the oxaliplatin-induced
inhibition of migration (Fig. 3A and
B). In addition, the inhibition of invasion by the GLP-1R
knockdown was reversed with oxaliplatin treatment (Fig. 3C and D). Due to the fact that tumor
invasion is associated with epithelial-mesenchymal transformation
(EMT), the present study examined whether GLP-1R could regulate the
expression of EMT-associated factors, such as CDH1, CDH2, SNAI1,
and SNAI2, and whether oxaliplatin treatment could reverse these
regulatory effects in ICC. The data showed that when comparing the
RBE and RBE-sh groups, GLP-1R knockdown in RBE cells significantly
decreased the mRNA levels of CDH2, SNAIl and SNAI2, and upregulated
CDH1 (Fig. 3E), while these effects
were reversed upon oxaliplatin treatment (Fig. 3E). Western blot analysis indicated
similar results for the protein levels of EMT-associated factors
(Fig. 3F). FoxO1 has been reported
to regulate tumor cell growth; however, the present study did not
investigate the effects of FOXO1 and its phosphorylation on ICC
cell proliferation. The present study only suppressed the
proliferation of ICC cells by using serum-free medium in the
Transwell assays. Taking into consideration that changes in tumor
growth may also influence the metastatic properties of ICC cells,
further investigation is required to determine whether the effects
of p-FoxO1 on metastasis depend on its regulation of tumor
growth.
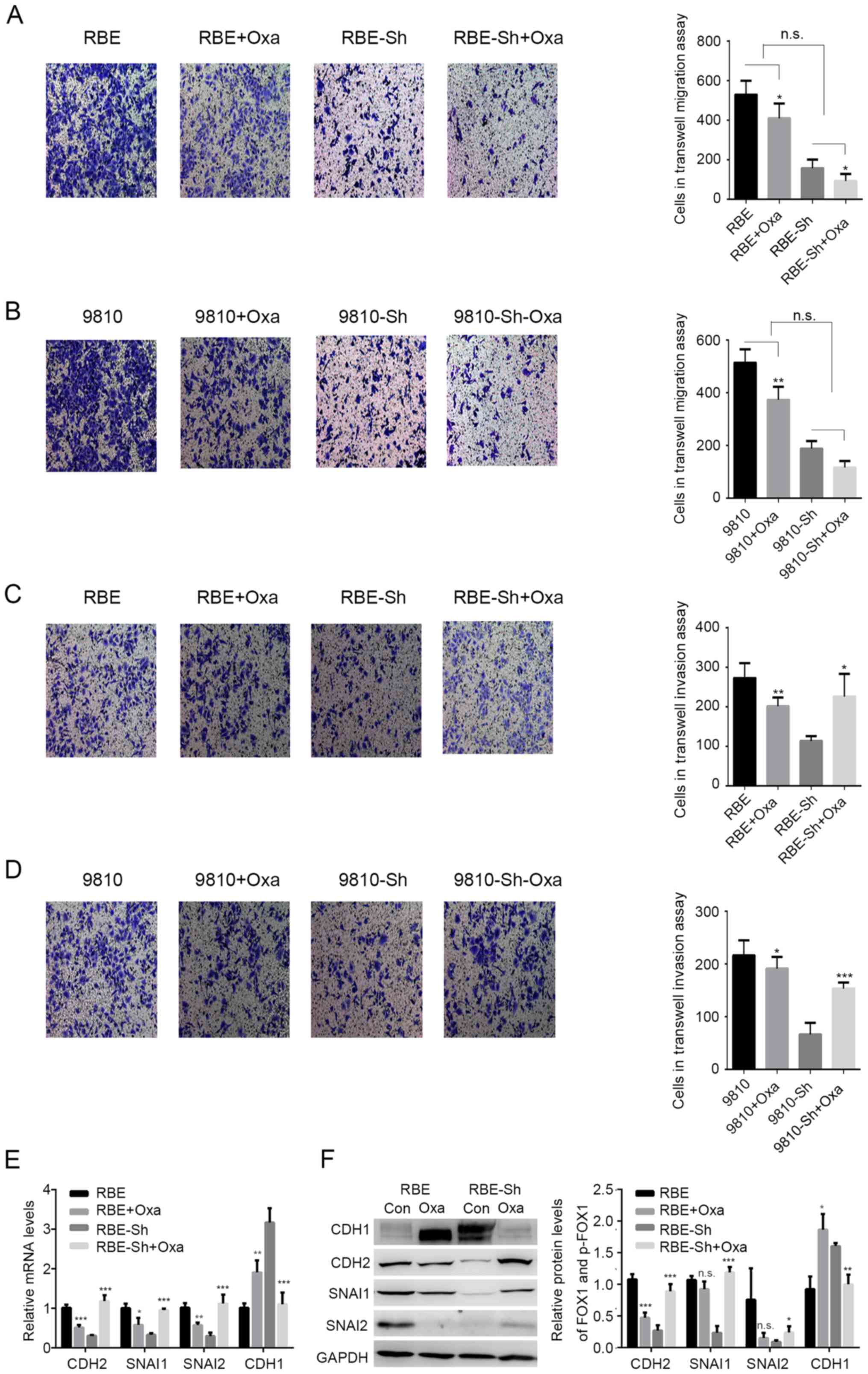 | Figure 3.Effects of GLP-1R on intrahepatic
cholangiocarcinoma invasion are reversed with oxaliplatin. (A-D)
Transwell assays were used to determine the effects of GLP-1R
silencing on migration and invasion of RBE and HCCC-9810 cells with
OXA treatment. (E and F) The mRNA and protein levels of CDH2,
SNAI1, SNAI2 and CDH1 in RBE cells with the indicated treatments.
CDH1, E-cadherin; CHD2, N-cadherin; GLP-1R, glucagon-like
peptide-1; OXA, oxaliplatin; si, small interfering RNA; n.s., not
significant. *P<0.05 vs. oxaliplatin treated, **P<0.01 vs.
oxaliplatin treated, ***P<0.001 vs. oxaliplatin treated. |
Oxaliplatin treatment reduces
GLP-1R-induced FoxO1 signaling
The present study investigated whether FoxO1
signaling is involved in oxaliplatin-mediated changes in GLP-1R
function. In contrast to the results of the GLP-1R knockdown
without oxaliplatin treatment (Fig.
2B), the mRNA and protein levels of FoxO1 were significantly
increased compared with the control, and the phosphorylation state
of FoxO1 was notably unaffected in the GLP-1R knockdown RBE cells
with oxaliplatin treatment (Fig. 4A and
B). In addition, the FoxO1 S256D mutant with oxaliplatin
treatment promoted the invasion of ICC cells, whereas
overexpression of WT FoxO1 with oxaliplatin significantly inhibited
invasion compared with the control (Fig.
4C), suggesting that an increase in unphosphorylated FoxO1 may
be the key factor in reversing the effects of GLP-1R on invasion.
An additional phosphorylation-deficient FoxO1 mutant, FoxO1 S256A,
was created. The present study reported that, compared with the
corresponding controls, the S256A mutation suppressed the invasion
of ICC cells with or without oxaliplatin (Fig. 4D).
Discussion
Despite the fact that chemotherapy is a
well-established cancer treatment, drug resistance has been
observed for classical cytotoxic drugs and drugs that target
specific molecules (26). Analyses
of the associations between gene-expression profiles of tumor
samples and the clinical responses of patients have revealed a
strong correlation between EMT-associated gene expression and drug
resistance (26). EMT is the process
in which epithelial cells lose their apical-basal polarity and
cell-cell adhesion properties, and transition to invasive
mesenchymal cells (27). Cells
undergoing EMT displayed reduced expression of epithelial genes,
including CDH1 (also known as E-cadherin), increased expression of
mesenchymal genes, including CDH2 (also known as N-cadherin), and
upregulated expression of a number of regulators of EMT, including
SNAIL factors (SNAI1, also known as Snail, and SNAI2, also known as
Slug) and FOX proteins (27–30). A number of these transcription
factors, including FOXC2 and FOXM1, have also been reported to
promote drug resistance (31,32).
FOXC2 was reported to increase the expression of ATP-binding
cassette transporters, which enhance drug efflux, thereby promoting
drug resistance (33). However,
various members of the FOX transcription factor superfamily have
the opposite effect on EMT-associated drug resistance. For example,
FOXF2 has been determined to suppress FOXC2-mediated EMT (34). In the present study, it was
demonstrated that the FOX transcription factor FoxO1, may inhibit
EMT, but promoted this process in oxaliplatin-treated ICC cells. In
addition, it was proposed that the phosphorylation of S256 in FoxO1
may be responsible for altering the function of FoxO1 during drug
treatment.
FoxO1 modulates numerous target genes, including
genes involved in apoptosis, cell cycle arrest and immune
regulation, which suggests that FoxO1 would inhibit cell
proliferation in cancer (35).
Upregulation of FoxO1 has been reported to inhibit invasion and
metastasis by reversing EMT in HCC (36). In this study, upregulation of FoxO1
in oxaliplatin-treated ICC cells was observed, and the data suggest
that unphosphorylated FoxO1 may reverse EMT-mediated invasion.
Previous studies have reported that the effects of FoxO1 depend on
its activation, which can be influenced by its abundance,
posttranscriptional modification, nuclear-cytoplasmic shuttling and
subcellular localization (35–38).
Among these activating factors, phosphorylation, particularly that
of S256, is critical for FoxO1 function (37). Since FoxO1 S256D and S256A mutations
conferred opposing effects on ICC invasion, we suggest that the
phosphorylation of S256 may inhibit or reverse the function of
FoxO1 in ICC. The phosphorylation of S256 may attenuate FoxO1
function by regulating its degradation (37–39);
however, increased phosphorylation of S256 in oxaliplatin-treated
ICC cells did not indicate reduced FoxO1 expression levels, and no
significant correlation between the FoxO1 level and its
phosphorylation in ICC tissues was reported. These data suggest
that phosphorylation of S256 affects the function of FoxO1 in ICC
via other mechanisms, which will be investigated in future
studies.
It has been reported that exendin-4 enhances the
phosphorylation of FoxO1 in liver and HCC cells (21). Geniposide, an agonist of GLP-1R, also
increased the levels of cytosolic phosphorylated FoxO1 in
pancreatic β-cells and primary cortical neurons to inhibit
apoptosis (40,41). Our data confirms the effects of
GLP-1R on the FoxO1 signaling pathway. In addition, GLP-1R has been
reported to regulate the nuclear translocation of activated FoxO1
(42). Since phosphorylation of S256
may modulate the transactivation and nuclear targeting of FoxO1
(37–39), it appears that GLP-1R functions in
ICC by regulating the phosphorylation of S256 in FoxO1. In
addition, despite that GLP-1-induced FoxO1 activation promotes
proliferation in early stage pancreatitis (42), FoxO1 signaling reversed the effects
of GLP-1 on proliferation in pancreatic cancer (43), indicating that GLP-1-induced FoxO1
activation serves different roles under different conditions. Our
data is in accordance with this conclusion and suggests that the
effects of GLP-1R-associated therapy may vary under different
conditions.
Incretin therapy, which involves GLP-1 receptor
agonists, has recently become more popular among patients with
tumors located in the digestive system (9). Incretin therapy provides effective
glycemic control with a low risk of hypoglycemia, as well as
improvements in lipid profiles, weight loss and insulin resistance
(44). However, GLP-1R activation
directly enhances proliferation and promotes cell survival in a
number of tissues and cells, including B cells, cardiomyocytes,
fibroblasts and neurons (45). In
accordance with these oncogenic effects, our data revealed that
upregulation of GLP-1R promoted EMT and enhanced tumor invasion.
Nevertheless, GLP-1R functions as a tumor suppressor in endometrial
cancer and pancreatic cancers (46,47).
Therefore, GLP-1R serves different roles in different types of
cancer, and further research should focus on the underlying
mechanisms of GLP-1R in specific cancers. Since the function of
GLP-1R was reversed during oxaliplatin treatment, we suggest that
agonists of GLP-1R have potential as chemotherapy boosters. For
patients that have received surgical or other treatments,
administration of these agonists requires further verification. In
addition, although gemcitabine and cisplatin are commonly used to
treat those with inoperable ICC, patients respond poorly to these
chemotherapies (48). Future studies
should focus on combinatorial treatments, including gemcitabine and
cisplatin or gemcitabine, cisplatin, and oxaliplatin. In addition,
oxaliplatin suppressed EMT in ICC cells and GLP-1R knockdown also
suppressed EMT in ICC cells. However, oxaliplatin partially
reversed GLP-1R knockdown-induced cell invasion. The potential
mechanism of GLP-1R requires further research.
In conclusion, this study indicated that GLP-1R
promotes ICC cell migration and invasion in vitro, and this
tumor-promoting effect depended on the upregulation of
EMT-associated proteins and was mediated by the FoxO1 signaling
pathway. The effects of GLP-1R on EMT and invasion were reversed
with oxaliplatin, which was associated with reduced phosphorylation
of S256 in FoxO1 and an increase in unphosphorylated FoxO1. Our
data also suggest that incretin-based therapies may increase the
risk of ICC metastasis; thus, such treatments should not be solely
used to treat patients with ICC. Additionally, GLP-1R agonists
could be used as boosters for certain types of chemotherapies.
The present study indicated GLP-1R is upregulated in
ICC, and incretin-based therapies may increase the risk of ICC
metastasis. Aberrant upregulation of GLP-1R promotes ICC cell
migration and invasion. Therefore, it should not be used solely for
the treatment of patients with diabetes and ICC. In addition,
oxaliplatin was proposed to reverse the incretin-mediated promotion
of tumor invasion by altering FoxO1 signaling in ICC.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Nature Science
Foundation of Ningxia (grants no. NZ14132, NGY2016122, and
XT201323).
Availability of data and material
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BC, WYZ, WCZ, PY, CT, GW, JL, JM, XW, YH, and QW
contributed to the study design, analysis, and interpretation of
the data. BC conceived the study. WYZ, WCZ, PY, CT and GW performed
the experiments. JL, JM, XW and YH performed the statistical
analyses. BC drafted the manuscript. QW supervised the study and
prepared the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Medical Ethics Committee of Ningxia Medical University (Yinchuan,
China) and was carried out in accordance with the approved
guidelines. Written informed consent was obtained from all of
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gatto M and Alvaro D: New insights on
cholangiocarcinoma. World J Gastrointest Oncol. 2:136–145. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jarnagin WR, Fong Y, DeMatteo RP, Gonen M,
Burke EC, Bodniewicz BS J, Youssef BA M, Klimstra D and Blumgart
LH: Staging, resectability, and outcome in 225 patients with hilar
cholangiocarcinoma. Ann Surg. 234:507–519. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rizvi S, Khan SA, Hallemeier CL, Kelley RK
and Gores GJ: Cholangiocarcinoma-evolving concepts and therapeutic
strategies. Nat Rev Clin Oncol. 15:95–111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Valle J, Wasan H, Palmer DH, Cunningham D,
Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira
SP, et al: Cisplatin plus gemcitabine versus gemcitabine for
biliary tract cancer. N Engl J Med. 362:1273–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tebay LE, Robertson H, Durant ST, Vitale
SR, Penning TM, Dinkova-Kostova AT and Hayes JD: Mechanisms of
activation of the transcription factor Nrf2 by redox stressors,
nutrient cues, and energy status and the pathways through which it
attenuates degenerative disease. Free Radic Biol Med. 88:108–146.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Orskov C, Rabenhøj L, Wettergren A, Kofod
H and Holst JJ: Tissue and plasma concentrations of amidated and
glycine-extended glucagon-like peptide I in humans. Diabetes.
43:535–539. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee YS and Jun HS: Anti-diabetic actions
of glucagon-like peptide-1 on pancreatic beta-cells. Metabolism.
63:9–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karaca M, Magnan C and Kargar C:
Functional pancreatic beta-cell mass: Involvement in type 2
diabetes and therapeutic intervention. Diabetes Metab. 35:77–84.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Drucker DJ: The biology of incretin
hormones. Cell Metab. 3:153–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tremblay AJ, Lamarche B, Deacon CF,
Weisnagel SJ and Couture P: Effects of sitagliptin therapy on
markers of low-grade inflammation and cell adhesion molecules in
patients with type 2 diabetes. Metabolism. 63:1141–1148. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Drucker DJ, Habener JF and Holst JJ:
Discovery, characterization, and clinical development of the
glucagon-like peptides. J Clin Invest. 127:4217–4227. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Elashoff M, Matveyenko AV, Gier B,
Elashoff R and Butler PC: Pancreatitis, pancreatic, and thyroid
cancer with glucagon-like peptide-1-based therapies.
Gastroenterology. 141:150–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koehler JA, Baggio LL, Yusta B, Longuet C,
Rowland KJ, Cao X, Holland D, Brubaker PL and Drucker DJ: GLP-1R
agonists promote normal and neoplastic intestinal growth through
mechanisms requiring Fgf7. Cell Metab. 21:379–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spranger J, Gundert-Remy U and
Stammschulte T: GLP-1-based therapies: The dilemma of uncertainty.
Gastroenterology. 141:20–23. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen BD, Zhao WC, Dong JD and Sima H:
Expression of GLP-1R protein and its clinical role in intrahepatic
cholangiocarcinoma tissues. Mol Biol Rep. 41:4313–4320. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen BD, Zhao WC, Jia QA, Zhou WY, Bu Y,
Wang ZZ, Wang F, Wu WJ and Wang Q: Effect of the GLP-1 analog
exendin-4 and oxaliplatin on intrahepatic cholangiocarcinoma cell
line and mouse model. Int J Mol Sci. 14:24293–24304. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Calnan DR and Brunet A: The FoxO code.
Oncogene. 27:2276–2288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hedrick SM: The cunning little vixen: Foxo
and the cycle of life and death. Nat Immunol. 10:1057–1063. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lam EW, Brosens JJ, Gomes AR and Koo CY:
Forkhead box proteins: Tuning forks for transcriptional harmony.
Nat Rev Cancer. 13:482–495. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deng Y, Wang F, Hughes T and Yu J: FOXOs
in cancer immunity: Knowns and unknowns. Semin Cancer Biol.
50:53–64. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee J, Hong SW, Chae SW, Kim DH, Choi JH,
Bae JC, Park SE, Rhee EJ, Park CY, Oh KW, et al: Exendin-4 improves
steatohepatitis by increasing Sirt1 expression in high-fat
diet-induced obese C57BL/6J mice. PLoS One. 7:e313942012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jackson JG, Pant V, Li Q, Chang LL,
Quintás-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et
al: p53-mediated senescence impairs the apoptotic response to
chemotherapy and clinical outcome in breast cancer. Cancer Cell.
21:793–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi W, Porten S, Kim S, Willis D, Plimack
ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, et al:
Identification of distinct basal and luminal subtypes of
muscle-invasive bladder cancer with different sensitivities to
frontline chemotherapy. Cancer Cell. 25:152–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ihle NT, Byers LA, Kim ES, Saintigny P,
Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, et al:
Effect of KRAS oncogene substitutions on protein behavior:
Implications for signaling and clinical outcome. J Natl Cancer
Inst. 104:228–239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia L, Huang W, Tian D, Zhu H, Qi X, Chen
Z, Zhang Y, Hu H, Fan D, Nie Y and Wu K: Overexpression of forkhead
box C1 promotes tumor metastasis and indicates poor prognosis in
hepatocellular carcinoma. Hepatology. 57:610–624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li W, Zhang Z, Liu X, Cheng X, Zhang Y,
Han X, Zhang Y, Liu S, Yang J, Xu B, et al: The FOXN3-NEAT1-SIN3A
repressor complex promotes progression of hormonally responsive
breast cancer. J Clin Invest. 127:3421–3440. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim T, Ha HI, Kim N, Yi O, Lee SH and Choi
Y: Adrm1 interacts with Atp6v0d2 and regulates osteoclast
differentiation. Biochem Biophys Res Commun. 390:585–590. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou Z, Zhang L, Xie B, Wang X, Yang X,
Ding N, Zhang J, Liu Q, Tan G, Feng D and Sun LQ: FOXC2 promotes
chemoresistance in nasopharyngeal carcinomas via induction of
epithelial mesenchymal transition. Cancer Lett. 363:137–145. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chiu WT, Huang YF, Tsai HY, Chen CC, Chang
CH, Huang SC, Hsu KF and Chou CY: FOXM1 confers to
epithelial-mesenchymal transition, stemness and chemoresistance in
epithelial ovarian carcinoma cells. Oncotarget. 6:2349–2365. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saxena M, Stephens MA, Pathak H and
Rangarajan A: Transcription factors that mediate
epithelial-mesenchymal transition lead to multidrug resistance by
upregulating ABC transporters. Cell Death Dis. 2:e1792011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cai J, Tian AX, Wang QS, Kong PZ, Du X, Li
XQ and Feng YM: FOXF2 suppresses the FOXC2-mediated
epithelial-mesenchymal transition and multidrug resistance of
basal-like breast cancer. Cancer Lett. 367:129–137. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xing YQ, Li A, Yang Y, Li XX, Zhang LN and
Guo HC: The regulation of FOXO1 and its role in disease
progression. Life Sci. 193:124–131. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dong T, Zhang Y, Chen Y, Liu P, An T,
Zhang J, Yang H, Zhu W and Yang X: FOXO1 inhibits the invasion and
metastasis of hepatocellular carcinoma by reversing ZEB2-induced
epithelial-mesenchymal transition. Oncotarget. 8:1703–1713.
2017.PubMed/NCBI
|
|
37
|
Zhang X, Gan L, Pan H, Guo S, He X, Olson
ST, Mesecar A, Adam S and Unterman TG: Phosphorylation of serine
256 suppresses transactivation by FKHR (FOXO1) by multiple
mechanisms. Direct and indirect effects on nuclear/cytoplasmic
shuttling and DNA binding. J Biol Chem. 277:45276–45284. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang H, Regan KM, Wang F, Wang D, Smith
DI, van Deursen JM and Tindall DJ: Skp2 inhibits FOXO1 in tumor
suppression through ubiquitin-mediated degradation. Proc Natl Acad
Sci USA. 102:1649–1654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rena G, Prescott AR, Guo S, Cohen P and
Unterman TG: Roles of the forkhead in rhabdomyosarcoma (FKHR)
phosphorylation sites in regulating 14-3-3 binding, transactivation
and nuclear targetting. Biochem J. 354:605–612. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu J, Yin F, Xiao H, Guo L and Gao X:
Glucagon-like peptide 1 receptor plays an essential role in
geniposide attenuating lipotoxicity-induced β-cell apoptosis.
Toxicol In Vitro. 26:1093–1097. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Y, Xia Z, Liu J and Yin F: Cell
signaling mechanisms by which geniposide regulates
insulin-degrading enzyme expression in primary cortical neurons.
CNS Neurol Disord Drug Targets. 14:370–377. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wewer Albrechtsen NJ, Albrechtsen R,
Bremholm L, Svendsen B, Kuhre RE, Poulsen SS, Christiansen CB,
Jensen EP, Janus C, Hilsted L, et al: Glucagon-like peptide 1
receptor signaling in acinar cells causes growth-dependent release
of pancreatic enzymes. Cell Rep. 17:2845–2856. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Roy SK, Chen Q, Fu J, Shankar S and
Srivastava RK: Resveratrol inhibits growth of orthotopic pancreatic
tumors through activation of FOXO transcription factors. PLoS One.
6:e251662011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Forst T, Weber MM and Pfutzner A:
Cardiovascular benefits of GLP-1-based herapies in patients with
diabetes mellitus type 2: Effects on endothelial and vascular
dysfunction beyond glycemic control. Exp Diabetes Res.
2012:6354722012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Buteau J, Roduit R, Susini S and Prentki
M: Glucagon-like peptide-1 promotes DNA synthesis, activates
phosphatidylinositol 3-kinase and increases transcription factor
pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding
activity in beta (INS-1)-cells. Diabetologia. 42:856–864. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kanda R, Hiraike H, Wada-Hiraike O,
Ichinose T, Nagasaka K, Sasajima Y, Ryo E, Fujii T, Osuga Y and
Ayabe T: Expression of the glucagon-like peptide-1 receptor and its
role in regulating autophagy in endometrial cancer. BMC Cancer.
18:6572018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhao H, Wang L, Wei R, Xiu D, Tao M, Ke J,
Liu Y, Yang J and Hong T: Activation of glucagon-like peptide-1
receptor inhibits tumourigenicity and metastasis of human
pancreatic cancer cells via PI3K/Akt pathway. Diabetes Obes Metab.
16:850–860. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gao Q, Yu GY, Shi JY, Li LH, Zhang WJ,
Wang ZC, Yang LX, Duan M, Zhao H, Wang XY, et al: Neddylation
pathway is up-regulated in human intrahepatic cholangiocarcinoma
and serves as a potential therapeutic target. Oncotarget.
5:7820–7832. 2014. View Article : Google Scholar : PubMed/NCBI
|