Introduction
Autophagy is an active degradative process that
removes or recycles bulk cytoplasmic constituents through the
endosomal and lysosomal fusion system, resulting in the formation
of autophagosomes in eukaryotic cells. The autophagic process is
robustly upregulated in response to cellular stress, such as
nutrient or cytokine depletion, hypoxia and oxidative damage, and
it is also pivotal to innate intracellular defense mechanisms
against certain pathogens. Autophagy has significant roles in
tissue development, differentiation and remodeling (1). It is also implicated in diseases such
as in the development of tumors, although its precise role is
ambiguous (2).
Melanoma is the most fatal form of skin cancer with
increasing incidence throughout the world. There are no efficacious
therapies for malignant melanoma at present (3). The alkylating agent dacarbazine,
administered as a single agent, remains the current standard
treatment. However, few patients are capable of achieving remission
from distant metastases and the 5-year survival rate is 10%. Thus,
new agents and/or therapeutic strategies with different action
targets need to be developed.
Rapamycin, a lipophilic macrolide antibiotic, was
originally identified as a fungicide and immunosuppressant
(4). However, studies have revealed
that rapamycin can potently arrest the growth of cells derived from
a broad spectrum of cancers (5).
Rapamycin has been shown to specifically inhibit its target,
mammalian target of rapamycin (mTOR), which plays a key role in
tumor development and progression. Rapamycin binds the immunophilin
FK506 binding protein (FKBP12) to form the FKBP12-rapamycin
complex, which then interacts with mTOR and inhibits the
mTOR-mediated phosphorylation of S6K1 and 4E-BP1. In addition,
rapamycin is the best characterized drug that enhances autophagy, a
process of ‘self-eating’ that involves both the death and survival
of cancer cells. Therefore, rapamycin may interfere with different
aspects of the tumor. Certain authors have demonstrated that
rapamycin inhibits lung metastasis of B16 melanoma cells through
downregulating alphav integrin expression and upregulating
apoptosis signaling; autophagy is not involved in the
rapamycin-mediated suppression of metastasis (6). However, there are few studies
concerning the effects of rapamycin on human melanoma and the
interaction with autophagy, thus the impact of rapamycin on M14
cells remains unclear.
Bcl-2 family proteins, which have either pro- or
anti-apoptotic activities, have been studied intensively for the
past decade owing to their significance in the regulation of
apoptosis, tumorigenesis and cellular responses to anti-cancer
therapy (7). Aberrant expression of
Bcl-2 family members is capable of inappropriately promoting or
preventing apoptosis. Bcl-2 is an anti-apoptotic member that
prevents the release of cytochrome c from the mitochondrial
intermembrane space (IMS) into the cytosol. Oppositely, Bax is a
cytosolic protein that translocates to the mitochondria and
participates in the release of cytochrome c in response to
apoptotic stimuli. There is a negative correlation between the
expression of Bcl-2 and Bax. In short, Bcl-2 overexpression leads
to cell survival and Bax overexpression results in cell death
(8).
Morever, Bcl-2 family proteins also target the
autophagy pathway. In this study, we set out to observe the
autophagy of M14 cells induced by rapamycin; to investigate the
effects of rapamycin on regulating the expression of Bcl-2 and Bax
and to identify whether rapamycin may be a promising strategy for
the effective treatment of melanoma.
Materials and methods
Cell culture
The human melanoma cell line M14 was obtained from
Fuxiang Bio-Technology Company (Shanghai, China). Cells were
maintained at 37°C and 5% CO2 in Dulbecco’s modified
Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with
10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin
(Gibco). Cells were inoculated at a density of 1×105
cells/ml and grown for 3 days to reach a phase of exponential
growth (log phase), and were then used for the majority of
experiments unless specified otherwise.
Autophagy induction and reagent
treatment
Cells were plated at a density of approximately
1×105 viable cells/well in 96-well microtiter plates.
For autophagy induction, cells were treated with or without
rapamycin (10, 50 or 100 nmol/l) for 24 h. Following treatment,
cells were analyzed as subsequently described.
Monodansylcadaverine (MDC) labeling
Cells were grown on chamber slides, washed with
phosphate-buffered saline (PBS) and fixed in 10% formalin solution
for 10 min. Autophagic vacuoles were labeled with MDC by incubating
cells with 0.05 mmol/l MDC (Sigma-Aldrich, St. Louis, MO, USA) in
PBS at 37°C for 10 min. Following incubation, cells were washed
three times with PBS and immediately analyzed under a fluorescence
microscope (BX50, Olympus).
Immunofluorescent staining of LC3B
M14 cells (1×105/well) were seeded on
glass cover slides in 24-well plates and left to attach overnight.
Cells were then treated for 24 h with 10, 50 or 100 nmol/l
rapamycin. Cells were then washed with PBS and fixed with 4%
paraformaldehyde in PBS for 15 min at room temperature.
Subsequently, they were permeabilized by 100 μg/ml digitonin
for 15 min, followed by PBS with 3% bovine serum albumin (BSA) for
1 h at room temperature. The anti-LC3B antibody was diluted to
1:200 in PBS, which contained 1% BSA, and then co-incubated with
cells overnight at 4°C. After washing twice with PBS, cells were
then incubated with Cy3-labeled goat anti-rabbit IgG (Sigma, St.
Louis, MO, USA) for 1 h at room temperature in the dark. Then,
cells were washed three times with PBS and stained with
4′,6-diamidino-2-phenylindole (DAPI; 10 μg/ml) for 5 min.
Finally, samples were imaged under a confocal fluorescence
microscope (BX50, Olympus).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
analyses
Cell viability was measured by MTT assay (Amresco,
Solon, Ohio, USA). M14 cells (1×105/100 μl) were
seeded in 96-well plates. Following treatment, 20 μl of MTT
solution (5 mg/ml) was added to each well and incubated (at 37°C
and 5% CO2) for 4 h. Next, the medium was removed and
the wells were allowed to dry. The MTT metabolic product was
resuspended in 200 μl of dimethylsulfoxide (DMSO) and placed
on a shaking table for 5 min. At this point, the absorbance
(optical density) was measured at 530 nm using a microplate reader.
The cell proliferation inhibition rate was calculated using the
equation: Inhibition rate of proliferation (%) =
(Acontrol−Aexperimental)/Acontrol
×100 (1).
Western blot analyses for Bcl-2 and
Bax
Cellular lysate was prepared using
radioimmunoprecipitation assay (RIPA) buffer with protease
inhibitors and quantified using the bicinchoninic acid (BCA)
protein assay (Pierce Biotechnology Inc., Rockford, IL, USA). Equal
amounts of protein were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
electrophoretically transferred to polyvinylidene fluoride (PVDF)
membranes (Millipore, Bedford, MA, USA) using a mini trans-blot
(Bio-Rad, Hercules, CA, USA). Membranes were then blocked with PBST
(PBS with 0.05% Tween-20) containing 5% non-fat dry milk for 1 h
and incubated at 4°C overnight with anti-Bcl-2 antibody (Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA), anti-Bax antibody (Cell
Signaling Technology, Inc., Beverly, MA, USA) in fresh blocking
buffer. Membranes were then washed with PBST and incubated with
horseradish peroxidase-conjugated secondary antibody (Santa Cruz
Biotechnology) for 1 h. The blots were developed using an enhanced
chemiluminescence (ECL) kit (Pierce). Protein levels were
normalized against β-actin (Sigma-Aldrich).
Flow cytometry with rhodamine 123 (Rh123)
staining
Mitochondrial membrane potential (MMP) was assessed
by the retention of Rh123, a membrane-permeable fluorescent
cationic dye. The uptake of Rh123 by mitochondria is proportional
to the MMP. Cells were incubated with Rh123 (0.25 nmol/l) in the
dark at room temperature for 20 min. After washing with PBS, the
cells were analyzed by FACScan (Becton-Dickinson, San Jose, CA,
USA) with excitation and emission wavelengths of 495 and 535 nm,
respectively. Cells treated with rapamycin (10, 50 and 100 nmol/l)
for 24 h were incubated with Rh123 (0.25 nmol/l) in the dark at
room temperature for 20 min. After washing with PBS, the change in
MMP was detected by Rh123 staining using flow cytometry.
Electron microscopy
Cells were harvested by scraping them from the
plates. They were then washed twice with PBS and fixed with 2%
paraformaldehyde/2% glutaraldehyde in 0.2 M sodium cacodylate
buffer (pH 7.4). Cell pellets were post-fixed with 1% (v/v) osmic
acid in sodium cacodylate and stained with 1% uranyl acetate.
Following dehydration, pellets were embedded in Durcopan (Fluka,
Sigma). Ultrathin sections were prepared using ULTRACUT S and
observed with a JEM 1010 transmission electron microscope. Images
were captured and are shown in Results.
Statistical analysis
Data were expressed as mean ± standard deviation.
Mean values were compared using a Student’s t-test for independent
variables. P<0.05 was considered statistically to indicate a
statistically significant difference. All statistical analyses were
performed with SPSS 11.0 (SPSS Inc., Chicago, IL, USA).
Results
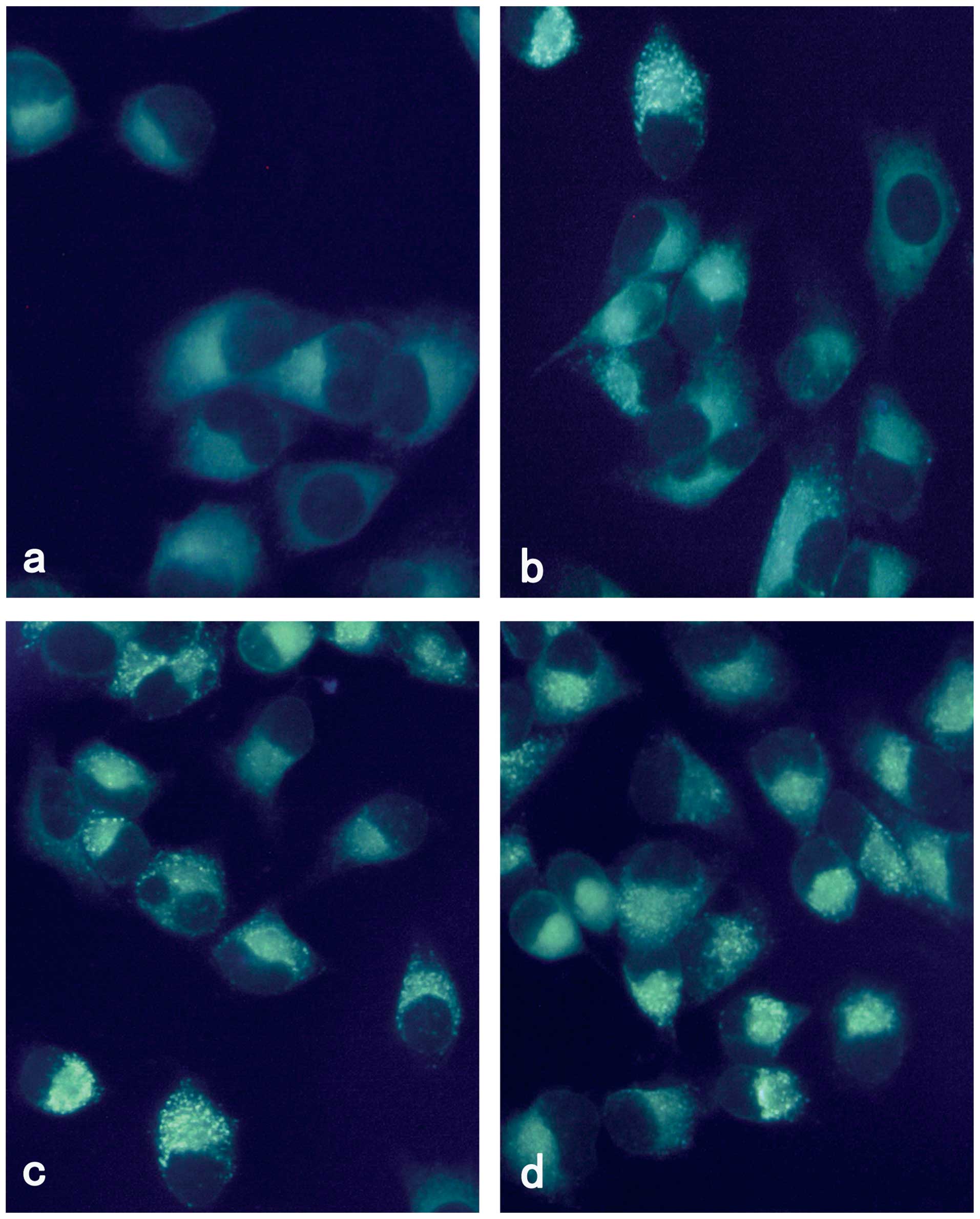
Vesicular accumulation of MDC following
rapamycin treatment
MDC is an autofluorescent compound and has been
proposed as a tracer for autophagic vacuoles. Thus, we studied the
incorporation of MDC into M14 cells using fluorescence microscopy.
As shown in Fig. 1, M14 cells
treated with rapamycin for 24 h demonstrated a punctate pattern of
MDC-labeled fluorescence. By contrast, uninfected cells exhibited a
diffused distribution of MDC-labeled fluorescence. Rapamycin
induced autophagy of M14 in a concentration-dependent manner.
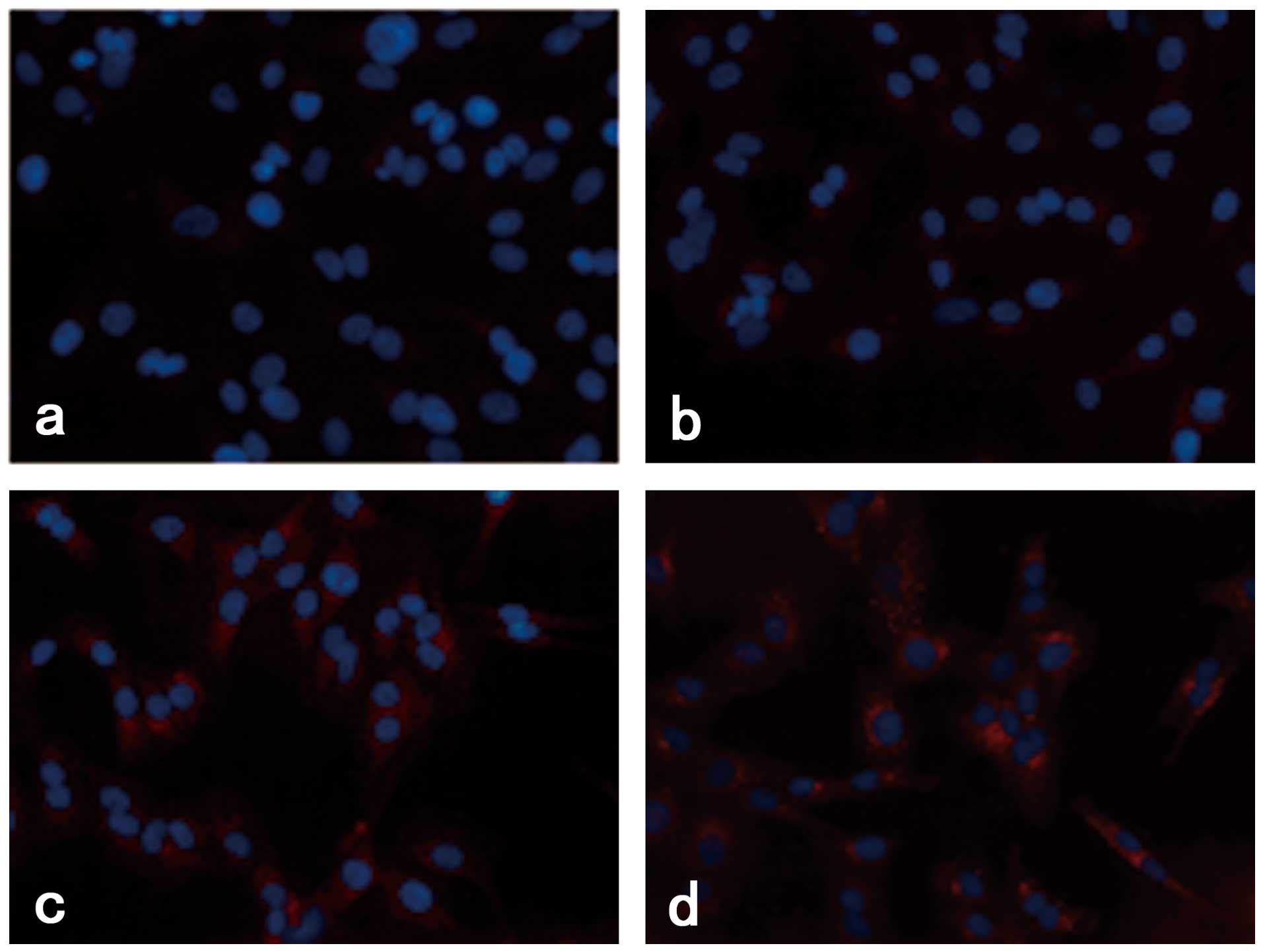
Rapamycin induced the accumulation of
LC3B fluorescence dots in M14 cells
We investigated the effect of rapamycin treatment on
the staining of LC3B protein, which is produced during
autophagosome formation. As demonstrated in Fig. 2, the quantity of LC3B fluorescence
dots increased with increasing rapamycin concentration.
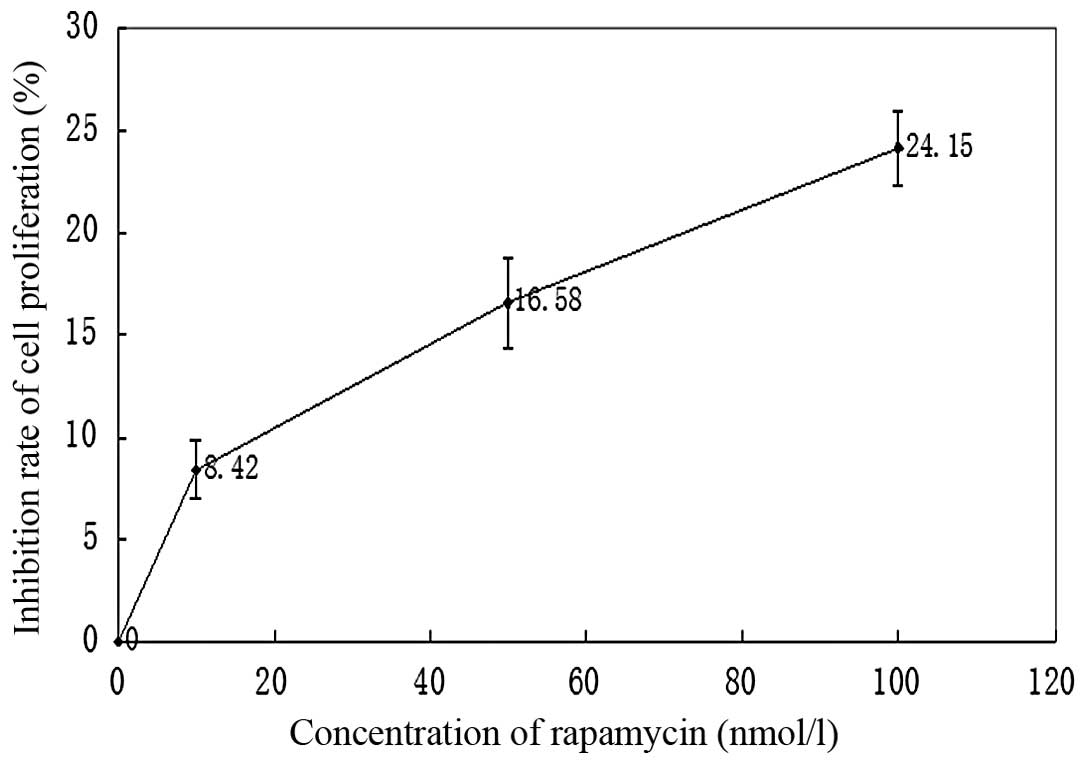
Effects of rapamycin treatment on the
proliferation of M14 cells
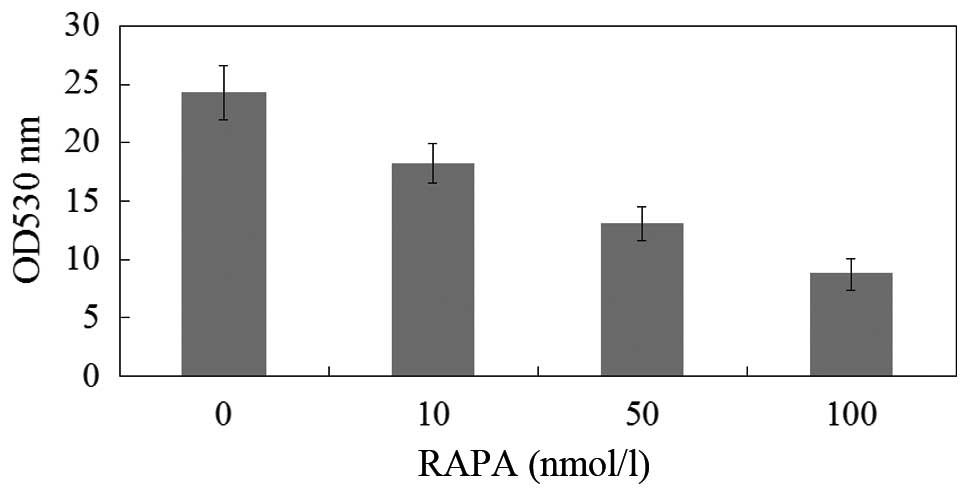
The proliferation of M14 cells was measured using
MTT assay and expressed as a ratio of color intensity from the
rapamycin treatment group to that of the DMSO-treated control
group. Data were obtained from three independent experimental
replicates. Fig. 3 and Table I demonstrated that as the rapamycin
concentration increased from 0–100 nmol/l, the strength of the
inhibitory effect increased.
 | Table IEffects of rapamycin treatment on the
proliferation of M14 cells (n=4, mean ± SD). |
Table I
Effects of rapamycin treatment on the
proliferation of M14 cells (n=4, mean ± SD).
| Group | Inhibition rate of
proliferation (%) |
|---|
| Control | 0 |
| Rapamycin | |
| 10 nmol/l | 8.42±2.88a |
| 50 nmol/l |
16.58±4.43b |
| 100 nmol/l |
24.15±3.69c |
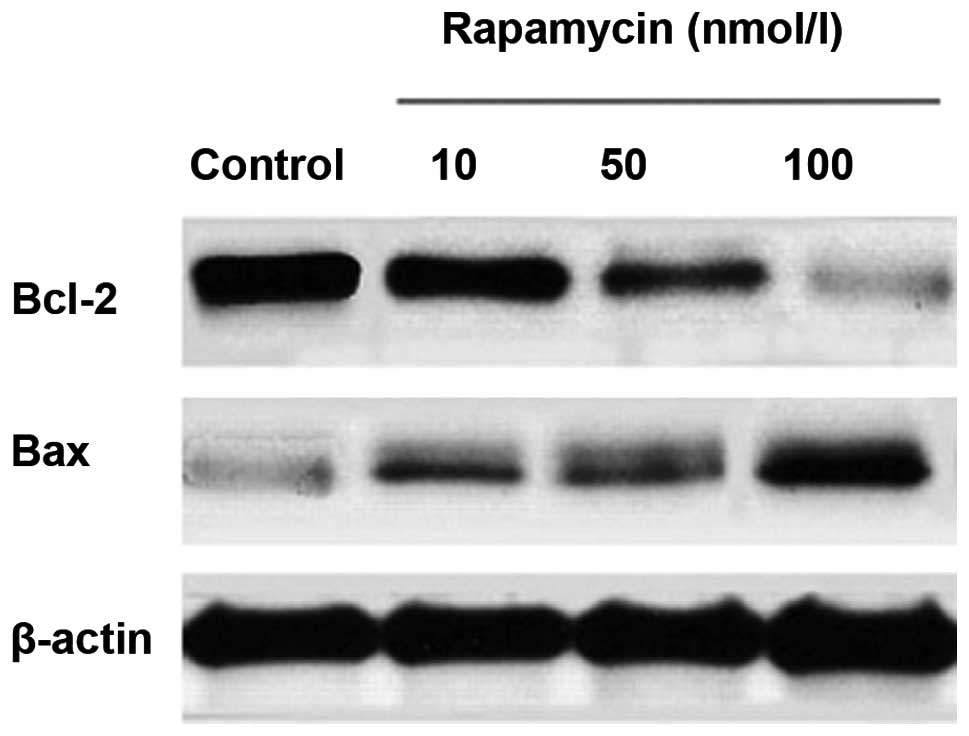
Effects of rapamycin treatment on Bcl-2
and Bax expression
M14 cells were treated with various concentrations
of rapamycin. Bcl-2 and Bax levels were assayed by western blot
analysis. Equal amounts of total cellular protein were resolved by
10% SDS-PAGE and β-actin was used as an internal control. As
revealed in Fig. 4 and Table II, there was a significant
dose-dependent reduction in the level of Bcl-2 expression after
rapamycin treatment for 24 h of 10, 50 or 100 nmol/l. By contrast,
the level of Bax expression increased.
| Table IIGray value of Bcl-2 and Bax. |
Table II
Gray value of Bcl-2 and Bax.
| Group | Bcl-2 | Bax |
|---|
| Control | 1 | 1 |
| Rapamycin | | |
| 10 nmol/la | 0.94765±0.042485 | 1.96733±0.097991 |
| 50 nmol/lb | 0.70357±0.043447 | 2.23067±0.372162 |
| 100 nmol/lc | 0.34717±0.048413 | 2.89067±0.208375 |
MMP decreased following rapamycin
treatment
Changes in mitochondrial function are capable of
launching autophagy. In the case of nutritional deficiencies, there
may be mitochondrial permeability transition pores (MPTP), which
are a marker of impaired mitochondrial function. Following this,
mitochondria are engulfed by lysosomes. A decline in MMP is a
morphological characteristic of mitochondrial function recession.
Rh123 is a cationic lipophilic fluorescent dye. It accumulates in
mitochondria and its intake is proportional to the MMP. When the
MMP decreases, Rhl23 in mitochondria leaks out and the fluorescence
intensity in the cells reduces. Thus, Rhl23 fluorescence intensity
reflects the MMP of mitochondria. As indicated in Fig. 5, the fluorescence intensity of Rh123
was significantly lower in the cells exposed to rapamycin relative
to the control group, and the fluorescence intensity decreased with
increasing rapamycin concentration.
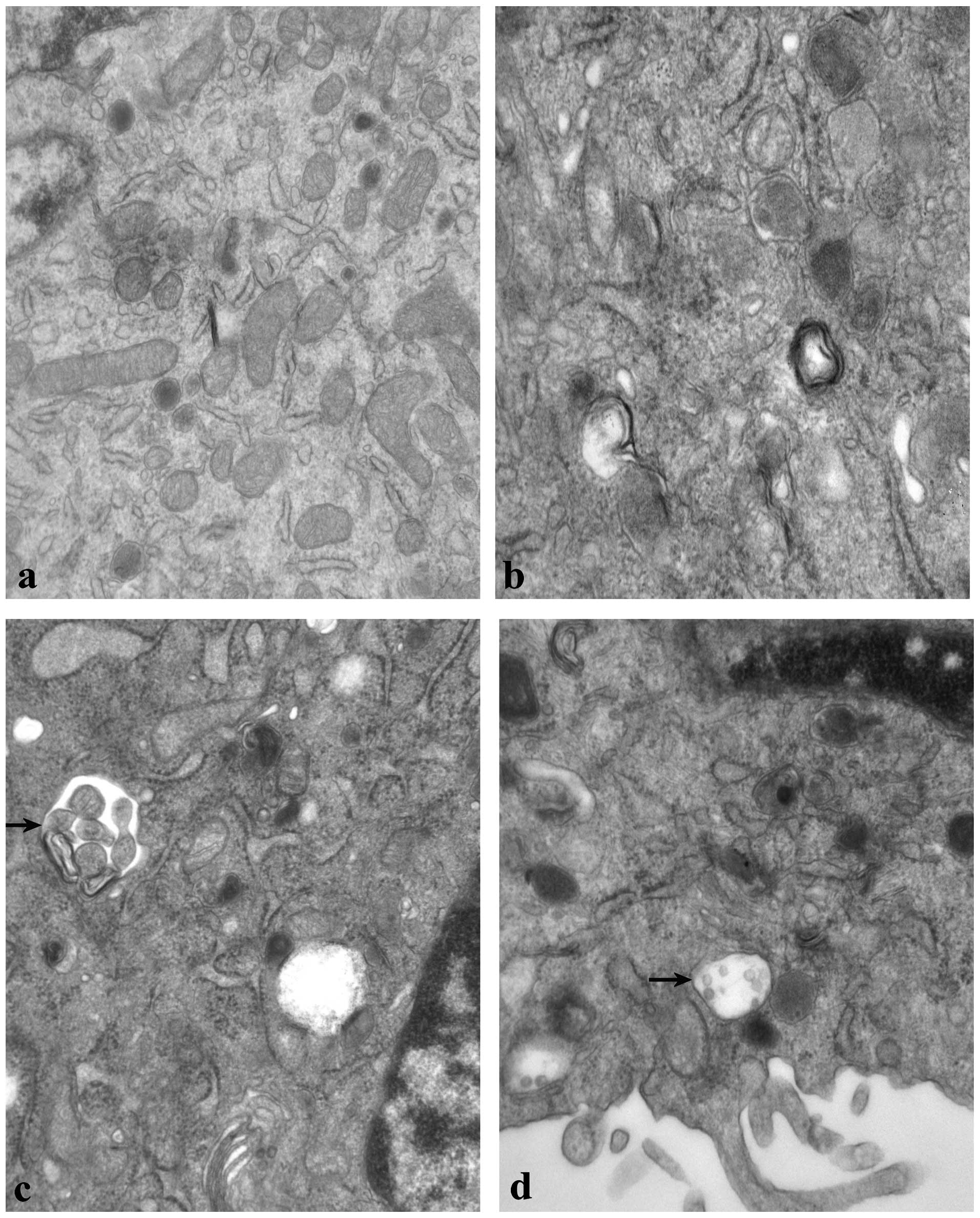
Ultrastructural examination of autophagy
following rapamycin treatment
As demonstrated in Fig.
6, the ultrastructure of the control group (6a) was normal
compared with the rapamycin group (6b-d). Double-membrane-bound
vesicles, swelling of the mitochondria and vacuolization of certain
cells were observed in M14 cells following treatment with 100
nmol/l rapamycin for 24 h. Additionally, abnormal mitochondria, an
increased number of lysosomes and autophagosomes were observed.
Discussion
Melanoma is the most fatal form of skin cancer with
increasing incidence throughout the world. There are no efficacious
therapies for malignant melanoma at present. It is possible to
explore new methods of treatment by modulating autophagy. For
example, this may be achieved via rapamycin, an autophagy inducer
that promotes autophagy by inhibiting mTOR (9). Interest in applying rapamycin in
cancer therapy was initiated due to observations of frequent Akt
activation in multiple types of cancer and identification of
TSC1/TSC2 as tumor suppressors that are inhibited by Akt. Cancer
therapy with rapamycin has been successfully implemented for kidney
cancer, with the approval of CCI-779 (temsirolimus) for the
treatment of renal clear cell carcinoma (RCC) (10). In RCC, rapamycin may be effective
due to its inhibition of the production of HIF-1α, a key
transcription factor that mediates oncogenic alterations in the
cellular metabolism (11,12). Aside from RCC, numerous clinical
trials are testing rapamycin in cancers linked to Akt activation,
such as glioblastoma and prostate cancer. Therefore, in this study,
we set out to investigate rapamycin-induced autophagy and its
possible mechanisms.
Autophagy is a process by which cells degrade
macromolecular intracellular material via sequestration into a
double-membranous structure, known as an autophagosome, which then
delivers the enclosed material to a lysosome for degradation.
Initially believed to be a system dedicated to the ‘recycling’ of
macromolecular material within the cell, autophagy is now known to
be involved in a multitude of cellular processes including
immunity, tumorigenesis, programmed cell death, the selective
degradation of organelles, aging and numerous neurodegenerative
conditions (13). There are
currently 33 identified autophagy-related genes shown to play a
role in autophagy, and many techniques are available to detect
autophagy, including transmission electron microscopy, half-life
assessments of long-lived proteins, detection of LC3
maturation/aggregation, fluorescence microscopy and co-localization
of mitochondrion or endoplasmic reticulum-specific markers with
lysosomal proteins (14). In this
study, we observed autophagy in M14 cells treated with various
concentrations of rapamycin for 24 h by MDC labeling, LC3B protein
staining and transmission electron microscopy. The results showed
that rapamycin induced autophagy in M14 cells in a
concentration-dependent manner. By using electron microscopy (the
most reliable method of observing cell ultrastructure) we observed
autophagosomes (dysfunctional mitochondria sequestered into a
double-membrane-bound vesicle) and mitochondria that had lost their
cristae in cells treated with rapamycin compared with those in the
control group. Mitochondria that were abnormal in appearance and
and an increased number of lysosomes were also observed.
Mitochondria are vital organelles for cellular
metabolism and bioenergetics, but they are also the key regulators
of cell death (15). Notably, in
many (if not all) paradigms of apoptosis, MMP represents the point
of no return in the cascade of events that ultimately leads to cell
death (16). MMP results in the
leakage of potentially toxic proteins from the mitochondrial IMS.
In addition, mitochondria play a role in stress responses and can
produce reactive oxygen species (ROS) when damaged. Selective
degradation of mitochondria by autophagy is also known as
‘mitophagy’ and is considered to be promoted by their functional
impairment and/or by MMP. Mitophagy may ensure the removal of
damaged and potentially dangerous mitochondria, thus acting as a
quality control mechanism. We used Rh123 to detect the changes in
MMP by flow cytometry and discovered that the fluorescence
intensity of Rh123 was significantly lower in the cells exposed to
rapamycin relative to the control group (P<0.05). Thus, combined
with the electron microscopy results, we conclude that rapamycin
may affect the regulation mechanism of mitochondria and cause
swelling and vacuolization of mitochondria.
The proliferation of M14 cells was measured by the
inhibition rate of proliferation using MTT assay (Table II). The results showed that the
proliferation of melanoma cells was significantly inhibited in the
rapamycin group (P<0.01). Compared with the other two treatment
groups, the 10 nmol/l group demonstrated notably higher gray values
(P<0.05); however, no significant differences were observed
between the 50 nmol/l and 100 nmol/l groups (P>0.05). Thus, it
is suggested that rapamycin possesses a biphasic effect. On one
hand it is capable of inducing autophagy, on the other hand it may
inhibit proliferation.
The correlation between autophagy and apoptosis is
complex and controversial (17). It
varies with cell type and stress stages. Depending on the cellular
context and stimulus, autophagy may be indispensable for apoptosis
by initiating the process (18). In
other cellular settings, autophagy may rather antagonize or delay
apoptosis and inhibition of autophagy may increase the sensitivity
of the cells to apoptotic signals (19). In certain cell systems, two
processes can occur independently. Numerous signaling pathway
overlaps are found between autophagy and apoptosis, including
various kinases such as PI3K, PKB/Akt, Bcl-2 family members, PTEN,
c-Myc and Ras.
Members of the Bcl-2 family proteins, including
Bcl-2, Bax and Bak, are thought to play regulatory roles in the
apoptotic execution of the cells (20). However, more and more studies have
revealed that Bcl-2 family proteins also target the autophagy
pathway. Further biochemical and genetic data has led to a
resurgence of interest in the role of autophagy in tumor
suppression (21). In addition to
the discovery that an autophagy execution protein, Beclin 1, is a
tumor suppressor protein, oncogenic signaling molecules are capable
of suppressing autophagy and tumor suppressors are able to
stimulate autophagy.
Bcl-2 and Beclin 1 interact in mammalian cells.
Firstly, Bcl-2 inhibits Beclin 1-dependent autophagy. To explore
the mechanism, an autophagy-competent cell line (HT-29 colon
carcinoma cells) that expresses endogenous Beclin 1 has been used.
In HT-29 cells, stable transfection of Bcl-2 inhibits
starvation-induced autophagy decreases the association of Beclin 1
with Vps34 and decreases the magnitude of Beclin 1-associated class
III phosphoinositide-3-kinase activity. This finding suggests that
Bcl-2 overexpression blocks the formation of the
autophagy-promoting Beclin 1-Vps34 complex. Additionally, Bcl-2
inhibits Beclin 1-dependent autophagic cell death. Bcl-2,
apoptosis-inhibiting gene and Bax, apoptosis inducing-gene, are two
important members of the Bcl-2 family; the ratio between the two
determines the survival of cells. There is a negative correlation
between the expression of Bcl-2 and Bax. Bcl-2 overexpression leads
to cell proliferation, while alternatively Bax leads to cell death.
In this study, we investigated the effects of rapamycin treatment
on Bcl-2 and Bax expression. As described previously, there was a
significant dose-dependent reduction in the level of Bcl-2
expression after rapamycin treatment for 24 h of 10, 50 or 100
nmol/l. By contrast, the level of Bax expression increased. This is
speculated to be caused by MPTP. Certain studies suggest that MPTP
is formed by the interaction with Bcl-2 family proteins and other
proteins outside the mitochondria; Bax may promote the opening of
MPTP. Besides, certain authors consider Bcl-2 protein to inhibit
the release of Ca2+ from the endoplasmic reticulum,
while Bax may promote this release. Rapamycin reduces the
expression of Bcl-2 and increases the expression of Bax, which
results in an overload of Ca2+ in the mitochondria and
promotes the opening of MPTP. Then, mitochondria swell, their outer
membranes collapse and exit into the cytoplasm, thus initiating
apoptosis.
The ability of Bcl-2 to inhibit autophagy through a
direct interaction with Beclin 1 is of particular interest with
regards to cancer. The role of autophagy in tumorigenesis and
cancer treatment is complex. Autophagy has roles in both tumor
prevention and survival, as well as in treatment resistance. As
autophagy protects cells from metabolic stress, it is speculated
that the upregulation of autophagy preserves cellular fitness and
genomic integrity, and thus prevents tumorigenesis (22). On the contrary, established tumor
cells utilize autophagy to survive stresses such as nutrient
limitation and hypoxia. Furthermore, tumor cells activate autophagy
as a stress response to survive cancer treatment. A recent study
has demonstrated that under certain situations, such as in
radiation-resistant melanoma, inhibition of autophagy may be
exploited to prevent resistance to treatment (23). Notably, modulation of autophagy has
significant potential in cancer diagnosis and treatment.
At present, there are few studies concerning the
effects of rapamycin on melanoma cells; we speculate that rapamycin
may be a promising strategy for the effective treatment of melanoma
by modulating autophagy and regulating the expression of Bcl-2
family proteins. However, in view of the biphasic effect of
rapamycin (in inducing autophagy and apoptosis) there is a
requirement to establish the appropriate concentration of rapamycin
that is capable of inducing mitophagy, promoting tumor cell
apoptosis and activating autophagy, so as to provide a new approach
to treating malignant melanoma.
Acknowledgements
This study was supported by the
Science and Technology Social Development Plan of Jiangsu Province
(No. BS2007072) and the National Natural Science Foundation of
China (No. 81171517).
References
|
1
|
Tang Y, Chen Y, Jiang H and Nie D:
Short-chain fatty acids induced autophagy serves as an adaptive
strategy for retarding mitochondria-mediated apoptotic cell death.
Cell Death Differ. 18:602–618. 2011. View Article : Google Scholar
|
|
2
|
Bialik S and Kimchi A: Autophagy and tumor
suppression: recent advances in understanding the link between
autophagic cell death pathways and tumor development. Adv Exp Med
Biol. 615:177–200. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ouyang D, Zhang Y, Xu L, Li J, Zha Q and
He X: Histone deacetylase inhibitor valproic acid sensitizes B16F10
melanoma cells to cucurbitacin B treatment. Acta Biochim Biophys
Sin. 43:487–495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mondesire WH, Jian W, Zhang H, et al:
Targeting mammalian target of rapamycin synergistically enhances
chemotherapy-induced cytotoxicity in breast cancer cells. Clin
Cancer Res. 10:7031–7042. 2004. View Article : Google Scholar
|
|
5
|
Wu Q, Kiguchi K, Kawamoto T, et al:
Therapeutic effect of rapamycin on gallbladder cancer in a
transgenic mouse model. Cancer Res. 67:3794–3800. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang Z, Lei Z, Li B, et al: Rapamycin
inhibits lung metastasis of B16 melanoma cells through
down-regulating alphav integrin expression and up-regulating
apoptosis signaling. Cancer Sci. 101:494–500. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cory S, Huang DC and Adams JM: The Bcl-2
family: roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Renna M, Jimenez-Sanchez M, Sarkar S and
Rubinsztein DC: Chemical inducers of autophagy that enhance the
clearance of mutant proteins in neurodegenerative diseases. J Biol
Chem. 285:11061–11067. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hudes G, Carducci M, Tomczak P, et al:
Temsirolimus, interferon alfa, or both for advanced renal-cell
carcinoma. N Engl J Med. 356:2271–2281. 2007. View Article : Google Scholar
|
|
11
|
Hudson CC, Liu M, Chiang GG, et al:
Regulation of hypoxia-inducible factor 1alpha expression and
function by the mammalian target of rapamycin. Mol Cell Biol.
22:7004–7014. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhong H, Chiles K, Feldser D, et al:
Modulation of hypoxia-inducible factor 1alpha expression by the
epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP
pathway in human prostate cancer cells: implications for tumor
angiogenesis and therapeutics. Cancer Res. 60:1541–1545. 2000.
|
|
13
|
Goldman SJ, Taylor R, Zhang Y and Jin S:
Autophagy and the degradation of mitochondria. Mitochondrion.
10:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tasdemir E, Galluzzi L, Maiuri MC, et al:
Methods for assessing autophagy and autophagic cell death. Methods
Mol Biol. 445:29–76. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fantin VR and Leder P: Mitochondriotoxic
compounds for cancer therapy. Oncogene. 25:4787–4797. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Laane E, Tamm KP, Buentke E, et al: Cell
death induced by dexamethasone in lymphoid leukemia is mediated
through initiation of autophagy. Cell Death Differ. 16:1018–1029.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi M, Wang HN, Xie ST, et al:
Antimicrobial peptaibols, novel suppressors of tumor cells,
targeted calcium-mediated apoptosis and autophagy in human
hepatocellular carcinoma cells. Mol Cancer. 9:262010. View Article : Google Scholar
|
|
20
|
Matsumoto A, Isomoto H, Nakayama M, et al:
Helicobacter pylori VacA reduces the cellular expression of STAT3
and pro-survival Bcl-2 family proteins, Bcl-2 and Bcl-XL, leading
to apoptosis in gastric epithelial cells. Dig Dis Sci. 56:999–1006.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pattingre S and Levine B: Bcl-2 inhibition
of autophagy: A new route to cancer? Cancer Res. 66:2885–2888.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eskelinen EL: The dual role of autophagy
in cancer. Curr Opin Pharmacol. 11:294–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ravikumar B, Sarkar S, Davies JE, et al:
Regulation of mammalian autophagy in physiology and
pathophysiology. Physiol Rev. 90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|