Introduction
Colorectal cancer is a major cause of cancer-related
mortality worldwide. The incidence of new cases has increased in
recent years, particularly in affluent societies, and colon cancer
is often highly metastatic and resistant to anticancer treatment
strategies (1,2). One of the key genetic defects that
confers resistance against colon cancer treatment is the mutation
of p53 (3). This key apoptotic
regulator and tumor suppressor mediates multiple responses to
anticancer agents by modulating p21-associated cell cycle control,
apoptosis and DNA repair (4).
Mutations in p53 have been shown to abolish the efficiency of
cancer treatment in vitro and in vivo, while
reintroduction of wild-type p53 was demonstrated to sensitize
p53-null cancer cells to therapeutic agents (5). In vitro and in vivo
studies have suggested that a mutation in p53 synergistically
interacts with hypoxia to elevate the expression of vascular
endothelial growth factor (VEGF; a tumor-associated angiogenesis
factor) in cancer cells. This is notable as VEGF is an important
factor in cancer cell proliferation and metastasis, and p53
inhibits various cancer cell survival signals, including VEGF
(6–9).
A number of phytochemicals have been reported to
activate p53, thereby inducing apoptosis and suppressing the
expression levels of VEGF and matrix metalloproteinase (MMP)-9,
which are elevated in colon cancer (10–12).
One such phytochemical, epigallocatechin-3-gallate (EGCG), the
major polyphenolic compound of green tea, has been reported to have
anti-proliferatory and apoptotic effects in a number of types of
cancer cells (13). VEGF is known
to support tumor growth and metastasis, and the status of VEGF has
been shown to predict the prognosis of various human tumors
(14). MMP-9 is one of the most
important proteins involved in cancer cell metastasis and the
status of MMP-9 has been shown to be overexpressed in various
cancer cells (15). However, no
previous study has examined the effects of EGCG on VEGF or MMP-9
and the involvement of p53 in this process.
The present study examined the effects of EGCG on
VEGF and MMP-9 protein expression in two colon cancer cell lines:
HCT-116 cells, which express wild-type p53, and HT-29 cells, which
express mutant p53. The effects of EGCG on migration and apoptosis
in the two cell lines and the possible involvement of AMP-activated
protein kinase (AMPK) activation in the response to EGCG were also
tested.
Materials and methods
Cells and reagents
The HCT116 and HT-29 human colon cancer cell lines
were purchased from the American Type Culture Collection (Manassas,
VA, USA) and were cultured in RPMI-1640 with 10% fetal bovine serum
(Gibco, Rockville, MD, USA). EGCG,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and Hoechst 33342 were obtained from Sigma (St. Louis, MO, USA).
Pifithrin-α and compound C were purchased from Calbiochem (San
Diego, CA, USA). Monoclonal antibodies specific for p53 and AMPKα1
were purchased from Cell Signaling Technology (Beverly, MA, USA).
VEGF and MMP-9 antibodies were purchased from Santa Cruz
Biotechnology, Inc., (Santa Cruz, CA, USA) and the β-actin antibody
was obtained from Sigma.
Cell proliferation measurements and
morphological examination
Cells seeded on 96-well microplates at
4×103 cells/well were incubated with test compounds at
50 and 100 μm for 48 h. Following incubation with the test
compound, the medium was removed and the cells were incubated with
100 μl MTT solution (2 mg/ml MTT in PBS) for 4 h. The samples were
then solubilized in DMSO. The purple formazan dye, converted from
MTT by viable cells, was quantified by absorbance at 560 nm. For
the morphological examination, the cells were grown on 6-well
plates, treated with flavonoids for 48 h and then examined under a
light microscope (×400 magnification).
Apoptosis detection
Apoptosis was measured using a FITC-Annexin V
apoptosis detection kit (BD Pharmingen™, San Diego, CA, USA) or
Hoechst 33342 chromatin staining dye. For Annexin V/PI staining
following treatment with selenium, the cells were harvested by
trypsinization, washed with ice-cold phosphate-buffered saline
(PBS) and suspended in a binding buffer at a density of
1×106 cells/ml. The cells were stained with Annexin
V-FITC and propidium iodide (PI) and analyzed by flow cytometry
(Becton-Dickinson Biosciences, Franklin Lakes, NJ, USA). To examine
chromatin condensation, the cells were stained with 10 μM Hoechst
33342 for 30 min and fixed with 3.7% formaldehyde for 15 min.
Changes in chromatin condensation were observed by fluorescence
microscopy (Olympus Optical Co., Tokyo, Japan).
In vitro wound healing assay
An in vitro wound healing assay was applied
to determine cell mortality caused by EGCG. This assay was
performed using a standard method (16) with certain modifications. Briefly,
1×105 HCT116 and HT-29 cells were seeded on a 6-well
plate in complete medium overnight to obtain a full confluent
monolayer. Subsequent to 12 h of starvation, a 20-μl pipette tip
was used to create a straight cell-free wound. Each well was washed
twice with PBS to remove any debris. The cells were then cultured
in serum-free medium in the absence or presence of 50–100 μM EGCG.
The distances between the two edges of the scratch were analyzed
quantitatively.
Western blot analysis
Following starvation for 12 h in serum-free medium,
the cells were seeded into 6-well plates and treated with test
compounds. Total proteins were extracted using a RIPA lysis buffer
[50 mM Tris-HCl (pH 8.0), 1% NP-40, 0.5% sodium deoxycholate, 150
mM NaCl and 1 mM PMSF] and subjected to western blot analysis with
specific antibodies. The proteins were then visualized by enhanced
chemiluminescence (Intron, Kyunggi, Korea) and detected using a LAS
4000 chemiluminescence detection system (Fuji, Tokyo, Japan).
Immunofluorescence staining
The cells were seeded on a 12-well plate with cover
glasses. Subsequent to treatment with 10–50 μm EGCG for 24 h, the
cells were fixed in 3.7% formaldehyde for 20 min at room
temperature (RT) and permeabilized in 0.2% Triton X-100 for 20 min
at RT. Then cells were blocked with 1% bovine serum albumin for 1
h. The cells were then incubated overnight with the primary
antibodies of AMPKα1 and VEGF. Subsequent to being washed, the
cells were incubated with Alexa 546-conjugated anti-rabbit IgG and
Alexa 488-conjugated anti-mouse IgG (both from Molecular Probes,
Eugene, OR, USA) for 1 h at RT. The cell nuclei were then stained
with 10 μM Hoechst 33342 for 10 min and observed using a confocal
microscope (Carl Zeiss, Thornwood, NY, USA).
Statistical analysis
Cell viability and migration rate data were
statistically analyzed using an unpaired t-test (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
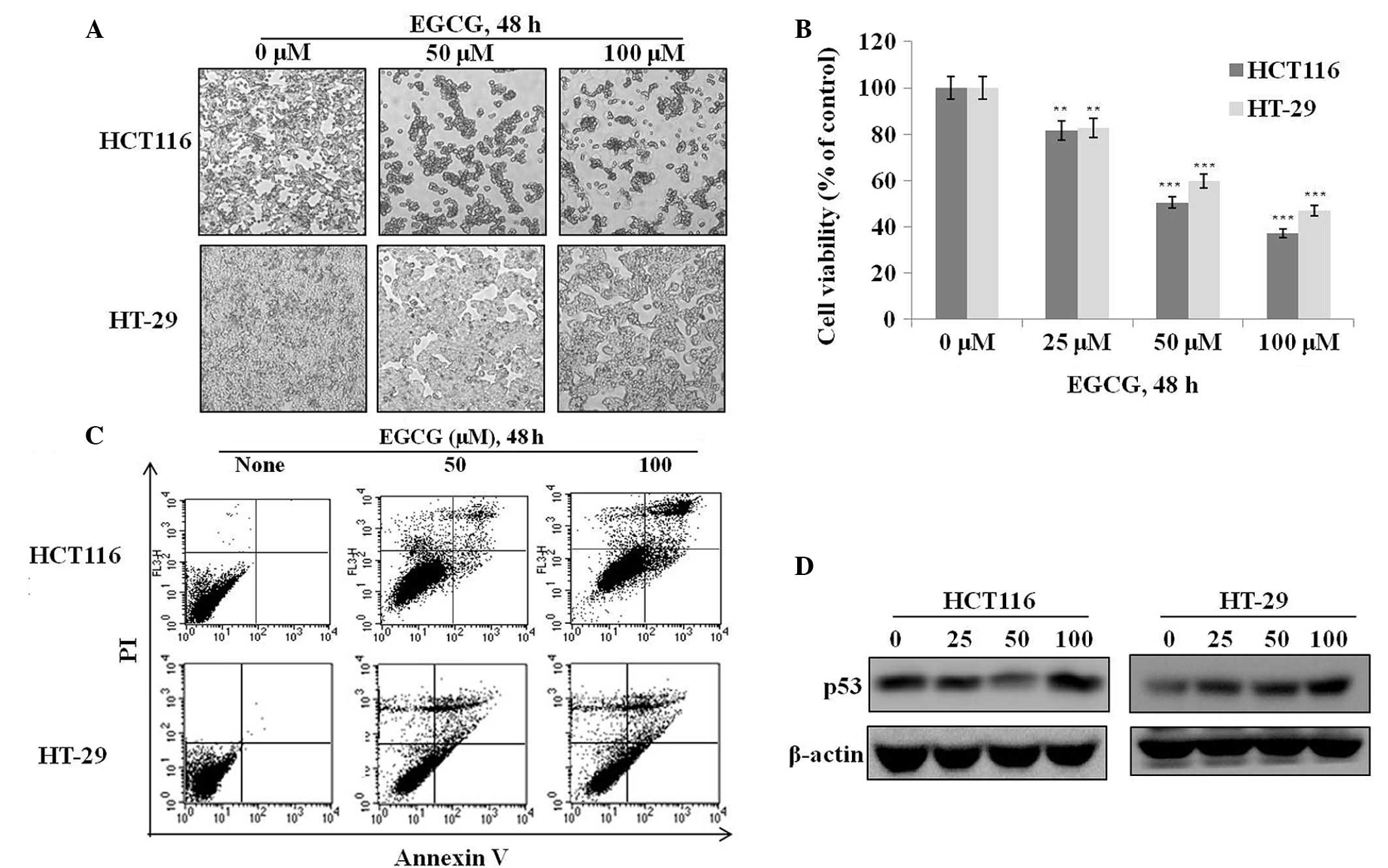
EGCG suppresses cell proliferation and
induces apoptosis in HT-29 and HCT116 cells
To investigate the effects of EGCG on cell
proliferation and apoptosis, HT-29 and HCT116 cells were treated
with various concentrations of EGCG for 48 h and their
morphological, proliferative and apoptotic characteristics were
observed. The majority of cells in the two EGCG-treated groups
shrank and became globular in shape compared to the controls
(Fig. 1A). An assessment of cell
viability by MTT assay showed that EGCG dose-dependently suppressed
the proliferation of the HT-29 and HCT116 cells (Fig. 1B). An examination of apoptosis by
Annexin V staining showed that EGCG dose-dependently increased
apoptotic cell death in the two cell types (Fig. 1C). These results suggest that the
growth inhibitory properties of EGCG may arise from p53-dependent
or -independent pathways. The ineffectiveness of EGCG in the
regulation of mutated p53 (HT-29 cells) compared with the increment
of p53 proteins with EGCG of HCT116 cells was shown (Fig. 1D). No previous study has clearly
shown that EGCG induces apoptosis through both p53-dependent and
p53-independent pathways. The present study reports that EGCG is
capable of inducing apoptosis without using the traditional p53
pathway.
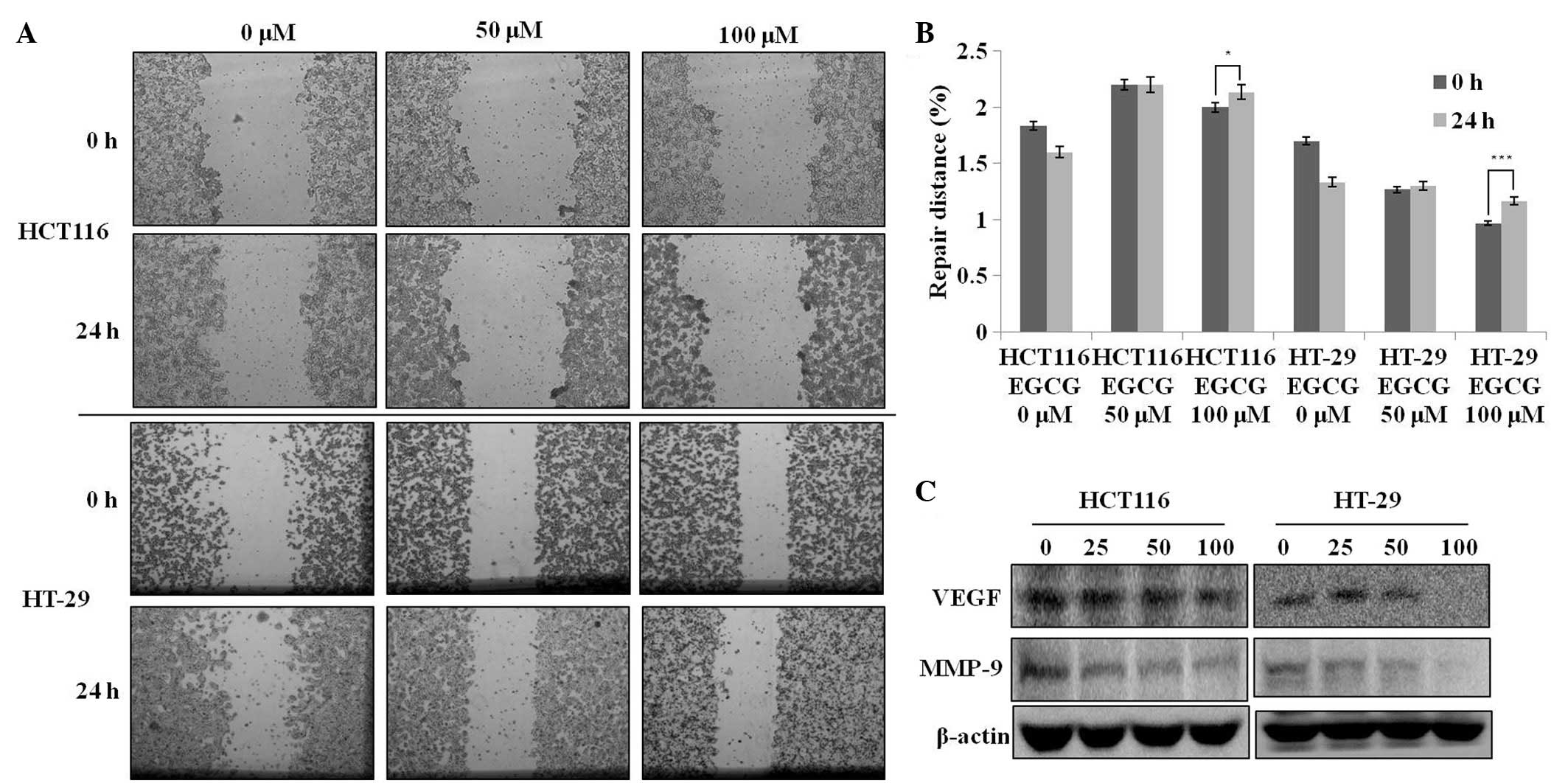
EGCG possesses migration-suppressing
potential
A wound-healing assay was used to evaluate the
effect of EGCG on metastatic activity (i.e., migration). The
treatment of the HCT116 and HT-29 cells with 50–100 μM EGCG for 24
h resulted in a significant reduction in the degree of wound
healing (Fig. 2A), indicating that
EGCG is able to inhibit metastatic activity in these two cell
lines. Next, the present study tested whether the
migration-suppressing activity of EGCG was associated with the
attenuation of MMP-9 and VEGF protein levels. EGCG treatment
dose-dependently decreased the protein expression levels of VEGF
and MMP-9 (Fig. 2C). These results
suggest that the migration-suppressing effect of EGCG is associated
with the inhibition of VEGF and MMP-9 protein expression regardless
of the p53 status in these cells.
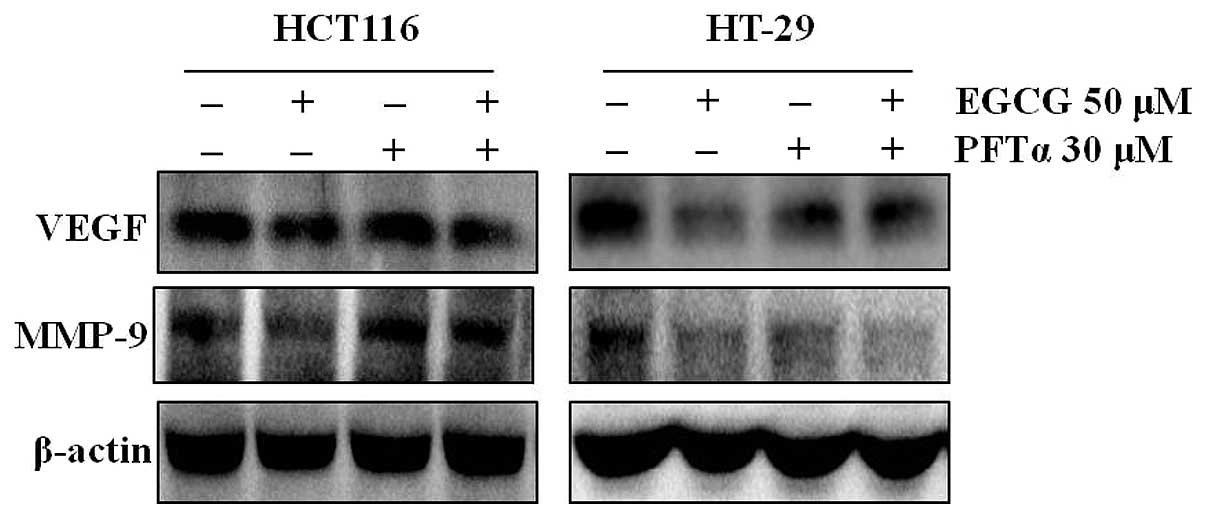
AMPK activation regulates VEGF and MMP-9
expression regardless of p53 status
To examine the involvement of p53 in the
EGCG-induced inhibition of VEGF and MMP-9 expression, the HT-29 and
HCT116 cells were treated with EGCG plus a specific inhibitor of
p53 (pifithrin-α). Inhibition of p53 by pifithrin-α abolished the
EGCG-induced inhibition of VEGF and MMP-9 in the HCT116 cells, but
not in the HT-29 cells (Fig.
3).
Discussion
A previous study showed that the adenovirus-mediated
gene transfer of wild-type p53 into p53-mutated cells is able to
inhibit VEGF expression (17).
Thus, the present results and those of the previous study indicate
that p53 is involved in the ability of EGCG to inhibit VEGF and
MMP-9 expression in p53-positive cells, but that EGCG is also able
to regulate VEGF and MMP-9 via a p53-independent pathway in cells
expressing mutant p53. Since other studies have suggested that EGCG
activates AMPK (18) and inhibits
VEGF (19), the EGCG-induced
regulation of VEGF expression was tested in the presence and
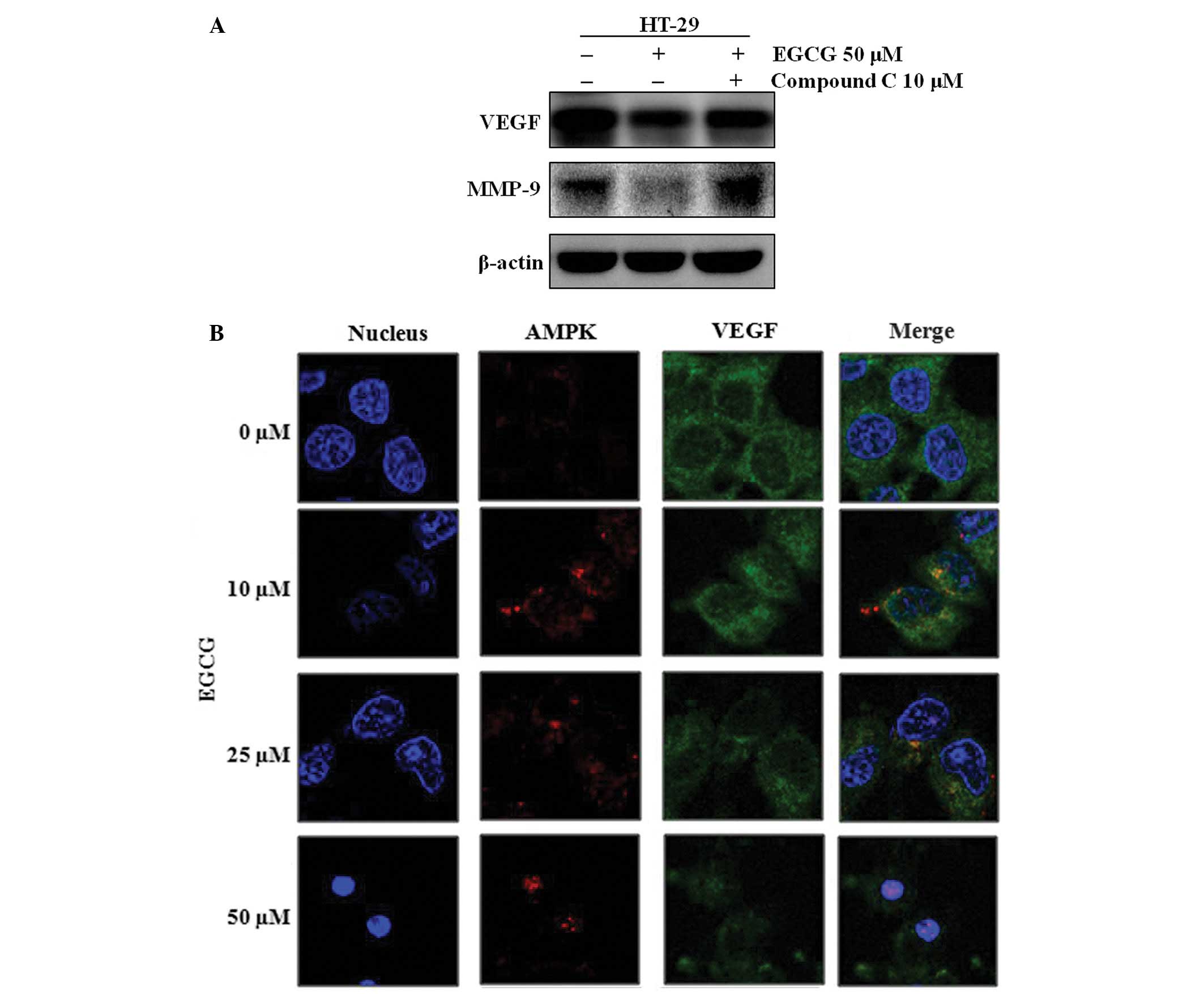
absence of the AMPK activity inhibitor, compound C. EGCG was no
longer able to downregulate VEGF and MMP-9 in the compound
C-treated HT-29 cells (Fig. 4A).
Furthermore, EGCG activated AMPK and inhibited VEGF expression in a
dose-dependent manner (Fig. 4B).
Using an immunofluorescence analysis with AMPK- and VEGF-specific
antibodies, it was observed that the regulatory effect of AMPK on
VEGF did not result from their direct binding, since AMPK and VEGF
did not co-localize (Fig. 4B).
These results indicate that although AMPK does not directly bind
VEGF, EGCG-activated AMPK controls VEGF.
In conclusion, the present study demonstrated that
EGCG inhibits colon cancer cell migration and induces apoptosis in
these cells regardless of the presence of functional p53. EGCG also
strongly inhibits VEGF and MMP-9 expression in HT-29 (p53 mutant)
and HCT116 (p53 wild-type) cells. This regulation appears to occur
through p53 in cells expressing wild-type p53, but in p53-mutant
cells this occurs via a p53-independent manner. Finally, although
direct binding was not observed between AMPK and VEGF, the present
results indicate that EGCG-induced AMPK activity regulates the
expression levels of VEGF and MMP-9.
Acknowledgements
This study was supported by the National Research
Foundation of Korea (KRF-2012-0021402) and the Korea Science and
Engineering Foundation (KOSEF) grant funded by the Korean
government (MEST; no. R01-2008-000-20131-0).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Murthy NS, Mukherjee S, Ray G and Ray A:
Dietary factors and cancer chemoprevention: an overview of
obesity-related malignancies. J Postgrad Med. 55:45–54. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rodrigues NR, Rowan A, Smith ME, Kerr IB,
Bodmer WF, Gannon JV and Lane DP: p53 mutations in colorectal
cancer. Proc Natl Acad Sci USA. 87:7555–7559. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu J and Zhang L: The transcriptional
targets of p53 in apoptosis control. Biochem Biophys Res Commun.
331:851–858. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Z and Sun Y: Targeting p53 for Novel
Anticancer Therapy. Transl Oncol. 3:1–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter
CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL and Bedi A:
Regulation of tumor angiogenesis by p53-induced degradation of
hypoxia-inducible factor 1alpha. Genes Dev. 14:34–44.
2000.PubMed/NCBI
|
|
7
|
Horiuchi A, Imai T, Shimizu M, Oka K, Wang
C, Nikaido T and Konishi I: Hypoxia-induced changes in the
expression of VEGF, HIF-1 alpha and cell cycle-related molecules in
ovarian cancer cells. Anticancer Res. 22:2697–2702. 2002.PubMed/NCBI
|
|
8
|
Montero E, Abreu C and Tonino P:
Relationship between VEGF and p53 expression and tumor cell
proliferation in human gastrointestinal carcinomas. J Cancer Res
Clin Oncol. 134:193–201. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oh SY, Kwon HC, Kim SH, Jang JS, Kim MC,
Kim KH, Han JY, Kim CO, Kim SJ, Jeong JS and Kim HJ:
Clinicopathologic significance of HIF-1alpha, p53, and VEGF
expression and preoperative serum VEGF level in gastric cancer. BMC
Cancer. 8:1232008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dorai T and Aggarwal BB: Role of
chemopreventive agents in cancer therapy. Cancer Lett. 215:129–140.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Narayanan BA: Chemopreventive agents
alters global gene expression pattern: predicting their mode of
action and targets. Curr Cancer Drug Targets. 6:711–727. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo RP, Zhong C, Shi M, Zhang CQ, Wei W,
Zhang YQ and Li JQ: Clinical value of apoptosis and angiogenesis
factors in estimating the prognosis of hepatocellular carcinoma. J
Cancer Res Clin Oncol. 132:547–555. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimizu M, Shirakami Y and Moriwaki H:
Targeting receptor tyrosine kinases for chemoprevention by green
tea catechin, EGCG. Int J Mol Sci. 9:1034–1049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saharinen P, Eklund L, Pulkki K, Bono P
and Alitalo K: VEGF and angiopoietin signaling in tumor
angiogenesis and metastasis. Trends Mol Med. 17:347–362. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stetler-Stevenson WG: Type IV collagenases
in tumor invasion and metastasis. Cancer Metastasis Rev. 9:289–303.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: a convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bouvet M, Ellis LM, Nishizaki M, Fujiwara
T, Liu W, Bucana CD, Fang B, Lee JJ and Roth JA:
Adenovirus-mediated wild-type p53 gene transfer down-regulates
vascular endothelial growth factor expression and inhibits
angiogenesis in human colon cancer. Cancer Res. 58:2288–2292.
1998.
|
|
18
|
Hwang JT, Ha J, Park IJ, Lee SK, Baik HW,
Kim YM and Park OJ: Apoptotic effect of EGCG in HT-29 colon cancer
cells via AMPK signal pathway. Cancer Lett. 247:115–121. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sartippour MR, Shao ZM, Heber D, Beatty P,
Zhang L, Liu C, Ellis L, Liu W, Go VL and Brooks MN: Green tea
inhibits vascular endothelial growth factor (VEGF) induction in
human breast cancer cells. J Nutr. 132:2307–2311. 2002.PubMed/NCBI
|