Introduction
Pituitary adenomas are the most common tumors that
arise from anterior pituitary cells in the sella turcica, which
represents up to 25% of brain tumors (1). Prolactin (PRL)secreting adenomas
(prolactinomas) and growth hormone (GH) secreting adenomas (GHomas)
are the most common types, accounting for 45% of all adenomas
(2). During the past decade,
significant advances have been made towards understanding the
molecular mechanisms that regulate the development of pituitary
adenomas and specific chromosomal alterations that lead to
pituitary adenoma pathogenesis. However, the pathogenetic
mechanisms that underlie pituitary adenomas are complex and remain
enigmatic (1).
Recently, our group found several new candidate
genes that suggested a role in the pathogenesis of prolactinomas
and GHomas using a human genome-wide bead-based fiber-optic array
(3,4). The HPGD is one of these genes, which
is located on chromosome 4 and encodes a 29-kDa enzyme named
15-hydroxyprostaglandin dehydrogenase (15-PGDH). The enzyme
catalyzes the oxidation of the 15(S)-hydroxyl group of
prostaglandins (PGs), resulting in the production of 15-keto-PGs
and 15-keto-lipoxins, which have greatly reduced biological
activities (5). Although previous
studies on the distribution and activity of 15-PGDH have focused
primarily on parturition and uterine biology, it is now recognized
as a tumor suppressor and demonstrates antitumorigenic activity in
various types of tumors including glioma, gastric, pancreatic,
colon, breast, lung prostate and bladder tumors (6–13).
COX-2, a rate-limiting enzyme in the arachidonic acid cascade,
plays a key role in prostaglandin E2 (PGE2)
biosynthesis. Overproduction of PGE2 stimulates
proliferation in various tumor cells, confers resistance to
apoptosis in cancerous or transformed cells, and accelerates
metastasis and angiogenesis. Excess PGE2 undergoes
metabolic inactivation catalyzed by 15-PGDH. 15-PGDH and COX-2 are
reciprocally regulated in many tumor cells, although the molecular
mechanism remains to be elucidated (13). Intense investigations of 15-PGDH
have been conducted in many kinds of tumors and cell lines.
However, its existence in the pituitary and its role in pituitary
tumorigenesis have not been investigated.
In the present study, we examined the expression of
15-PGDH mRNA in clinical specimens and normal pituitaries and
investigated the effects and possible mechanisms of 15-PGDH by
transfecting GH3 cells with an expression plasmid encoding 15-PGDH
in order to evaluate 15-PGDH effects on pituitary
tumorigenesis.
Materials and methods
Clinical tissue specimens and normal
pituitaries
Thirteen GHomas and eleven prolactinoma specimens
were obtained from transsphenoidal surgery at Tiantan Hospital.
Each tumor was classified according to the 2004 World Health
Organization Classification of Endocrine Tumors guidelines
(14). Due to the lack of a better
control tissue available for human semi-quantitative real-time PCR
(qPCR) study, normal pituitaries were chosen as control group
despite the fact that normal pituitaries are not patient-matched.
Five normal pituitaries were taken from autopsy cases that showed
no evidence of endocrine diseases at 8–12 h after sudden death. All
tissues were collected after written consent was obtained. The
gender and age of the control group and of the patient groups are
presented in Table I.
 | Table ICharacteristics of the three
groups. |
Table I
Characteristics of the three
groups.
| Control group | GHomas group | Prolactinomas
group |
|---|
| No. of patients | 5 | 13 | 11 |
| Gender
(male/female) | 3/2 | 6/7 | 6/5 |
| Age (years) | 21–43 | 26–59 | 24–56 |
| Average age (mean ±
SE) | 31.2±9.86 | 44.23±9.68 | 41.8±10.08 |
Cell culture, plasmid construction and
transfection
The rat pituitary tumor-derived cell line GH3 is
capable of secreting both PRL and GH. 15-PGDH expression levels
were low in GH3 cells (data not shown). Therefore, GH3 cells were
employed in this study and obtained from the Cell Center of the
School of Basic Medicine, Peking Union Medical College. The cells
were cultured in DMEM/F-12 medium supplemented with 10% fetal
bovine serum (FBS) (Gibco, Invitrogen, Grand Island, NY, USA), and
incubated at 37°C in a humidified atmosphere of 5%
CO2.
The recombinant flag-tagged pcDNA3-PGDH encoding rat
15-PGDH cDNA was constructed in our laboratory. Briefly, a
wild-type 15-PGDH cDNA was amplified from normal rat lung cDNA
(amplification primer sequences were forward
5′-CGGGATCCATGCACGTGAACGGCAAAGTGGC-3′, and reverse
5′-CCGCTCGAGTCATGGGGCTTTCGAAAAGG ATG-3′). The amplified 15-PGDH
cDNA fragment and plasmid were digested by BamHI and
XhoI, and then ligated using T4-ligase into the
HI-XhoI site of flag-tagged pcDNA 3.0 (kindly donated by Dr
Xiang He). The recombinant vector named flag-tagged pcDNA3-PGDH was
confirmed by restriction enzyme digestion and sequencing.
Lipofectamine 2000 transfection reagent was supplied
by Invitrogen Life Technologies. Transfection of the recombinant
vector or control empty vector was performed in accordance with the
manufacturer’s protocol. Briefly, cells were transferred to
complete medium before transfection, and then a mixture of vector
and Lipofectamine was added to the 25-cm2 plates. After
incubation for 4 h, the cells were incubated with fresh medium for
the desired time period.
Plotting of cell growth curve
To obtain flag-tagged pcDNA3-PGDH transfected cells
and the control group (cells transfected with empty flag-tagged
pcDNA3.0 and cells without transfection), cells were seeded onto
24-well culture plates (1×104 cells/well). After
transfection, three wells of each group were trypsinized and
harvested every 12 h, and the viability and cell number of the
samples were quantified under a light microscope after staining
with trypan blue.
Cell inhibitory rate assays using
WST-8
GH3 cells in log phase growth were washed twice with
PBS, and plated on 96-well plates (5,000 cells/well) in 100 μl
phenol red-free DMEM/F12 medium containing 10% FBS. Forty-eight
hours after transfection, cell proliferation was assessed with the
WST-8 cell staining kit (Neuronbc, Beijing, China). Briefly, 10 μl
WST-8 solution was added to each well, followed by incubation for 4
h. The absorbance of each well was measured at 450 nm in a
SpectraMax M5 multi-detection microplate reader (Molecular Devices,
Sunnyvale, CA, USA). The cell inhibitory rate was calculated using
the following equation: Inhibition rate (%) = (ODcontrol
group − ODdrug group)/ODcontrol group ×
100% (OD, optical density).
PGE2, PRL and GH enzyme-linked
immunosorbent assay (ELISA)
Cells treated with empty vector, flag-tagged
pcDNA3-PGDH plasmid or control solvent were plated onto 24-well
plates (3×104 cell/well), and then cultured for 48 h in
phenol red-free medium. The culture medium was collected from each
well and centrifuged at 120 × g for 5 min at 4°C. The supernatants
were frozen at −80°C until assay.
Secreted PGE2, PRL and GH levels in the
supernatants were measured using the ELISA kit (RapidBio, West
Hills, CA, USA). The concentrations were calculated using a
straight line regression equation of the standard curve with the
standard density and the OD value. The concentrations were then
normalized to viable cell numbers and expressed as % of
control.
Measurement of apoptosis using propidium
iodide (PI) and Annexin V double-staining flow cytometry
After transfection with flag-tagged pcDNA3-PGDH for
48 h, the cells were digested with trypsin to make a single-cell
suspension, rinsed 3 times with PBS, resuspended in buffer, and
adjusted to a density of 1×106 cells/ml. PI and Annexin
V were purchased from BD Pharmingen (San Jose, CA, USA). To 100 μl
of cell suspension in each tube was added 15 μl of
fluorescein-isothiocyanate-conjugated Annexin V and 10 μl of PI (20
μg/ml). The samples were stained at 4°C for 20 min, and flow
cytometry was used to examine cell apoptosis. A total of
104 cells were assayed for each sample.
SYBR-Green based qPCR analysis
The relative expression levels of several relevant
mRNAs were tested in frozen clinical specimens and GH3 cells
transfected for 48 h. Total RNA was extracted using TRIzol Reagent
(Invitrogen, Carlsbad, CA, USA) in accordance with the
manufacturer’s protocol and reverse-transcribed into cDNA using 1.5
μg of total RNA according to Quantscript RT Kit protocol (Tiangen
Biotech, Beijing, China). The primers are listed in Table II. The qPCR reaction was performed
using the SYBR-Green PCR kit on a Bio-Rad iCycler IQ Real-Time PCR
Detection System according to the manufacturer’s instructions in a
20 μl reaction containing the following: Master Mix (2X) 10 μl,
ROXII (50X) 0.4 μl, primer F/R (0.4 μl each, 10 μmol/l), sample
cDNA (1 μl), and MilliQ H2O (7.8 μl). The amplification
conditions were 94°C for 15 min, followed by 40 cycles of 94°C for
15 sec, 60 or 62°C for 30 sec, and 72°C for 30 sec. The relative
expression levels of the analyzed genes were normalized to β-actin.
The calculations were based on the cycle threshold (CT) value using
the 2−ΔΔCT method for quantification (15).
| Table IIPrimers used in SYBR-Green-based
semi-quantitative real-time PCR (qPCR) analysis. |
Table II
Primers used in SYBR-Green-based
semi-quantitative real-time PCR (qPCR) analysis.
| Gene | Size (bp) | Forward sequence
(5′→3′) | Reverse sequence
(5′→3′) |
|---|
| Human PGDH | 228 |
TCTGTTCATCCAGTGCGATGT |
ATAATGATGCCGCCTTCACCT |
| Human COX-2 | 193 |
TAAACAGACATTTATTTCCAGAC |
GAAAGAAATAGTCAATATGCTTG |
| Human β-actin | 188 |
AGAAAATCTGGCACCACACC |
AGAGGCGTACAGGGATAGCA |
| Rat COX-2 | 233 |
CCGGGTTGCTGGGGGAAGGA |
CCACCAGCAGGGCGGGATACAG |
| Rat MMP-9 | 114 |
AAACATGCTGAAACCGGACC |
GATCATCTCGGCTACCCTACCT |
| Rat Bcl-2 | 261 |
CACTGGCTTGACTGGCTGAA |
CACAGACCTGGTTCGTGCTC |
| Rat β-actin | 104 |
TGACAGGATGCAGAAGGAGA |
TAGAGCCACCAATCCACACA |
| Rat PRL | 238 |
AGCCAAGTGTCAGCCCGGAAAG |
TGGCCTTGGCAATAAACTCACGA |
| Rat GH | 251 |
TATTGGGCAGATCCTCAAGC |
CAAAGTGTAGGGGTGGCAGT |
Western blot analysis
The GH3 cells were transfected for 48 h, and the
protein extracted from the cells was assayed using western blot
analysis. Bcl-2 and β-actin polyclonal antibodies were purchased
from Abcam (Cambridge, UK). COX-2 and MMP-9 polyclonal antibodies
were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The flag-tagged PGDH protein in GH3 cells exhibited a
molecular weight of about 29 kDa. Monoclonal anti-flag antibodies
(Sigma-Aldrich, St. Louis, MO, USA) were used to detect flag-tagged
PGDH. Horseradish peroxidase-labeled secondary antibody was
purchased from Jackson ImmunoResearch (West Grove, PA, USA).
Briefly, experimental cells were lysed with cell lysis buffer, and
their protein concentrations were determined using the BCA protein
assay before separation by gel electrophoresis. Equal amounts of
proteins were subjected to electrophoresis and then transferred
onto polyvinylidene fluoride membranes. After transfer, the
membranes were rinsed and incubated with blocking buffer (5%
non-fat milk in TBST) for 1–2 h at room temperature. Membranes were
then incubated overnight with primary antibodies at 4°C, followed
by three 10-min washes with TBST, and then incubation with
secondary antibodies at room temperature for 1 h. After three
10-min washes with TBST, the antibody-antigen complex was detected
with an enhanced chemiluminescence detection system (Amersham
Pharmacia Biotech, Piscataway, NJ, USA).
Statistical analysis
All data are presented as mean ± standard error of
the mean (SEM). All experiments were performed at least three
times. Statistical analyses were performed using one-way ANOVA, the
Student’s t-test or the Kruskal-Wallis test (SPSS 13.0) and
factorial design ANOVA (SAS 9.0). P<0.05 was assumed to be
significant.
Results
Expression of 15-PGDH and COX-2 mRNA in
clinical specimens
For further verification of our previous microarray
results (3,4), 24 clinical specimens were analyzed
with qPCR. There were no statistical differences in the age and
gender distribution among the three groups (Table I, P>0.05). Possible effects of
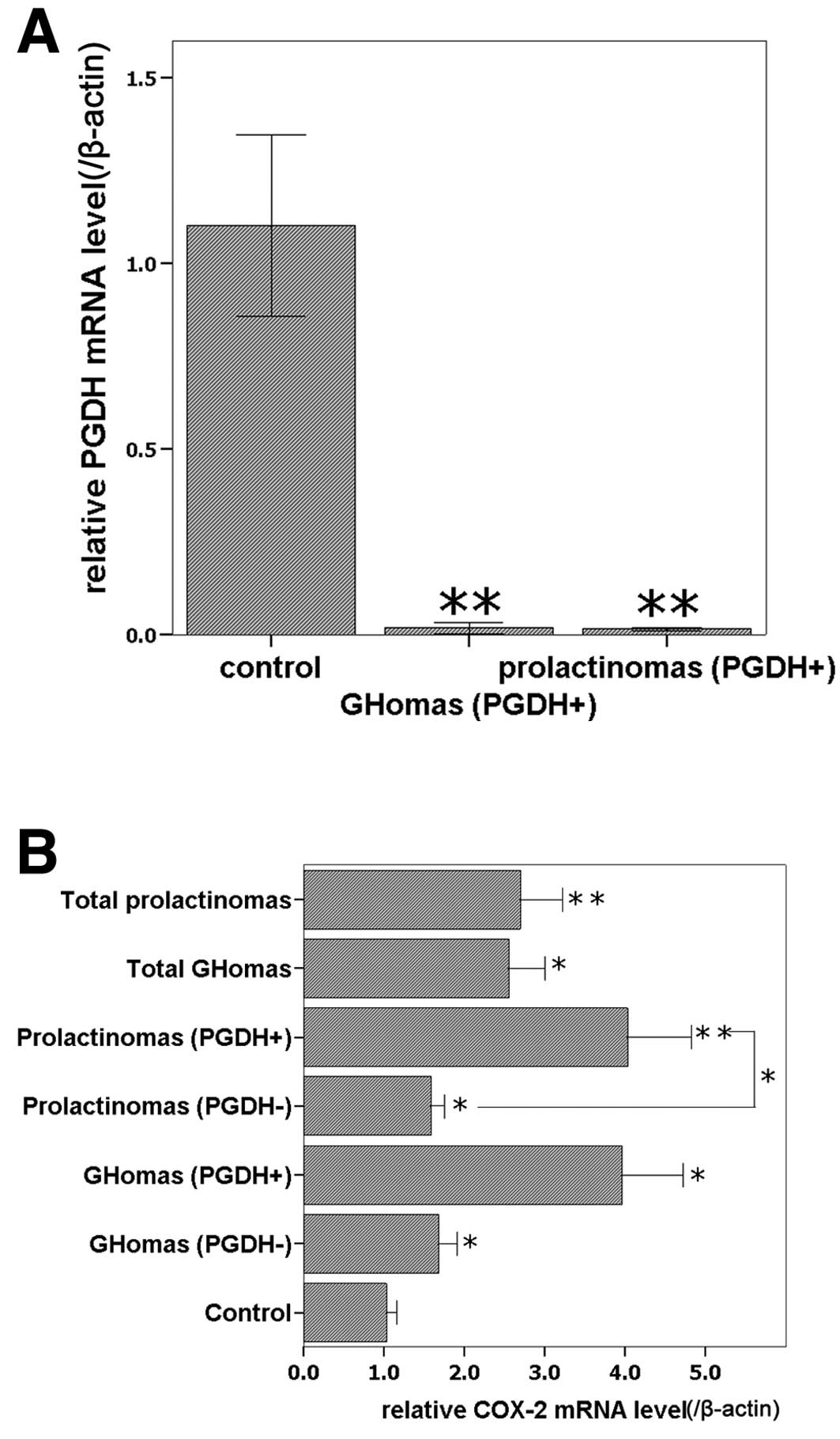
age and gender were ruled out. 15-PGDH mRNA was detected in all the
normal controls, while it was detected in 5 of 13 GHomas, and 5 of
11 prolactinomas according to the melting curve. The 15-PGDH
expression level was higher in normal control than in both the
GHoma and prolactinoma groups (P<0.05, Fig. 1A) and there was no significant
difference in 15-PGDH levels between the GHomas group and the
prolactinomas group (P>0.05). COX-2 mRNA was detected in all
specimens. All pituitary adenoma groups expressed higher COX-2 mRNA
levels than the normal control group (P<0.05), and the subgroups
of 15-PGDH-positive prolactinomas expressed higher COX-2 mRNA
levels than the corresponding 15-PGDH-negative subgroups
(P<0.05); although no significant difference was identified
between 15-PGDH-positive GHomas and 15-PGDH-negative GHomas
(P>0.05, Fig. 1B), COX-2 mRNA
level were higher in 15-PGDH-positive GHomas.
Inhibitory effects of 15-PGDH on GH3 cell
growth and proliferation
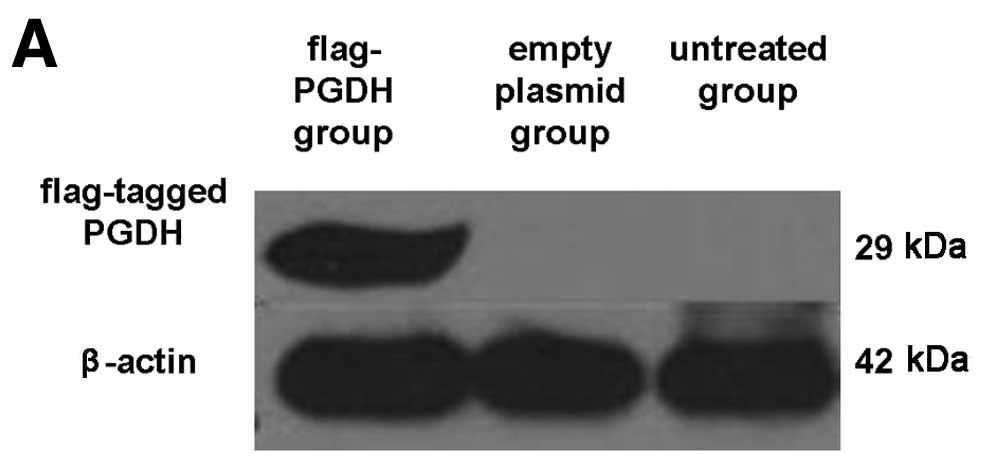
To observe the biological activity of 15-PGDH in GH3
cells, the flag-tagged pcDNA3-PGDH vector was transfected into GH3
cells. Forty-eight hours after transfection, 15-PGDH proteins were
detected with anti-flag antibody in the cells (Fig. 2A). The GH3 cell growth curve plotted
for different treatment methods and different time points is shown
in Fig. 2B. It indicates that
forced overexpression of 15-PGDH suppressed cell growth 24 h after
transfection (P<0.05).
As shown by the WST-8 assay results (Fig. 2C), transfection of GH3 cells with
the 15-PGDH expression vector inhibited cell proliferation by
32.29±9.71% compared with untreated cells 48 h after transfection.
Transfection with the empty expression vector generated a similar
proliferation profile as the untreated cells, indicating that the
suppression of tumor cell growth is specifically caused by
15-PGDH.
Overexpressed 15-PGDH promote apoptosis
of GH3 cells
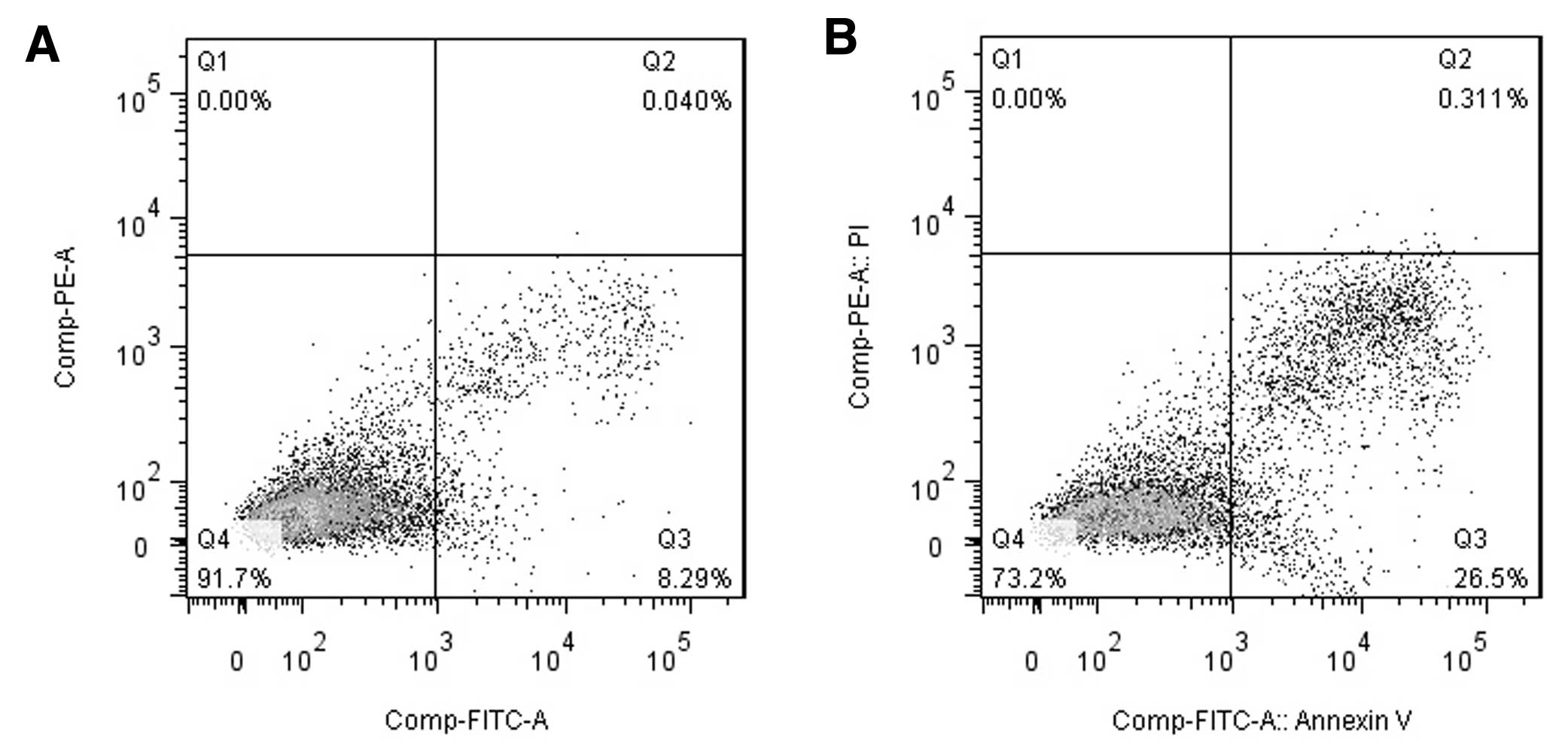
The flow cytometry results used to examine apoptosis
are shown in Fig. 3. Compared with
the control group (untreated cells), the overexpression of 15-PGDH
resulted in increased numbers of both early-stage (8.4±0.38 vs.
22.15±4.67%, P<0.01) and late-stage apoptotic cells (0.12±0.08
vs. 0.68±0.19%, P<0.05).
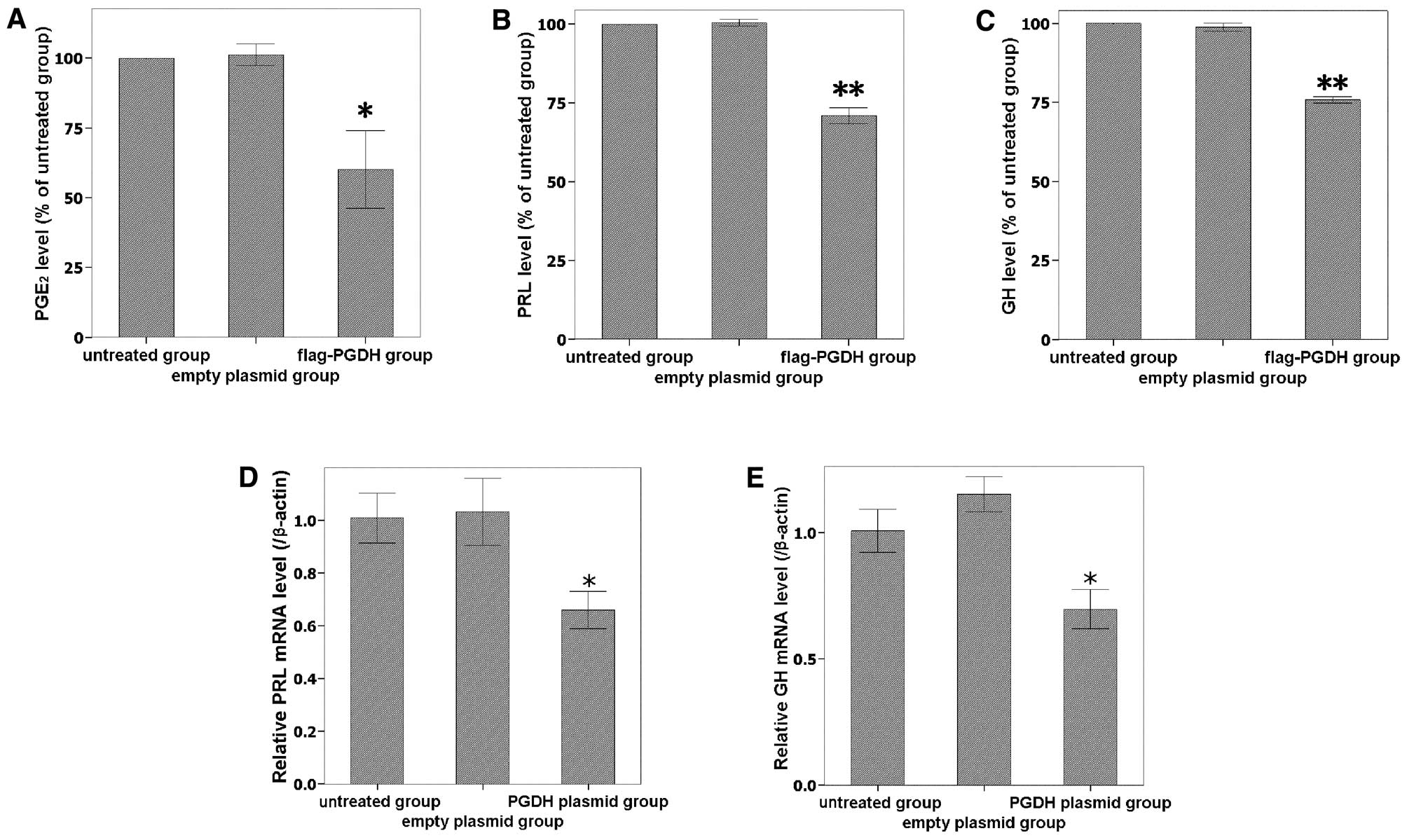
Levels of PGE2, PRL and GH are
reduced by overexpressing 15-PGDH
To further investigate the activity of overexpressed
15-PGDH, PGE2 levels in the culture medium were measured
using an ELISA assay. As the expression of 15-PGDH protein
increased, PGE2 levels significantly decreased (Fig. 4A). The secreted PGE2
levels of transfected cells were 60.1±23.91% of the untreated
control group (P<0.05). PRL and GH expression were downregulated
by 15-PGDH at both the secreted protein (Fig. 4B and C) and mRNA levels (Fig. 4D and E). Taken together, the data
indicate that 15-PGDH protein was expressed and had a biological
activity in GH3 cells after transfection.
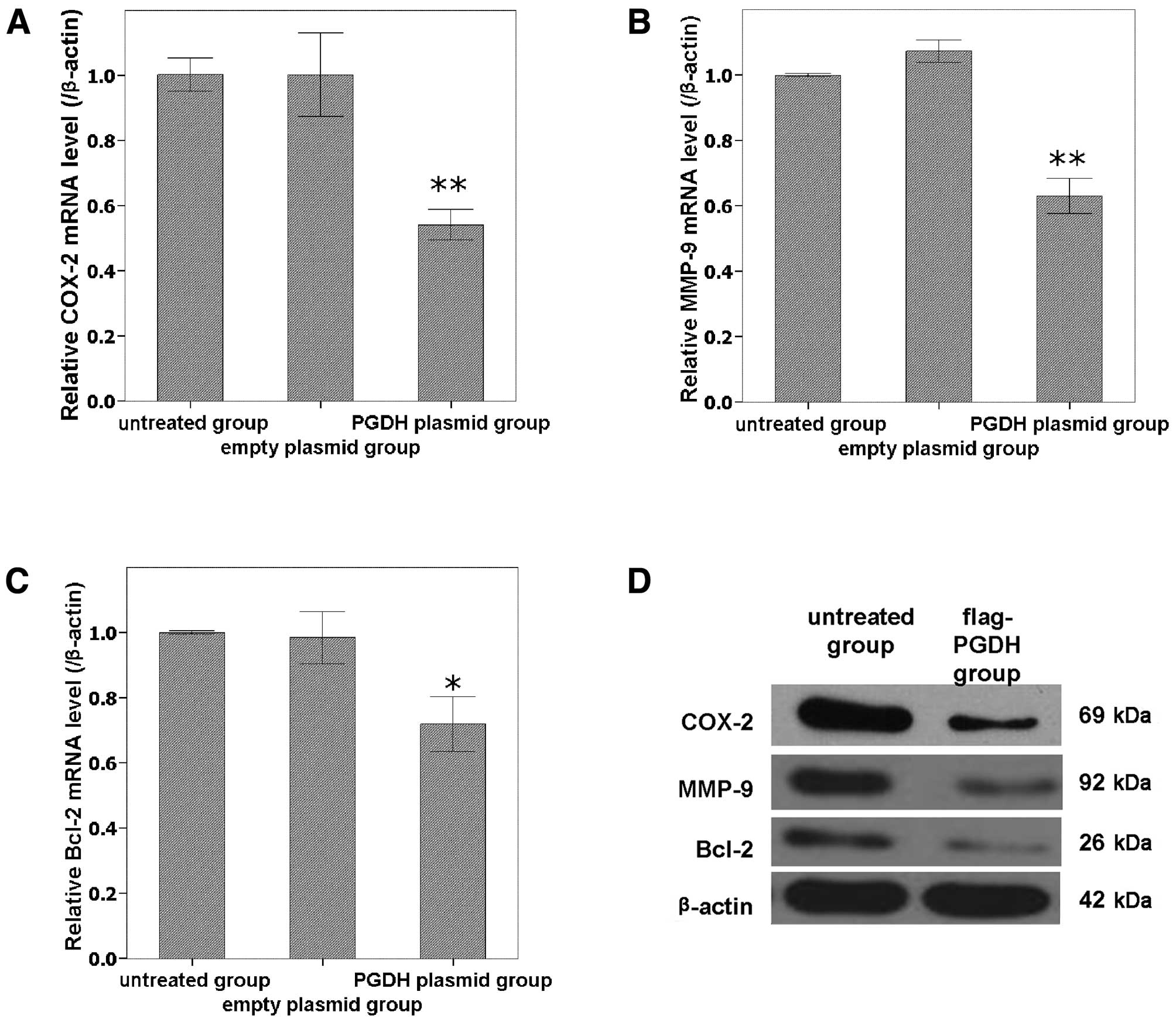
Expressions of COX-2, MMP-9 and Bcl-2
were downregulated by overexpression of 15-PGDH in GH3 cells
To analyze the possible molecular mechanism of
15-PGDH, the relevant genes including COX-2, MMP-9 and Bcl-2 were
analyzed using qPCR and western blot analysis. COX-2, MMP-9 and
Bcl-2 were all downregulated at both the mRNA (Fig. 5A-C) and protein (Fig. 5D) levels by the forced
overexpression of 15-PGDH compared with untreated cells
(P<0.05).
Discussion
15-PGDH is a member of the short chain
dehydrogenase/reductase family which includes more than 60
different enzymes. The fact that 15-PGDH may function as a tumor
suppressor has been exploited to examine the therapeutic efficacy
in vivo of 15-PGDH-mediated cancer therapy (16). Although 15-PGDH appears to be
widespread in mammalian tissues with relatively high activity in
lung, kidney and placenta, there is no other research in relation
to 15-PGDH and pituitary tumors until now. Low expression of
15-PGDH in pituitary tumors was reported by our group using
fiber-optic BeadArray (3,4). In this study, we increased the number
of specimens and tested the relative expression of 15-PGDH using
qPCR. We confirmed that 15-PGDH expression is low in both
prolactinomas and GHomas. In addition, 15-PGDH was completely lost
in some pituitary adenomas. As an inverse regulating enzyme of
15-PGDH, COX-2 was highly expressed in all pituitary adenomas,
especially in 15-PGDH-negative specimens. Forced overexpression of
15-PGDH in GH3 cells resulted in significant growth inhibition and
promotion of apoptosis and necrosis. These results indicate the
importance of 15-PGDH in controlling the growth of pituitary tumor
cells and pituitary adenoma pathogenesis. Accumulating evidence
indicates that the downregulation of 15-PGDH in tumors is due to
transcriptional repression and epigenetic silencing (10,17,18).
Studying epigenetic mechanisms in clinical specimens and tumor
cells may shed new light on therapeutic strategies for pituitary
tumors.
COX-2 has been implicated in regulating
tumorigenesis because it is overexpressed in many tumors, including
pituitary adenomas (19).
Expression of COX-2 has a wide range of biological activities,
including increased angiogenesis, cellular proliferation and
apoptosis inhibition. A strong correlation has been found between
COX-2 expression and vascularization in pituitary tumors (19). Recently, R-flurbiprofen, a novel
non-steroidal anti-inflammatory drug, was shown to decrease cell
proliferation and to induce apoptosis in pituitary adenoma cells
in vitro, which may function via COX-2 downregulation
through inhibition of the upstream target, nuclear factor-κB
(NF-κB) (20). COX-2 was
downregulated by 15-PGDH in GH3 cells in the present study, which
suggests that 15-PGDH functions via the COX-2 pathway in pituitary
tumor cells. It strengthens the rationale for application of
15-PGDH to control pituitary tumors.
It was reported that PGE2 enhanced PRL
mRNA expression and PRL secretion in human endometrial stromal
cells (21) and that PRL has a
stimulatory effect on PGs in hamster Leydig cells (22). Therefore, PGE2 and PRL
may be reciprocally regulated in tumor cells. Furthermore,
PGE2 stimulates the release of GH and PRL from cultured
bovine anterior pituitary cells (23). In the present study, PGE2
secretion was reduced in the culture medium. Both mRNA expression
and secretion of PRL and GH were also reduced after 15-PGDH
overexpression. Taken together, it is possible that 15-PGDH may
reduce PRL and GH by degrading PGE2, and PGE2
may be involved in endocrine disorders caused by prolactinomas or
GHomas.
The matrix metalloproteinases (MMPs) are a family of
zinc-dependent endopeptidases that mediate the degradation of the
extracellular matrix. Vascularization and apoptosis was found to be
linked to MMP-9 (24,25). GH3 cell proliferation and hormone
secretion were inhibited in the presence of the broad range
metalloproteinase inhibitor, batimastat (26). Taken together, these reports and our
results support the notion that 15-PGDH inhibited MMP-9 expression
and hormone secretion. It is possible that the inhibition of
hormone secretion and the promotion of apoptosis in GH3 cells by
15-PGDH may be related to MMP-9 suppression.
The Bcl-2 family of proteins regulates various steps
in apoptosis. Some of the members of this gene family, such as
Bcl-2, block cell death. More than 70% of pituitary adenomas
express Bcl-2 (27,28). In this study, overexpression of
15-PGDH induced cell apoptosis and necrosis as confirmed by flow
cytometry, and also downregulated Bcl-2 expression. Therefore, the
death-promoting effects of 15-PGDH in GH3 cells may be partly
exerted via Bcl-2.
In summary, we verified that 15-PGDH expression is
lost or greatly reduced in prolactinomas and GHomas. Transfection
of a rat 15-PGDH expression vector into GH3 cells resulted in a
substantial inhibition of tumor cell growth, promoted apoptosis and
inhibited hormone secretion. The data indicate that 15-PGDH may be
regarded as a tumor suppressor in pituitary adenomas. The
functional mechanism of 15-PGDH is attributed to the degeneration
of PGE2 and the inhibition of COX-2, MMP-9 and
Bcl-2.
Acknowledgements
We are indebted to Dr Xiang He of the Academy of
Military Medical Sciences for kindly providing the flag-tagged
pcDNA3.0 vector.
References
|
1
|
Asa SL and Ezzat S: The pathogenesis of
pituitary tumors. Annu Rev Pathol. 4:97–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osamura RY, Kajiya H, Takei M, et al:
Pathology of the human pituitary adenomas. Histochem Cell Biol.
130:495–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang Z, Gui S and Zhang Y: Analysis of
differential gene expression by fiber-optic BeadArray and pathway
in prolactinomas. Endocrine. 38:360–368. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Z, Gui S and Zhang Y: Analysis of
differential gene expression by bead-based fiber-optic array in
growth-hormone-secreting pituitary adenomas. Exp Ther Med.
1:905–910. 2010.PubMed/NCBI
|
|
5
|
Tai HH, Cho H, Tong M and Ding Y:
NAD+-linked 15-hydroxyprostaglandin dehydrogenase:
structure and biological functions. Curr Pharm Des. 12:955–962.
2006.
|
|
6
|
Tseng-Rogenski S, Gee J, Ignatoski KW, et
al: Loss of 15-hydroxyprostaglandin dehydrogenase expression
contributes to bladder cancer progression. Am J Pathol.
176:1462–1468. 2010. View Article : Google Scholar
|
|
7
|
Liu Z, Wang X, Lu Y and Han S: Expression
of 15-PGDH is downregulated by COX-2 in gastric cancer.
Carcinogenesis. 29:1219–1227. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lodygin D, Epanchintsev A, Menssen A,
Diebold J and Hermeking H: Functional epigenomics identifies genes
frequently silenced in prostate cancer. Cancer Res. 65:4218–4227.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tai HH, Tong M and Ding Y:
15-hydroxyprostaglandin dehydrogenase (15-PGDH) and lung cancer.
Prostaglandins Other Lipid Mediat. 83:203–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wolf I, O’Kelly J, Rubinek T, et al:
15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of
human breast cancer. Cancer Res. 66:7818–7823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Myung SJ, Rerko RM, Yan M, et al:
15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of
colon tumorigenesis. Proc Natl Acad Sci USA. 103:12098–12102. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wakimoto N, Wolf I, Yin D, et al:
Non-steroidal anti-inflammatory drugs suppress glioma via
15-hydroxyprostaglandin dehydrogenase. Cancer Res. 68:6978–6986.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tai HH: Prostaglandin catabolic enzymes as
tumor suppressors. Cancer Metastasis Rev. 30:409–417. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DeLellis RA, Lloyd RV, Heitz PU and Eng C:
Pathology and Genetics of Tumours of Endocrine Organs. WHO
Classification of Tumours. The International Agency for Research on
Cancer; Lyon: 2004
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaliberova LN, Kusmartsev SA,
Krendelchtchikova V, et al: Experimental cancer therapy using
restoration of NAD+-linked 15-hydroxyprostaglandin
dehydrogenase expression. Mol Cancer Ther. 8:3130–3139. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Greenland KJ, Jantke I, Jenatschke S,
Bracken KE, Vinson C and Gellersen B: The human
NAD+-dependent 15-hydroxyprostaglandin dehydrogenase
gene promoter is controlled by Ets and activating protein-1
transcription factors and progesterone. Endocrinology. 141:581–597.
2000.
|
|
18
|
Yang L, Amann JM, Kikuchi T, et al:
Inhibition of epidermal growth factor receptor signaling elevates
15-hydroxyprostaglandin dehydrogenase in non-small cell lung
cancer. Cancer Res. 67:5587–5593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vidal S, Kovacs K, Bell D, Horvath E,
Scheithauer BW and Lloyd RV: Cyclooxygenase-2 expression in human
pituitary tumors. Cancer. 97:2814–2821. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu JK, Patel SK, Gillespie DL, Whang K
and Couldwell WT: R-flurbiprofen, a novel nonsteroidal
anti-inflammatory drug, decreases cell proliferation and induces
apoptosis in pituitary adenoma cells in vitro. J Neurooncol.
106:561–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frank GR, Brar AK, Cedars MI and
Handwerger S: Prostaglandin E2 enhances human endometrial stromal
cell differentiation. Endocrinology. 134:258–263. 1994.PubMed/NCBI
|
|
22
|
Matzkin ME, Ambao V, Carino MH, et al:
Prolactin (PRL) induction of cyclooxygenase 2 (COX2) expression and
prostaglandin (PG) production in hamster Leydig cells. Mol Cell
Endocrinol. 348:33–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hashizume T and Kanematsu S: Effects of
prostaglandins E2, F2α, and D2 on
the release of growth hormone, prolactin, and luteinizing hormone
from cultured bovine anterior pituitary cells. J Reprod Dev.
41:41–46. 1995.
|
|
24
|
Canete SR, Gui YH, Linask KK and Muschel
RJ: MMP-9 (gelatinase B) mRNA is expressed during mouse
neurogenesis and may be associated with vascularization. Brain Res
Dev Brain Res. 88:37–52. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Turner HE, Nagy Z, Esiri MM, Harris AL and
Wass JA: Role of matrix metalloproteinase 9 in pituitary tumor
behavior. J Clin Endocrinol Metab. 85:2931–2935. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paez PM, Ledda MF, Goldberg V, et al: High
levels of matrix metalloproteinases regulate proliferation and
hormone secretion in pituitary cells. J Clin Endocrinol Metab.
85:263–269. 2000.PubMed/NCBI
|
|
27
|
Kulig E, Jin L, Qian X, et al: Apoptosis
in non-tumorous and neoplastic human pituitaries: expression of the
Bcl-2 family of proteins. Am J Pathol. 154:767–774. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ozer E, Canda MS, Ulukus C, Guray M and
Erbayraktar S: Expression of Bcl-2, Bax and p53 proteins in
pituitary adenomas: an immunohistochemical study. Tumori. 89:54–59.
2003.PubMed/NCBI
|