Introduction
Neurofibromatosis type-1 (NF-1) is a common,
autosomal dominant neurocutaneous disorder with an incidence of
1:3,000 worldwide, which primarily involves the skin, bone and
nervous system (1). The standard
clinical diagnostic criteria for NF-1 have existed since 1987, with
≥2 criteria required for a positive diagnosis. The criteria are
summarized as follows: ≥6 ‘café-au-lait’ spots, size >5 mm in
prepubertal children or >15 mm in post pubertal individuals,
axillary or inguinal freckling, ≥2 cutaneous neurofibromas, ≥1
plexiform neurofibroma, ≥2 Lisch nodules, a characteristic bone
lesion (sphenoid wing dysplasia, dysplasia of the long bones),
optic nerve glioma and a first degree relative with NF-1 (1,2).
Patients with NF-1 have an 8–13% risk of developing malignant
peripheral nerve sheath tumors (MPNSTs) (1,2). MPNSTs
are unusual and aggressive malignant soft-tissue tumors comprising
5–10% of all soft-tissue sarcomas, and ~50% of all MPNST cases are
associated with NF-1 (3). In
addition, MPNST can also develop sporadically or be associated with
exposure to radiation (3,4). It has been reported that there is no
sex or ethnicity predilection for MPNST (4). MPNST usually develops in the deep soft
tissues and tends to appear in the extremities, trunk and the head
or neck region; however, it can also rarely occur in superficial
locations, including the skin or subcutaneous tissues (5,6).
Histologically, MPNSTs are categorized into the most common
conventional type plus three specific subtypes, including
rhabdomyoblastic, glandular and epithelioid tumors (7). As a subset of MPNSTs, the epithelioid
variant of MPNST (eMPNST) is histologically characterized by the
predominance (≥50%) of epithelioid tumor cells and diffuse
positivity for S-100 proteins; however, eMPNST is rare and accounts
for <5% of all MPNSTs (8).
Furthermore, the association between eMPNST and NF-1 compared with
conventional MPNST is extremely rare and occurs in <2% of all
MPNSTs (9). Although extensive
clinicopathological studies have been conducted on eMPNST, it is
often difficult to provide an accurate diagnosis, since its
histological characteristics are similar to that of poorly
differentiated cancer, malignant melanoma and other soft-tissue
tumors (8). In addition, the
uncommon association of eMPNST with NF-1 increases the difficultly
to obtain a differential diagnosis. However, eMPNST displays
consistent, uniform, strong and diffuse positive staining for S-100
(10). Staining for second-line
melanoma markers (including HMB45, melan A, tyrosinase, and MITF)
and vascular markers is usually negative (8). Therefore, immunohistochemical
examination is required to diagnose eMPNST and rule out other
possibilities (8). Ki-67 has a well
characterized expression pattern and is known to be associated with
cell proliferation (11). The
classifications of neuroendocrine tumors include the Ki-67 index,
high proliferation rates indicate poor prognosis and rapid tumor
growth (11). MPNST has a high
probability rate of local recurrence and distant metastasis, with a
poor prognosis. A local recurrence rate of 40–65% and a 5-year
survival rate of 23–69% have been reported (3,12).
However, biological complications, including recurrence, metastasis
or mortality rate in patients with eMPNST remain unclear due to the
small group of patients involved, the lack of large-scale clinical
research and the variation in survival rates. In addition, the
majority of the published literature primarily focuses on the
clinicopathological characteristics of patients (13–15). The
present study describes the case of a 71-year-old woman with a
family history of NF-1 who was diagnosed with eMPNST and suffered
twice from recurrence, with tumors located on the lower back. The
present study also includes a review of the literature focusing on
the clinicopathological differential diagnosis and prognosis of
eMPNST.
Case report
In November 2016, a 71-year-old woman was admitted
to the Department of General Surgery of The First Affiliated
Hospital of Nanchang University (Nanchang, China) with an enlarged
and painful mass on the lower back, which had been surgical excised
twice in October 2014 and September 2015 at the Jiangxi Provincial
People's Hospital (Nanchang, China). The slides and details for the
specimens from October 2014 and September 2015 were available and
confirmed the diagnosis of an MPNST. The mass recurred at the same
site following each excision within a follow-up period of 6 months.
The patient noticed the third mass at 5 months after the second
operation and noted that it was gradually enlarging. Furthermore,
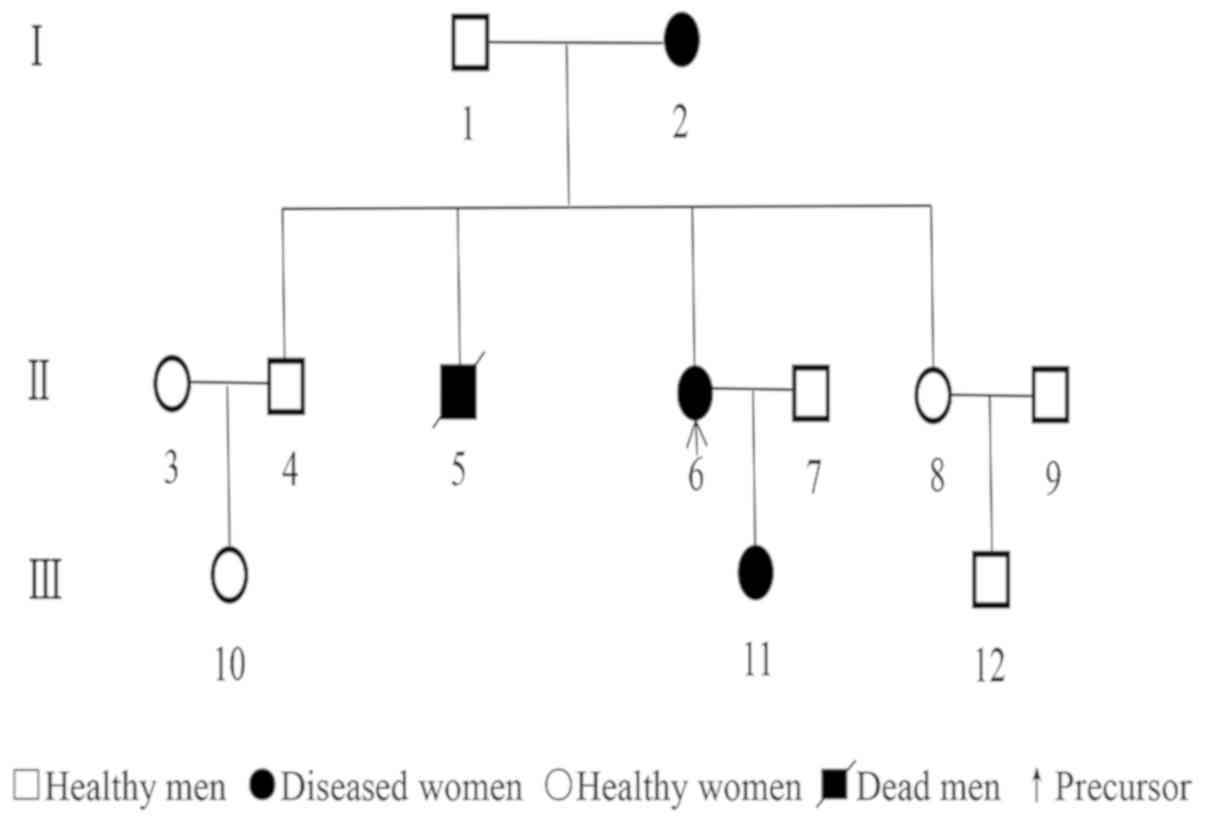
the patient was from a family with a history of NF-1, and the
records indicated that 12 people over three generations, including
the patient's grandmother and mother, had suffered from NF-1. Among
the family members, 4 suffered from NF-1 that developed during
adolescence. The patients' older brother died at the age of 68 from
other diseases, whereas the other members of the family remained
alive. The diagnosis of all patients was confirmed by medical
examinations (Fig. 1). There was no
familial history of other major diseases. Physical examinations of
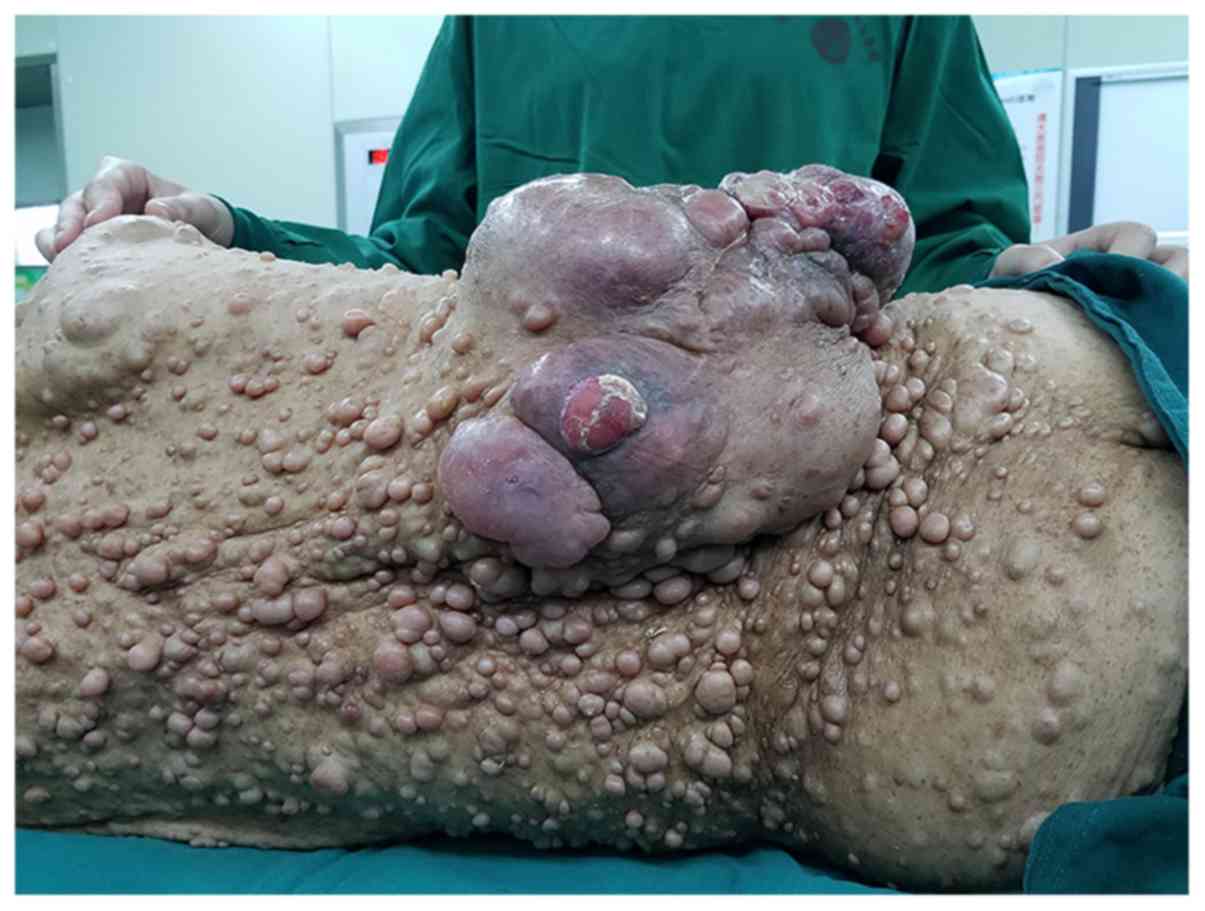
the present patient revealed multiple nodules of different sizes
throughout the entire body combined with numerous ‘café-au-lait’
spots. There was also a hard, slightly erythematous, irregular,
highly swollen mass, with local surface ulceration and suppuration
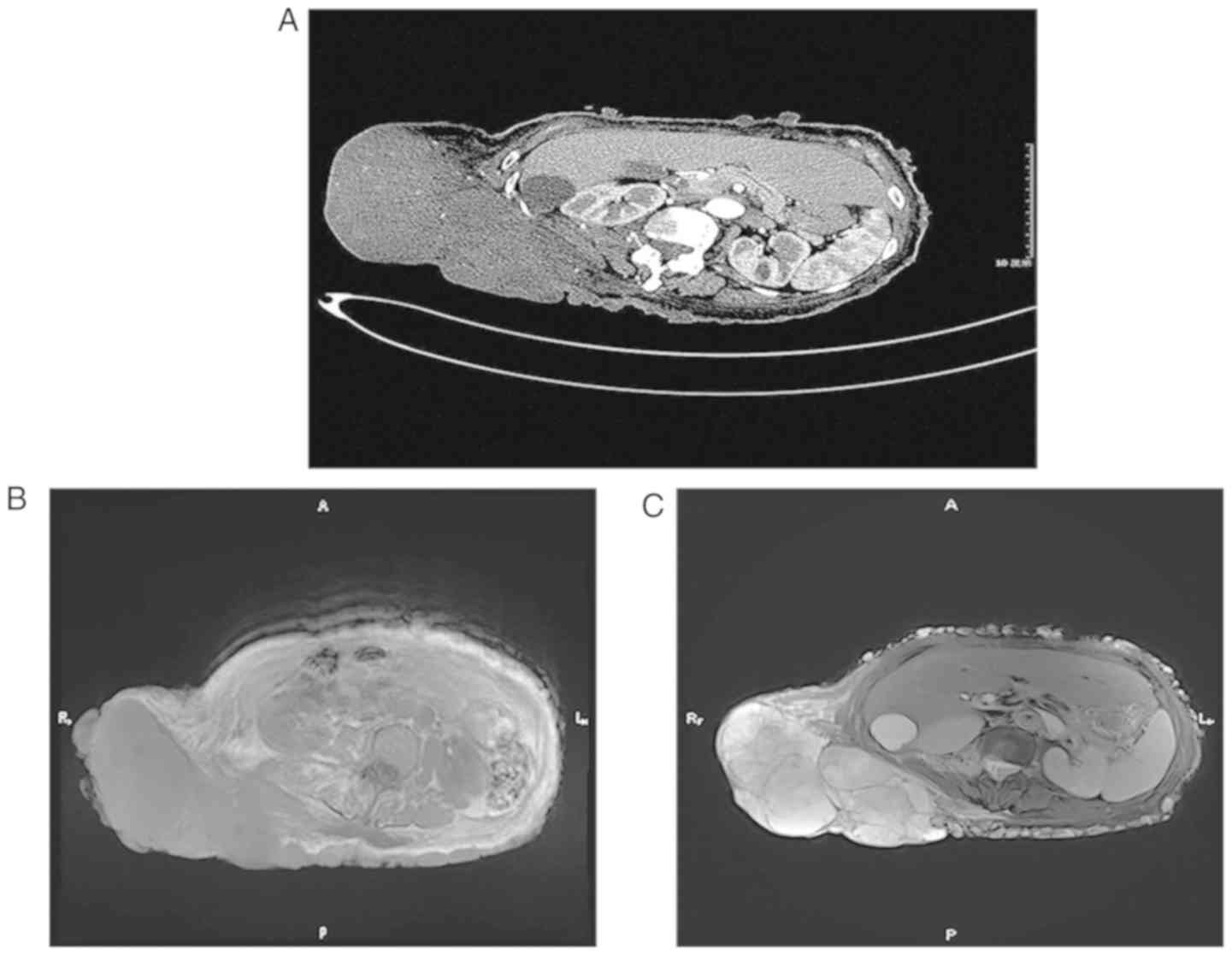
on the lower back, which measured 20×13×8 cm in size (Fig. 2). Computed tomography (CT) of the
chest, abdomen and pelvis revealed a mass with a maximum diameter
of 18 cm, characterized as a heterogeneously dense soft-tissue mass
with an irregularity border on the lower back. According to the
contrast-enhanced CT scans, there was mild to moderate
non-homogeneous enhancement, and no sign of distant metastases was
observed (Fig. 3A). Magnetic
resonance imaging (MRI) revealed that the mass had low signal
intensity on T1-weighted images (Fig.
3B) and high signal intensity on T2-weighted images (Fig. 3C). Additional laboratory
investigations did not reveal any other abnormalities, other than a
low level of hemoglobin at 91 g/l (normal range, 115–150 g/l).
Comprehensive analysis of the imaging examination and the medical
history of the patient were indicative of the presence of a
recurrent malignant tumor on the back. During surgery, the
physicians decided to perform an en bloc resection of the tumor
with a negative surgical margin. Gross examination of the mass
surface revealed a gray-white, fleshy tumor weighing ~1,470 g and
containing solid and cystic regions. Histologically, tumor tissue
was fixed with 10% neutral formalin for 24 h at room temperature
and embedded in paraffin. After being cut into slices (4 µm thick)
using a microtome (BQ-318D; Bona Medical Technology, Hubei, China).
The paraffin sections were dewaxed by washing with xylene three
times, 5 min each time, and hydrated in 100, 95, 85 and 75% ethanol
solutions for 3 min in each. These slices were incubated in
hematoxylin solution for 5 min at room temperature and rinsed with
running water. Subsequently, the slices were immersed in eosin
solution for 3 min at room temperature and washed again with
running water. After dehydration, the slices were mounted with
neutral resin and viewed under a light microscope (CX22; Olympus
Corporation, Tokyo, Japan) for pathological analysis
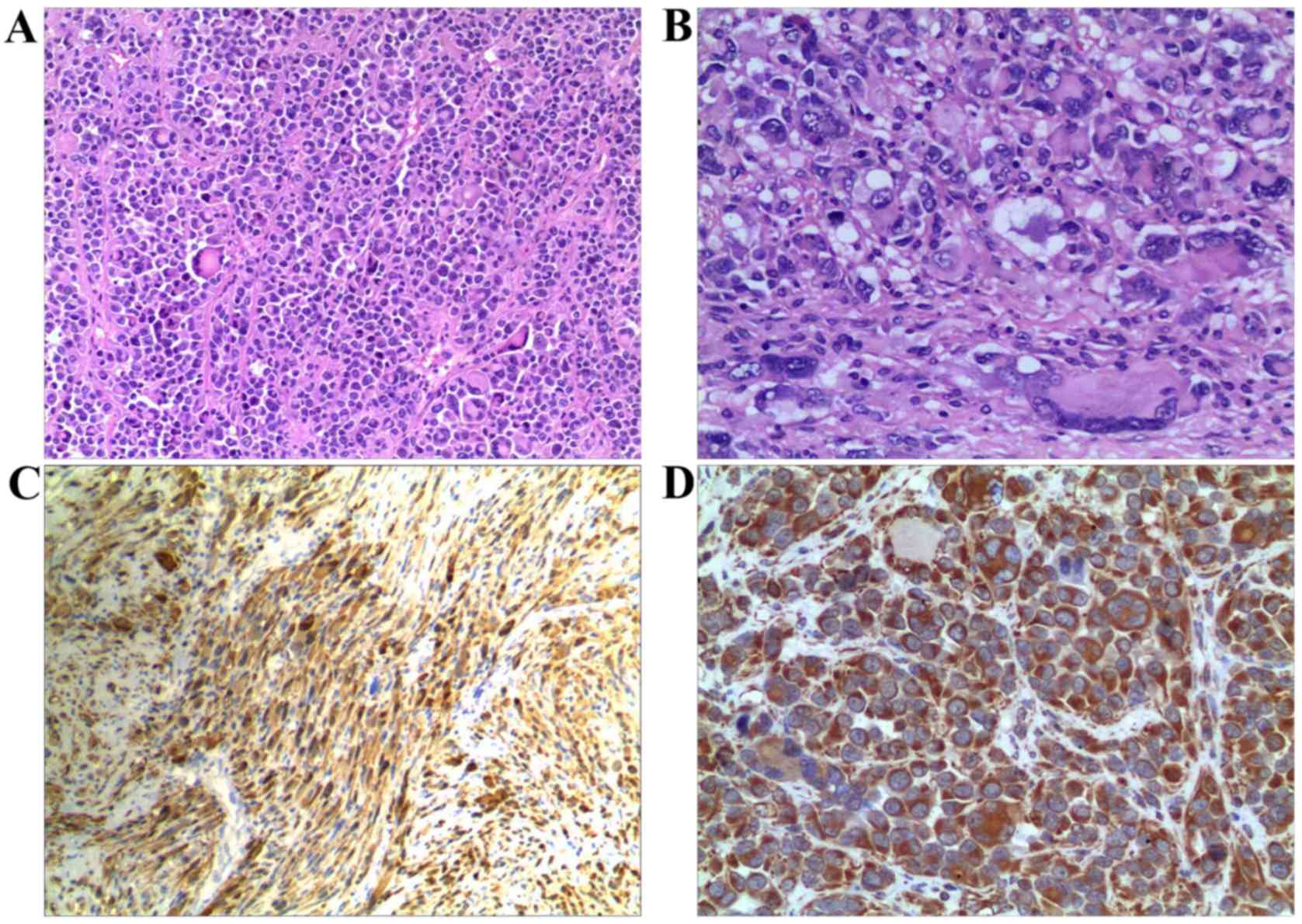
(magnification, ×100 and ×400). The tumor was primarily composed of
cells with an epithelioid morphology arranged in a nesting and
diffuse growth pattern, containing a small proportion of
interspersed spindle cell components (Fig. 4A). The polygonal or round-shaped
epithelioid cells had large, round, vesicular nuclei with
basophilic nucleoli and abundant eosinophilic cytoplasm, and 10
mitotic figures per 10 high-power fields (HPF) (Fig. 4B). The morphology of the cells from
these sections was similar to that observed in the cells from the
previous surgical excision at the Jiangxi Provincial People's
Hospital. The edge of the resection margin of the tumor was
negative. Immunohistochemical analysis was performed on the
4-µm-thick formalin-fixed paraffin-embedded tissue sections. Tissue
sections were deparaffinized in xylene, and hydrated in graded
ethanol solutions. Endogenous peroxidase activity was blocked by
incubating the sections in 3% hydrogen peroxide at room temperature
for 10 min. Antigen retrieval was performed using boiling sodium
citrate buffer (pH 6.0) in a microwave for 15 min. Nonspecific
binding was blocked in 5% bovine serum albumin (BSA) in PBS for 60
min at room temperature. The primary antibodies used in the lesions
were, anti-S100 (cat. no. Z0311; 1:600), vimentin (cat. no. V9;
1:400), cluster of differentiation (CD) 34 (cat. no. Qbend10;
1:50), cytokeratin (cat. no. MNF116; 1:100), smooth muscle actin
(SMA) (cat. no. 1A4; 1:400), melan-A (cat. no. A103; 1:50), HMB45
(cat. no. HMB45; 1:100), melanocyte inducing transcription factor
(MITF) (cat. no. D5; 1:100), myogenin (cat. no. F5D; 1:50),
epithelial membrane antigen (EMA) (cat. no. E29; 1:100),
neurofilament (cat. no. 2F11; 1:100), desmin (cat. no. D33; 1:100)
and Ki-67 (cat. no. MIB-1; 1:100) all from Dako; Agilent
Technologies GmbH (Waldbronn, Germany). The samples were incubated
with the primary antibodies overnight at 4°C in bovine serum
albumin. Subsequently the samples were incubated with a horseradish
peroxidase-conjugated goat anti-mouse immunoglobulin G secondary
antibody (cat. no. K5007; 1:1,000; Dako; Agileant Technologies,
GmbH) for 30 min at 37°C. The 3,3-diaminobenzidine chromogenic
liquid was added to the sections and incubated for 10 min at room
temperature followed by stopping with water. After restaining with
hematoxylin, the sections were dehydrated using a series of ethanol
solutions (75, 85, 95, 100 and 100% for 3 min in each), and treated
with xylene. The sections were sealed with neutral gum and observed
using a light microscope (magnification, ×200). The fraction of
proliferating cells (positive for Ki-67) was calculated based on a
count of at least 500 tumor cells in the relatively dense
concentration of positive cancer nuclei (hot spot). The percentage
of positive cells were calculated and used as the Ki-67 index.
Immunohistochemical staining revealed that tumor cells exhibited
strong and diffuse positive staining for S-100 (Fig. 4C) and vimentin (Fig. 4D), whereas CD34, cytokeratin, SMA,
melan-A, HMB45, MITF, myogenin, EMA, neurofilament and desmin were
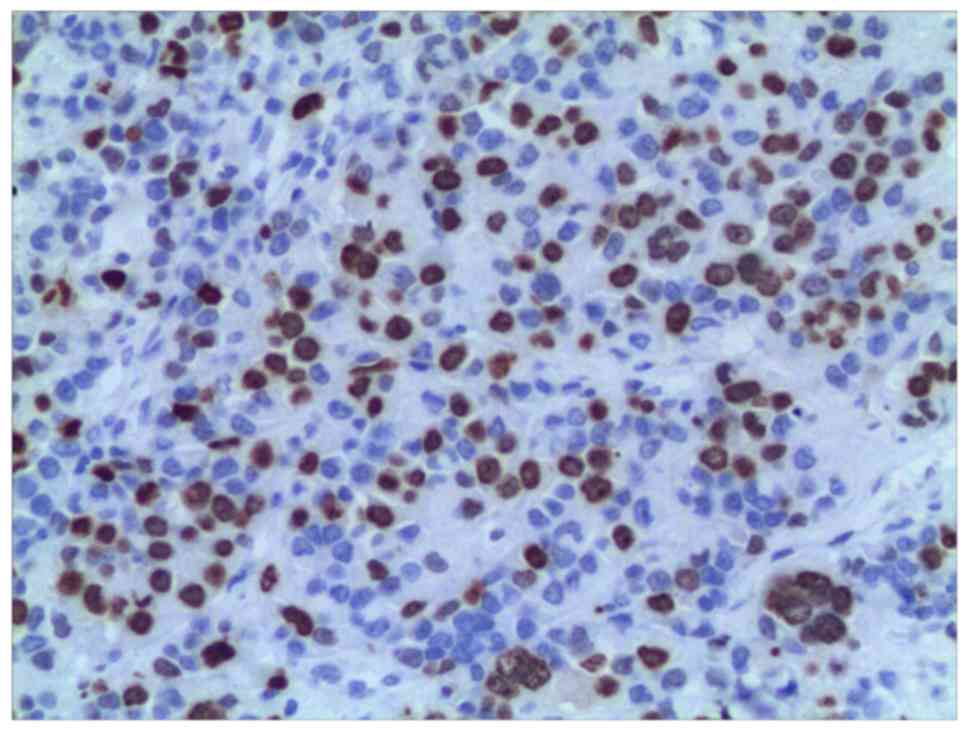
negative. The Ki-67 labeling index was 40% (Fig. 5). The final pathological diagnosis
was eMPNST arising in the peripheral nerves. The patient recovered
uneventfully following the operation and was subsequently referred
to the Department of Oncology for further treatment due to the
multiple recurrences and highly invasive nature of the tumor. A
course of adjuvant radiotherapy, with a dose of 60 Gray in 30
fractions, was administered over a period of 6 weeks. At the end of
all treatments, examinations for potential recurrence or metastases
were performed, including CT of the chest and abdomen, a whole-body
bone scan and a brain MRI, and the results were normal.
Postoperative follow-up at 2 years revealed no evidence of
recurrence and metastatic disease, or residual side effects from
therapy. Despite the repeated relapses, metastasis has not been
detected in the patient in the 4 years since the initial operation
in 2014. However, the recovery of the patient continues to be
evaluated.
Discussion
The eMPNST is a distinct subtype of MPNST that
accounts for <5% of all cases. MPNST commonly affects patients
aged between 40 and 70 years old, with no sex predilection
(16). eMPNSTs primarily involve the
trunk and lower extremities of the body, and are more superficial
than MPNSTs (8). The association
between eMPNST and NF-1 is rare and occurs in <2% of all MPNSTs
(9). To the best of our knowledge,
only 5 cases in the English literature describing eMPNST associated
with NF-1 have been reported (6,13,15,17,18).
A characteristic histological feature of eMPNST is the predominance
(≥50%) or prevalence of epithelioid tumor cells arranged in
multinodular growth pattern, with myxoid and/or fibrous stroma, and
usually admixed with a diverse proportion of spindle cell
components (8,13,14).
Tumor cells surrounded by hyalinized or myxoid stroma frequently
grow in cords, strands, nests or sheets (8). Polygonal epithelioid cells have
characteristic round or oval-shaped nuclei, vesicular chromatin,
variably prominent basophilic nucleoli and an abundant eosinophilic
or amphophilic cytoplasm, with well-defined cell membranes
(19). The nuclear atypia is usually
moderate to severe and widespread, and the mitotic activity that
reflects the degree of nuclear atypia is consistently present, with
a mean number of 7/10 HPF (13). The
immunohistochemical characteristics of eMPNSTs usually exhibit
diffuse positivity for S-100 proteins and vimentin, whereas focal
and patchy positive S-100 staining is rare (8,13,20). The
SMARCB1 (INI1/BAF47/SNF5) gene is located on the chromosome 22
(22q11.2) and functions as a tumor suppressor gene, deletions
and/or mutations of SMARCB1 gene resulting in the loss of nuclear
integrase interactor 1 (INI1) protein expression have been detected
in eMPNST (14). A previous study
reported that eMPNSTs exhibit a loss of expression of INI1 ranging
from 50–67% (14). Negative staining
for second-line melanoma markers, including HMB45, melan A and
MIFT, is crucial for distinguishing eMPNSTs from melanoma. Notably,
focal positive staining for melan A and HMB45, and negative
staining for S-100 in eMPNST cases have also been reported
(13,19). In addition, eMPNST is rarely
associated with NF-1, and occurs in a variety of anatomical
locations. Subsequently, a precise diagnosis of eMPNST can be
difficult. The main differential diagnosis of eMPNST comprises
other skin tumors with an epithelioid morphology, including
malignant melanoma, metastatic poorly differentiated carcinoma,
epithelioid sarcoma and myoepithelial carcinoma (8). In addition, distinction from other
differentiations of MPNST, including glandular MPNST and
rhabdomyoblastic MPNST, is also critical. Malignant melanoma or
metastatic melanoma may be characterized histologically by an
epithelioid phenotype, and often exhibits diffuse positivity for
S-100 protein, which makes it harder to distinguish from eMPNST
(13,14). However, melanoma-associated markers,
including melan A, HMB45, tyrosinase and MITF, are generally absent
in eMPNST, in contrast with melanoma of epithelioid morphology
(21). In particular, MITF and S-100
positive staining, which, to the best of our knowledge, has not yet
been detected in eMPNSTs, is therefore considered to be highly
specific for melanoma (21). In
addition, malignant melanoma commonly exhibits expression of INI1,
whereas the loss of INI1 nuclear expression is observed in 50–67%
of eMPNST cases (20). Observation
of the tumor ultrastructure by electron microscope can provide a
solution to the difficulties associated with differential
diagnosis, since the presence of atypical junctional components or
melanin pigmentation can be indicative of melanoma (13). Careful examination to exclude the
history of melanoma or the clinical presence of melanoma is also
crucial in order to differentiate eMPNST from malignant melanoma in
the majority of cases. Epithelioid sarcoma can be differentiated
from eMPNST through immunohistochemical analysis. For example,
epithelioid sarcoma is positive for cytokeratin and negative for
S-100, and exhibits a loss in INI1 expression (8). In addition, CD34 is positive in ~50% of
epithelioid sarcomas (8,13). Negative staining results for
cytokeratins can serve in distinguishing eMPNSTs from metastatic
poorly differentiated carcinoma, with a lack of staining or focal
positivity in the former, and positivity observed in the latter
(20). Myoepithelial carcinoma
shares similar morphological characteristics with eMPNST, and is
predominantly comprised of epithelioid cells. However, unlike
eMPNST, myoepithelial carcinoma is positive for multiple
cytokeratins, including CK-AE1/AE3, CAM 5.2 and/or EMA (18). Notably, myoepithelial carcinoma
exhibits unusually strong and multifocal S-100 positivity and a
loss of INI1 expression of up to 40% (8). Malignant triton tumor (MTT) is a
variant of MPNST with rhabdomyoblastic differentiation (22,23). The
immunohistochemical characteristics of MTT overlap with those of
eMPNST; however, it can be distinguished from eMPNST by the desmin,
myo-D1 and myogenin positivity of rhabdomyoblasts in MMT, and
consistent negativity in eMPNST (23). Glandular MPNST is an MPNST subset
with glandular differentiation. Histologically, the majority of
glandular PNSTs are composed of a malignant spindle cell component
and a benign glandular component (24). Unlike eMPNST, glands stain positive
for epithelial markers, including keratins and epithelial membrane
antigen, and neuroendocrine markers, including somatostatin,
chromogranin, calcitonin and Leu-7 (23,24).
Immunohistochemistry is therefore a useful technique to
differentiate eMPNSTs from other differential diagnoses of MPNST.
The useful immunohistochemical features of differential diagnosis
are summarized in Tables I and
II. The patient from the present
study exhibited clinical characteristics of NF-1, and the histology
and immunophenotype were consistent with an eMPNST diagnosis.
 | Table I.Immunophenotypic features used for
the differential diagnosis of tumors with epithelioid
morphology. |
Table I.
Immunophenotypic features used for
the differential diagnosis of tumors with epithelioid
morphology.
| Protein | Malignant
melanoma | Poorly
differentiated carcinoma | Epithelioid
sarcoma | Myoepithelial
carcinoma | eMPNST |
|---|
| S-100 | + | − | − | + | + (strong,
diffuse) |
| Vimentin | + | − | + | + | + (strong,
diffuse) |
| HMB45 | + | − | − | − | − |
| Melan A | + | − | − | − | − |
| SMA | − | − | ± | ± | − |
| Desmin | − | − | ± | ± | − |
| CD34 | − | − | ± | − | − |
| CK (AE1/AE3) | − | + | + | + | +/- |
| EMA | − | + | + | + | +/- |
| SMARCB1/INI-1 | Retained | Retained | Loss (~90%) | Loss (~40%) | Loss (50–67%) |
| Table II.Immunophenotypic features used to
distinguish an eMPNST from other divergent differentiations of
MPNST. |
Table II.
Immunophenotypic features used to
distinguish an eMPNST from other divergent differentiations of
MPNST.
| Protein | Conventional
MPNST | Glandular
MPNST | MTT | eMPNST |
|---|
| S-100 | + (focal) | + | + | + (strong,
diffuse) |
| Vimentin | + | + | + | + (strong,
diffuse) |
| Desmin | − | − | + | − |
| MyoD1 | − | − | + | − |
| Myogenin | − | − | + | − |
| CK (Pan) | − | + | − | +/- |
| EMA | − | + | − | +/- |
| CEA | − | + | − | − |
MPNSTs are commonly considered to be aggressive
malignant soft-tissue tumors. The primary treatment for MPNST is a
complete surgical resection with negative margins, and the surgical
margin status is a crucial prognostic factor for MPNST (3). Wong et al (25) reported that the 5-year survival rates
of patients with MPNST who received surgical treatment with and
without negative margins were 67 and 22%, respectively. However,
complete resection with negative margins cannot always be achieved
due to unacceptable complications, including severe loss of
vascular function. Gachiani et al (26) reported that amongst 34 patients with
MPNST who received surgical tumor resection over a period of 40
years, successful complete excision only occurred in 16 patients
(47%). Previous studies have recommended the use of postoperative
radiation therapy to treat MPNST in order to decrease the incidence
of local recurrences, especially in tumors that exhibit the
following characteristics: Lesions >5 cm in size, high tumor
grade, unresectable lesions and positive margins (25,27).
Basso-Ricci (27) reported that
among 25 patients with MPNST who were followed up for >3 years,
14 were free of disease following combined radiological-surgical
treatment, representing a rate that was higher than that found in
the other patient cohort who underwent surgery only. Wong et
al (25) reported that patients
who received irradiation compared with patients who did not had
5-year overall survival rates of 72 and 50%, respectively. However,
the risk-benefit profile of adjuvant radiation therapy must be
carefully discussed with all patients, as it increases the risk of
radiation-induced sarcomas (4).
MPNSTs are typically considered to be insensitive to chemotherapy.
The role of chemotherapy in MPNSTs is commonly limited to the
management of metastatic MPNST in patients with tumors that cannot
be resected (28). The outcome of
MPNST treatment remains poor, with a reported local recurrence rate
between 40–65% and an overall 5-year survival rate <50%
(3,12). As a subset of MPNSTs; however, the
biological features and prognosis in patients with eMPNST remain
unclear due to the small patient cohorts, the lack of large-scale
clinical research and the variation in survival rates. The
clinicopathological characteristics of eMPNST from the
aforementioned clinical studies and case reports are summarized in
Table III. The largest study,
including 63 cases, was presented by Jo and Fletcher (13). The study reported that follow-up data
were available for 31 cases over a median duration of 26 months,
and that 9 patients developed local recurrence and 4 patients
succumbed to metastatic disease. It was concluded that there is a
lower risk for recurrence, metastasis and disease-associated
mortality in patients with eMPNST compared with that in patients
with conventional MPNST. The study by Laskin et al (19) analyzed 26 patients with eMPNSTs,
including 16 patients (61.5%) with tumors in superficial locations
and 10 patients (38.5%) with tumors in deep-seated locations. The
results revealed that eMPNSTs in superficial locations are less
aggressive than the deep-seated tumors. The patient included in the
present study experienced two recurrences, one at 6 months after
the initial surgery, and the other one at 5 months after the second
surgery. Postoperative follow-up at 2 years revealed no evidence of
recurrent disease following the final surgery, which may be due to
the use of a wide resection combined with radiotherapy at a dose of
60 Gy. Furthermore, metastasis has not occurred in the patient in
the 4-year period following initial surgery. In the present study,
the behavior of the superficial eMPNST was favorable, despite
repeated recurrences. Similarly, Misago et al (29) reported that in a patient with
superficial eMPNST located on the back, who underwent three
operations and had relapses following two previous surgeries, no
evidence of recurrence was observed at ~2 years following final
surgery. Despite repeated recurrences, the patient did not exhibit
any signs of metastasis for ~8 years following the first operation.
Superficial eMPNST is generally considered to have benign clinical
behavior (13), which may be due to
its early clinical manifestation and detection of recurrence, the
superficial location and the fact that it can be clinically treated
at an early stage by radical excision with free margins. In
addition, other sarcomas, including superficial/cutaneous
leiomyosarcoma, have a more favorable prognosis than their
deep-seated counterparts (30).
However, previous studies and case reports on eMPNSTs suggested
that superficial eMPNSTs may not have such favorable clinical
progression. For example, in the study by Rekhi et al
(15), data from the follow-up
performed on 8 patients with superficial eMPNST over a median
duration of 26 months revealed that tumors exhibit the same
potentially aggressive biological behaviors regardless of tumor
depth. Although most patients in the study had undergone surgical
resection, >50% of surgical margin statuses were either positive
or unclear, which could affect the accuracy of the conclusion.
Tsuchiya et al (31) reported
that a patient with superficial eMPNST on the face, who had
undergone three surgical excisions and three metastases during a
period of 12 months, succumbed to multiple metastases to the lung,
liver and brain 13 months after the initial surgery. In the present
study, lesions were present on the cheek near the corner of the
eye, and the difficulty in completely excising the tumor, and the
skin defects which remained following surgical removal of the tumor
required a skin graft from other areas of the body each time. These
reasons may have resulted in the tumor not being removed completely
with an adequate surgical margin. The primary treatment for MPNST
consisted of urgent radical excision with a free margin. However,
it is technically challenging to obtain a free margin resection,
due to the large size of the tumor or the difficulty in accessing
the anatomical location of the tumor, including the head and neck
area. This may explain the poor prognosis of MPNSTs of the head and
neck (32,33). Although superficial eMPNSTs may carry
a good prognosis, they have the potential for recurrence and
metastasis, and long-term follow-up for every patient is therefore
essential.
| Table III.Summary of the clinicopathological
features of eMPNST in previous clinical studies and case
reports. |
Table III.
Summary of the clinicopathological
features of eMPNST in previous clinical studies and case
reports.
| First author/s | Year | Cases, n | Location (n/total,
%) | NF1 (n/total) | Treatment (n/total,
%) | Follow-up
(n/months) | Prognosis (n) | IHC positivity
(n/total, %) | (Refs.) |
|---|
| Jo and
Fletcher | 2015 | 63 | Lower extremity
(30/63, 48); upper extremity (6/63, 10); neck (4/63, 6); trunk
(16/63, 25); lip (2/63, 3) visceral (5/63, 8) | Yes (1/63) | Surgery (50/63,
79); RT/CT (13/63, 21) | Available follow-up
data (n=31); median, 36 months | A/NED (n=22); Recur
(n=9); Meta (n=5); DOD (n=4) | S-100+ (63/63,
100); S-100 (+strong, diffuse; 55/63, 87); GFAP+ (24/40, 60); EMA+
(4/29; 14); MARCB1/INI1 lost (35/52, 67) | (13) |
| Laskin et al
region (11/26, 42); arm (9/26, 35); hip/shoulder (2/26, 8);
hand/foot (3/26, 12); trunk (1/26, 4) | 1991 | 26 | Leg/inguinal | No | Surgery (26/26,
100) | Available follow-up
data (n=24); Median, 36 months | A/NED (n=18); Recur
(n=3); Meta (n=4); DOD (n=3) | S100+ (20/25, 80);
S-100 (+strong, diffuse; 14/20, 70); NSE+ (11/17, 65) | (19) |
| Rekhi et al | 2017 | 11 | Axilla (3/11, 27);
leg (4/11, 36); elbow (1/11, 9); subscapular (1/11, 9); mediastinum
(1/11, 9); popliteal fossa (1/11, 9) | Yes (1/11) | Surgery (10/10,
100); RT (1/10, 10%) | Available follow-up
data (n=8); median, 23 months | A/NED (n=2); Recur
(n=4); Meta (n=4); DOD (n=2) | S-100+ (11/11,
100); S-100 (+strong, diffuse; 9/11,82); EMA+; (3/7, 43); CK
(AE1/AE3)+ (2/7, 29); HMB45+ (1/11, 9); SMARCB1/INI1 lost (3/11,
27) | (15) |
| Luzar et al | 2015 | 11 | Arm (1/11, 9); leg
(5/11, 45); trunk (5/11, 45) | No | Surgery (11/11,
100) | Available follow-up
data (n=9); median, 53 months | A/NED (n=4); Recur
(n=2); Meta (n=3); DOD (n=3) | S-100 (+strong,
diffuse; 11/11, 100); GFAP (1/11, 9); Melan A (1/11, 9);
SMARCB1/INI1 lost (3/6, 50) | (14) |
| Dodd et al | 1997 | 2 | Abdominal wall
(1/2, 50); foot (1/2, 50) | No | Surgery (2/2, 100);
RT/CT (1/2, 50) | Available follow-up
data (n=1); 14 months | Meta (n=1); DOD
(n=1) | S-100+ (2/2,
100) | (34) |
| Gupta et al | 2017 | 1 | Abdominal wall | Yes (1/1) | FNAC | 1 month | Meta; DOD | S-100 (+diffuse);
vimentin (+diffuse) | (17) |
| Jiwani et al | 2016 | 1 | Back | No | FNAC | NA | NA | S-100 (+strong,
diffuse); vimentin (+strong, diffuse); D2-40 (+strong, diffuse);
CD57 (+strong, diffuse) | (16) |
| Linos and
Warren | 2015 | 1 | Back | No | Surgery | 45 months | A/NED | S-100 (+strong,
diffuse) | (35) |
| Kusumoto et al | 2015 | 1 | Axilla | No | Incision
biopsy | 2.5 months | Meta; DOD | S-100 (+strong,
diffuse); vimentin (+strong, diffuse); NSE (+strong, diffuse) | (9) |
| Kuzmik et
al | 2013 | 1 | Vestibular
nerve | No | Surgery; RT | 2 months | Meta; DOD | S-100+;
vimentin+ | (36) |
| Natasha et
al | 2011 | 1 | Uterine corpus | No | Surgery; RT;
CT | 6 months | Meta | S-100 (+strong,
diffuse); vimentin (+strong, diffuse) | (37) |
| Minagawa et
al | 2011 | 1 | Foot | No | Surgery; CT | 12 months | Meta; A/NED | S-100+ | (38) |
| Crystal et
al | 2009 | 1 | Back | Yes (1/1) | Incision
biopsy | NA | NA | S-100+ | (18) |
| Allison et
al | 2005 | 1 | Wrist | Yes (1/1) | Surgery | 36 months | A/NED | S-100 (+strong,
diffuse) | (6) |
| Matsuda et
al | 2005 | 1 | Thigh | No | Incision
biopsy | NA | Meta | S-100+; vimentin+;
NSE+; GFAP+; MBP+ | (39) |
| Izycka-Swieszewska
et al | 2005 | 1 | Maxillary
sinus | No | Incision
biopsy | 6 months | DOD | S-100+; vimentin+;
Cam 5.2+; NSE+ | (7) |
| Tsuchiya et
al | 2004 | 1 | Face | No | Surgery | 13 months | Meta; DOD | S-100+; vimentin+;
NGFR+ | (31) |
| Reis-Filho et
al | 2002 | 1 | Presacral area | No | FNAC | 2 months | Meta; DOD | S-100+; vimentin+;
GFAP+; EMA+ | (40) |
| Eltoum et
al | 1999 | 1 | Urinary
bladder | No | Surgery; RT | 2 months | Recur; Meta | S-100 (+strong,
diffuse); vimentin (+strong, diffuse); NSE+ | (41) |
| Lee et
al | 1997 | 1 | Vulva | No | Surgery | 16 months | A/NED | S-100+; vimentin+;
NSE+ | (42) |
| Misago et
al | 1996 | 1 | Back | No | Surgery | 96 months | Recur; A/NED | S-100+;
vimentin+ | (29) |
In conclusion, eMPNST is histologically
characterized by the predominance of epithelioid tumor cells, with
diffuse positivity for S-100 protein, and is rarely associated with
NF-1. eMPNST is a diagnostically challenging neoplasm with an
extensive differential diagnosis. Appropriate clinical and
histological evaluation with a suitable immunohistochemical panel
is crucial to determine the correct diagnosis of this rare tumor.
The principal treatment for eMPNST consists of a complete surgical
resection with negative margins; however, subsequent radiotherapy
may be crucial to reduce the risk of local recurrences. eMPNSTs are
frequently identified in superficial locations and this location
confers a good prognosis. However, eMPNSTs may have the potential
for recurrence and metastasis. Long-term patient follow-up is
therefore crucial.
Acknowledgements
The authors would like to thank Dr YeYuan Chen
(Department of Radiology, The First Affiliated Hospital of Nanchang
University) for the imaging data and description.
Funding
The present study was supported by The Project Fund
of Technology Department of Jiangxi Province, China (grant no.
20141040).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
PD, JZ, CH, MYY, YXL, JSZ and QHT participated in
the treatment of this patient and helped to draft the manuscript.
ZDZ performed the pathological examination and immunohistochemical
staining. PD and JZ performed the data analyses and wrote the
manuscript. JSZ and QHT gave final approval of the version to be
submitted. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics Review
Committee of The First Affiliated Hospital of Nanchang University
(Nanchang, China).
Patients consent for publication
Written informed consent was provided by the
patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tonsgard JH: Clinical manifestations and
management of neurofibromatosis type 1. Semin Pediatr Neurol.
13:2–7. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferner RE and Gutmann DH:
Neurofibromatosis type 1 (NF1): Diagnosis and management. Handb
Clin Neurol. 115:939–955. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
James AW, Shurell E, Singh A, Dry SM and
Eilber FC: Malignant peripheral nerve sheath tumor. Surg Oncol Clin
N Am. 25:789–802. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Farid M, Demicco EG, Garcia R, Ahn L,
Merola PR, Cioffi A and Maki RG: Malignant peripheral nerve sheath
tumors. Oncologist. 19:193–201. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kahn J, Gillespie A, Ondos J, Dombi E,
Camphausen K, Widemann BC and Kaushal A: Radiation therapy in
management of sporadic and neurofibromatosis type 1-associated
malignant peripheral nerve sheath tumors. Front Oncol. 4:3242014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Allison KH, Patel RM, Goldblum JR and
Rubin BP: Superficial malignant peripheral nerve sheath tumor: A
rare and challenging diagnosis. Am J Clin Patho. 124:685–692. 2005.
View Article : Google Scholar
|
|
7
|
Izycka-Swieszewska E, Drogoszewska B,
Filipowicz J, Szurowska E, Kaminski M and Jaskiewicz K: Epithelioid
malignant peripheral nerve sheath tumor involving maxillary sinus.
Neuropathology. 25:341–345. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luzar B and Falconieri G: Cutaneous
malignant peripheral nerve sheath tumor. Surg Pathol Clin.
10:337–343. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kusumoto E, Yamaguchi S, Sugiyama M, Ota
M, Tsutsumi N, Kimura Y, Sakaguchi Y, Kusumoto T, Ikejiri K,
Nakayama Y and Momosaki S: Huge epithelioid malignant peripheral
nerve sheath tumor in the left axilla: A case report. Surg Case
Rep. 1:642015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thway K: Malignant peripheral nerve sheath
tumor: Pathology and genetics. Ann Diagn Pathol. 18:109–116. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Graefe C, Eichhorn L, Wurst P, Kleiner J,
Heine A, Panetas I, Abdulla Z, Hoeft A, Frede S, Kurts C, et al:
Optimized Ki-67 staining in murine cells: A tool to determine cell
proliferation. Mol Biol Rep. May 15–2019.(Epub ahead of print).
doi: 10.1007/s11033-019-04851-2. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hagi T, Nakamura T, Yokoji A, Matsumine A
and Sudo A: Medullary metastasis of a malignant peripheral nerve
sheath tumor: A case report. Oncol Lett. 12:1906–1908. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jo VY and Fletcher CD: Epithelioid
malignant peripheral nerve sheath tumor: Clinicopathologic analysis
of 63 cases. Am J Surg Pathol. 39:673–682. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luzar B, Shanesmith R, Ramakrishnan R,
Fisher C and Calonje E: Cutaneous epithelioid malignant peripheral
nerve sheath tumour: A clinicopathological analysis of 11 cases.
Histopathology. 68:286–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rekhi B, Kosemehmetoglu K, Tezel GG and
Dervisoglu S: Clinicopathologic features and immunohistochemical
spectrum of 11 cases of epithelioid malignant peripheral nerve
sheath tumors, including INI1/SMARCB1 results and BRAF V600E
analysis. Apmis. 125:679–689. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiwani S, Gokden M, Lindberg M, Ali S and
Jeffus S: Fine-needle aspiration cytology of epithelioid malignant
peripheral nerve sheath tumor: A case report and review of the
literature. Diagn Cytopathol. 44:226–231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gupta RK, Saran RK, Ghuliani D, Garg L and
Das A: Metastatic epithelioid malignant peripheral nerve sheath
tumor in a known case of neurofibromatosis-1, cytomorphological
appearance, and critical analysis of immunohistochemistry. Indian J
Med Paediatr Oncol. 38:387–390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Crystal T, Najwa S, Owen LG, Malone JC and
Billings SD: Cutaneous malignant peripheral nerve sheath tumors. J
Cutan Pathol. 36:896–900. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Laskin WB, Weiss SW and Bratthauer GL:
Epithelioid variant of malignant peripheral nerve sheath tumor
(malignant epithelioid schwannoma). Am J Surg Pathol. 15:1136–1145.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hornick JL, Paola DC and Fletcher CD: Loss
of INI1 expression is characteristic of both conventional and
proximal-type epithelioid sarcoma. Am J Surg Pathol. 33:542–550.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miettinen M, Fernandez M, Franssila K,
Gatalica Z, Lasota J and Sarlomo-Rikala M: Microphthalmia
transcription factor in the immunohistochemical diagnosis of
metastatic melanoma: Comparison with four other melanoma markers.
Am J Surg Pathol. 25:205–211. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li G, Liu C, Liu Y, Xu F, Su Z, Wang Y,
Ren S, Deng T, Huang D, Tian Y and Qiu Y: Analysis of clinical
features and prognosis of malignant triton tumor: A report of two
cases and literature review. Oncol Lett. 10:3551–3556. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khin T and Cyril F: Malignant peripheral
nerve sheath tumor: Pathology and genetics. Ann Diagn Pathol.
18:109–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nagasaka T, Lai R, Sone M, Nakashima T and
Nakashima N: Glandular malignant peripheral nerve sheath tumor: An
unusual case showing histologically malignant glands. Arch Pathol
Lab Med. 124:1364–1368. 2000.PubMed/NCBI
|
|
25
|
Wong WW, Hirose T, Scheithauer BW, Schild
SE and Gunderson LL: Malignant peripheral nerve sheath tumor:
Analysis of treatment outcome. Int J Radiat Oncol Biol Phys.
42:351–360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gachiani J, Kim D, Nelson A and Kline D:
Surgical management of malignant peripheral nerve sheath tumors.
Neurosurg Focus. 22:E132007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Basso-Ricci S: Therapy of malignant
schwannomas: Usefulness of an integrated radiologic. Surgical
therapy. J Neurosurg Sci. 33:253–257. 1989.PubMed/NCBI
|
|
28
|
Ferner RE and Gutmann DH: International
consensus statement on malignant peripheral nerve sheath tumors in
neurofibromatosis. Cancer Res. 62:1573–1577. 2002.PubMed/NCBI
|
|
29
|
Misago N, Ishii Y and Kohda H: Malignant
peripheral nerve sheath tumor of the skin: A superficial form of
this tumor. J Cutan Pathol. 23:182–188. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fields JP and Helwig EB: Leiomyosarcoma of
the skin and subcutaneous tissue. Cancer. 47:1561981. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsuchiya D, Takamura H, Saito K, Kashiwa
H, Maeda K and Yamashita H: Immunohistochemical diagnosis of a rare
case of epithelioid malignant peripheral nerve sheath tumor with
multiple metastases. Jpn J Ophthalmol. 48:565–569. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Harald V, Noriaki N, Eva W, Wenzel J,
Bieber T, Wendtner CM, Reinhard G and Schmid-Wendtner MH: Malignant
peripheral nerve sheath tumor of the scalp: Case report and review
of the literature. Dermatol Surg. 37:1684–1688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hanai U, Akamatsu T, Kobayashi M, Tsunoda
Y, Hirabayashi K, Baba T, Atsumi H and Matsumae M: A case of
occipital malignant peripheral nerve sheath tumor with
neurofibromatosis type 1. Tokai J Exp Clin Med. 41:130–134.
2016.PubMed/NCBI
|
|
34
|
Dodd LG, Scully S and Layfield LJ:
Fine-needle aspiration of epithelioid malignant peripheral nerve
sheath tumor (epithelioid malignant schwannoma). Diagn Cytopathol.
17:200–204. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Linos K and Warren S: A misdiagnosed
melanoma: A case of cutaneous epithelioid malignant peripheral
nerve sheath tumor. Dermatol Online J. 21:13030/qt319176×b.
2015.PubMed/NCBI
|
|
36
|
Kuzmik GA, Michaelides EM, Chiang VL,
Nonaka Y, Fukushima T, Vortmeyer AO and Bulsara KR: Rapidly
progressive epithelioid malignant peripheral nerve sheath tumor of
the vestibular nerve. Otol Neurotol. 34:17392013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Natasha G, Bharat R, Pallavi S and
Jambhekar NA: Epithelioid malignant peripheral nerve sheath tumor
of the uterine corpus. Ann Diagn Pathol. 15:441–445. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Minagawa T, Shioya R, Sato C, Shichinohe
R, Yasui G, Ishikawa K and Takahashi H: Advanced epithelioid
malignant peripheral nerve sheath tumor showing complete response
to combined surgery and chemotherapy: A case report. Case Rep Oncol
Med. 2011:7053452011.PubMed/NCBI
|
|
39
|
Matsuda Y, Saoo K, Hosokawa K, Yamakawa K,
Yokohira M, Zeng Y, Takeuchi H, Iwai J, Shirai T, Obika K and
Imaida K: Epithelioid malignant peripheral nerve sheath tumor.
Report of a case with inflammatory infiltration. Pathol Res Pract.
201:355–360. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Reis-Filho JS, Pope LZ, Balderrama CM,
Fillus-Neto J and Schmitt FC: Epithelioid malignant peripheral
nerve sheath tumour: Case report and review of the previously
published cases. Cytopathology. 13:54–63. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Eltoum IA, Moore RJ, Cook W, Crowe DR,
Rodgers WH and Siegal GP: Epithelioid variant of malignant
peripheral nerve sheath tumor (malignant schwannoma) of the urinary
bladder. Ann Diagn Pathol. 3:3041999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee YS, Choi YJ, Kang CS, Kang SJ, Kim BK
and Shim SI: Purely epithelioid malignant peripheral nerve sheath
tumor of the vulva. J Korean Med Sci. 12:78–81. 1997. View Article : Google Scholar : PubMed/NCBI
|