Introduction
Although a number of cytostatic drugs with different
mechanisms of action are used in the treatment of breast cancer,
intrinsic or acquired resistance is a common problem culminating in
the treatment failure (1). Taxanes
and anthracyclines are widely used and their antitumour activity
contributes to apoptosis in cancer cells through interaction via
several intracellular functions (2,3). For
instance, the taxane paclitaxel exerts its apoptotic effects by
stabilising the microtubuli thus preventing them from disassembling
(4). It is also a potent inhibitor
of chromosomal replication resulting in late Gap 2 (G2) or mitotic
(M) block of the cell cycle (5).
Paclitaxel is generally used as a single agent as well as in
combination therapy with other chemotherapeutic agents to treat
early and advanced stage breast cancer (6). The reported overall response rate of
paclitaxel is 21 to 54% when used as a first-line single agent
therapy. Anthracyclines such as epirubicin, on the other hand, act
in part by inhibiting DNA and RNA synthesis as well as by inducing
permanent double strand DNA breaks by inhibiting the enzyme
topoisomerase II thereby promoting G2-blockage (7,8).
Anthracycline-based polychemotherapy reportedly reduces the
mortality rate by 20 to 38% (9).
However, anthracyclines are associated with a higher rate of acute
and late adverse effects, such as increased risk of cardiotoxicity
and secondary leukaemia (10).
In order to achieve an individualised antitumour
therapy, targeted therapy directed against specific cellular
molecules has been developed. The monoclonal antibody trastuzumab
is the first targeted therapy directed against the extracellular
domain of the human epidermal growth factor receptor 2 (HER2).
Trastuzumab is often used in treatment of patients with an
amplification/overexpression of HER2. Binding of trastuzumab to the
HER2-receptor induces a G1 block, thus reducing both the
proliferation and survival advantages of the tumours (11–15).
Trastuzumab has a modest overall response rate ranging from 15 to
30% when used as a neoadjuvant single agent (16). In clinical settings, trastuzumab is
predominantly administered in combination with chemotherapy drugs
such as taxanes to improve disease-free and overall survival
(17–19). Nevertheless, even when combining
targeted therapy and chemotherapy, treatment failure remains an
ongoing problem in the clinic, indicating the complexity of the
mechanisms involved in drug efficiency.
In recent years, a hypothesis has emerged,
identifying a new role of the mitochondria as an important
regulator of the cell cycle by connecting mitochondrial function
and energetic status with cell cycle progression (20–26).
This supports the theory that altered mitochondrial function along
with deregulated energy metabolism are key for cancer cells to
sustain uncontrolled cell proliferation and avoid apoptosis
(27).
We previously described novel findings of a commonly
deleted gene in HER2-positive breast cancer (28). This gene encodes the mitochondrial
inner membrane solute carrier protein SLC25A43. However, the
complete function of SLC25A43 along with the substrate carried by
this membrane transporter remains unknown. We noted that lower
expression of SLC25A43 in the tumours correlated significantly with
a lower S phase fraction in HER2-positive breast cancer (28).
The aim of this study was to investigate the
possible role of the mitochondrial protein SLC25A43 in drug
sensitivity in vitro. Our results showed that knockdown (KD)
of SLC25A43 in different breast cell lines altered the
sensitivity to the cytostatic drugs, as demonstrated by altered
cell viability and altered distribution and regulation of cell
cycle phases. The findings presented herein support the theory of a
mitochondrial role in drug susceptibility.
Materials and methods
Cell culturing
The immortalized breast epithelial cell line MCF10A,
the HER2-negative breast adenocarcinoma cell line MCF7 and the
HER2-positive breast cancer cell line BT-474 were all obtained from
the American Type Culture Collection (Manassas, VA, USA). MCF10A
was cultured in D-MEM/F-12 supplemented with 10% FBS, 10 μg/ml
insulin, 20 ng/ml H-EGF and 0.5 μg/ml hydrocortisone. MCF7 was
cultured in Eagle's minimum essential medium (MEM) supplemented
with 10% FBS and 10 μg/ml insulin, and BT-474 was cultured in
RPMI-1640 supplemented with 10% FBS and 10 μg/ml insulin. Cells
were cultured in a humidified atmosphere at 37°C with 5%
CO2.
The cells were seeded at a density of
25×103 cells/cm2, 24 h before transfection.
Transfection was performed using Lipofectamine™ 2000 (Invitrogen,
Carlsbad, CA, USA) and scrambled siRNA (siCtrl) or target-specific
siRNA [Hs_LOC203427_2 (Qiagen Sciences, Germantown, MD, USA)]
(siSLC) for KD, according to the manufacturer's recommendations.
The obtained SLC25A43 mRNA KD was 90% in MCF10A, 90% in MCF7
and 75% in BT-474, and was stable in all cell lines for a minimum
of 96 h.
For all cytotoxicity assays, cells were exposed to
the drugs 24 h after transfection and incubated for 72 h, using 16
or 160 nM paclitaxel or 2.5 or 10 μM epirubicin (Actavis,
Hafnarfjordur, Iceland). BT-474 cells were also subjected to
exposure of 10 or 100 μg/ml trastuzumab (Roche AB, Stockholm,
Sweden) or a combination of trastuzumab (10 μg/ml) and paclitaxel
(16 nM), referred to as T/P. As a drug-free control for all
experiments, cells were transfected and cultured in medium without
cytostatic drugs. Incubation with epirubicin was terminated after 1
h by replacing the medium with fresh medium.
Flow cytometry assays
Determination of viable cells
Cell viability was determined by incubating
collected cells in the culture medium together with the trypsinized
cells using 0.25 μg 7-AAD (BD Biosciences, San Jose, CA, USA) for
10 min at room temperature and protected from light, according to
the manufacturer's protocol.
Inhibition of cell proliferation
assay
Measurement of cell proliferation was carried out
using PKH67 Green Fluorescent Cell Linker (Sigma-Aldrich, St.
Louis, MO, USA) according to the manufacturer's protocol. PKH67 is
a green fluorochrome that incorporates into the cell membrane
without affecting cell viability. Following cell division,
fluorescence intensity is decreased due to dilution of the
fluorochrome. The cells were stained with PKH67 at time of
seeding.
Cell cycle phase analysis
Analysis of cell cycle phase distribution was
performed as previously described (29) on isolated cell nuclei using 100
μg/ml propidium iodide (PI) (Sigma-Aldrich) for DNA-staining.
Cell cycle regulation assay with Ki-67
and p21
The expression of Ki-67 and p21 was analysed after
72 h of exposure with 16 nM paclitaxel, 2.5 μM epirubicin, 10 μg/ml
trastuzumab or a combination of trastuzumab (10 μg/ml) and
paclitaxel (16 nM), as indicated. Pelleted cells were resuspended
for 10 min with an ice cold lysing solution containing 0.1% Igepal
CA-630 in wash buffer (1% FBS in PBS) to isolate cell nuclei. The
nuclei were then washed once with ice cold Wash buffer before
adding antibodies against p21 Alexa Fluor® 488 (1:50,
clone 12D1; Cell Signaling Technology, Inc., Danvers, MA, USA) and
Ki-67 PE (clone 56; BD Biosciences) to one tube and isotype control
to a second tube [IgG isotype for Alexa Fluor 488 (1:50) and IgG1
isotype for PE]. All tubes were supplemented with 0.5 μg 7-AAD (BD
Biosciences) for DNA staining and incubated for 15 min. The nuclei
were then diluted with PBS and stored on ice prior to analysis. All
incubation steps were performed on ice.
The flow cytometry analyses were performed 96 h
after transfection using an EPICS Altra equipped with an argon
laser (488 nm) and EXPO 32 software (Beckman-Coulter, Fullerton,
CA, USA). During acquisition, 10,000 events were collected. For
analysis of cell viability, PKH67 and the expression of cell cycle
proteins, Kaluza software (Beckman-Coulter) was used. Cell cycle
phase distribution was analysed using ModFit LT v3.2 (Verity
Software House, USA). To analyse the expression of Ki-67 and p21 a
gate was set on the 7-AAD histogram to exclude sub-G0/G1 events and
then the isotype control was used to determine the cut-off for
positivity. The positivity was expressed as a percentage of
positive cells.
Statistical analysis
The Mann-Whitney test was used to assess statistical
significance in all assays. P<0.05 was considered to indicate a
statistically significant difference. SPSS 17.0 statistical
software for Windows (SPSS Inc., Chicago, IL, USA) was used for all
tests.
Results
Reduced paclitaxel efficacy following
SLC25A43 knockdown
Our experiments described the effects of SLC25A43 on
drug-related cellular outcome and sensitivity utilising
siRNA-mediated mRNA KD in in vitro models. The non-malignant
cell line MCF10A and the two cell lines MCF7 and BT-474, with
different clinically relevant breast cancer phenotypes, were used.
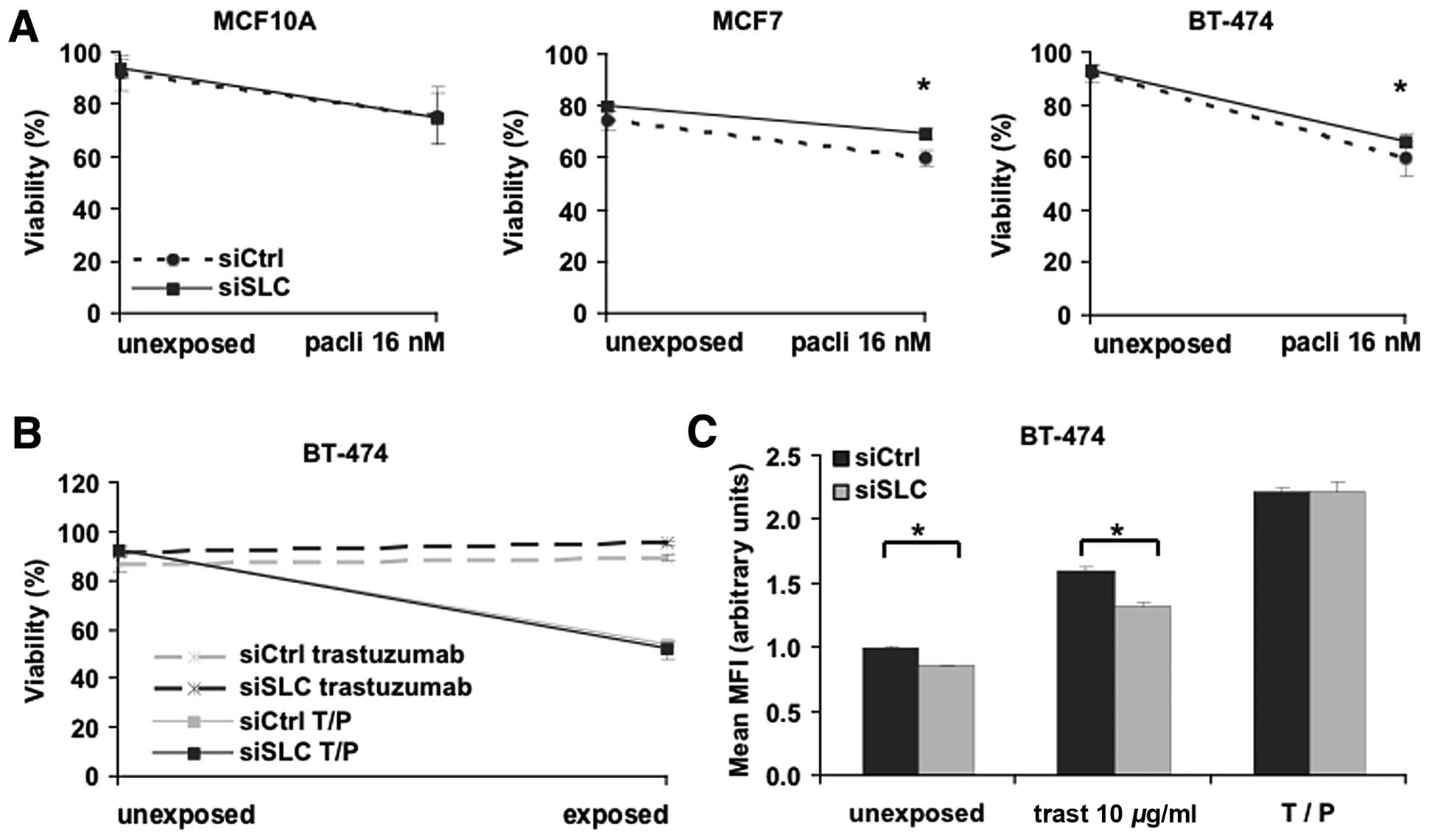
To evaluate the effects of SLC25A43 KD on cytotoxicity, we
analysed the viability of MCF10A, MCF7 and BT-474 cells after 72-h
exposure to paclitaxel or epirubicin. The sensitivity for
paclitaxel was not altered in MCF10A cells after KD of
SLC25A43 (Fig. 1A). By
contrast, KD of SLC25A43 resulted in a significant reduction
of cytotoxic effects by 16 nM paclitaxel in the two breast cancer
cell lines MCF7 and BT-474 compared to siCtrl (Fig. 1A). The reduction in cytotoxicity was
also observed at the higher concentration of paclitaxel in MCF7
cells (P<0.05). Contrary to the paclitaxel exposure, KD of
SLC25A43 did not influence the cytotoxic effect of
epirubicin in any cell line (data not shown).
The HER2-positive breast cancer cell line, BT-474,
was further investigated through exposure with the targeting drug
trastuzumab. Trastuzumab alone does not directly induce cell death
in vitro; however, it inhibits proliferation and is an
important agent when treating HER2-positive breast tumours
(30). We therefore measured both
the viability and inhibition of proliferation of BT-474 cells
subjected to exposure of either trastuzumab (10 μg/ml) or a
combination-dose of trastuzumab (10 μg/ml) and paclitaxel (16 nM)
(T/P) (Fig. 1B and C). As expected,
trastuzumab alone did not induce cell death (Fig. 1D). However, the reduced cytotoxic
effect of paclitaxel after SLC25A43 KD was no longer present
when paclitaxel was combined with trastuzumab. Measuring
proliferation of BT-474 cells after trastuzumab exposure revealed
that SLC25A43 KD contributes to a significantly lower
inhibition of cell proliferation (Fig.
1C). This effect was, however, eliminated when combining
trastuzumab and paclitaxel. These data show that KD of
SLC25A43 influences the efficacy of paclitaxel and
trastuzumab in the breast cancer cell lines.
SLC25A43 affects the cell cycle phase
distribution upon drug exposure
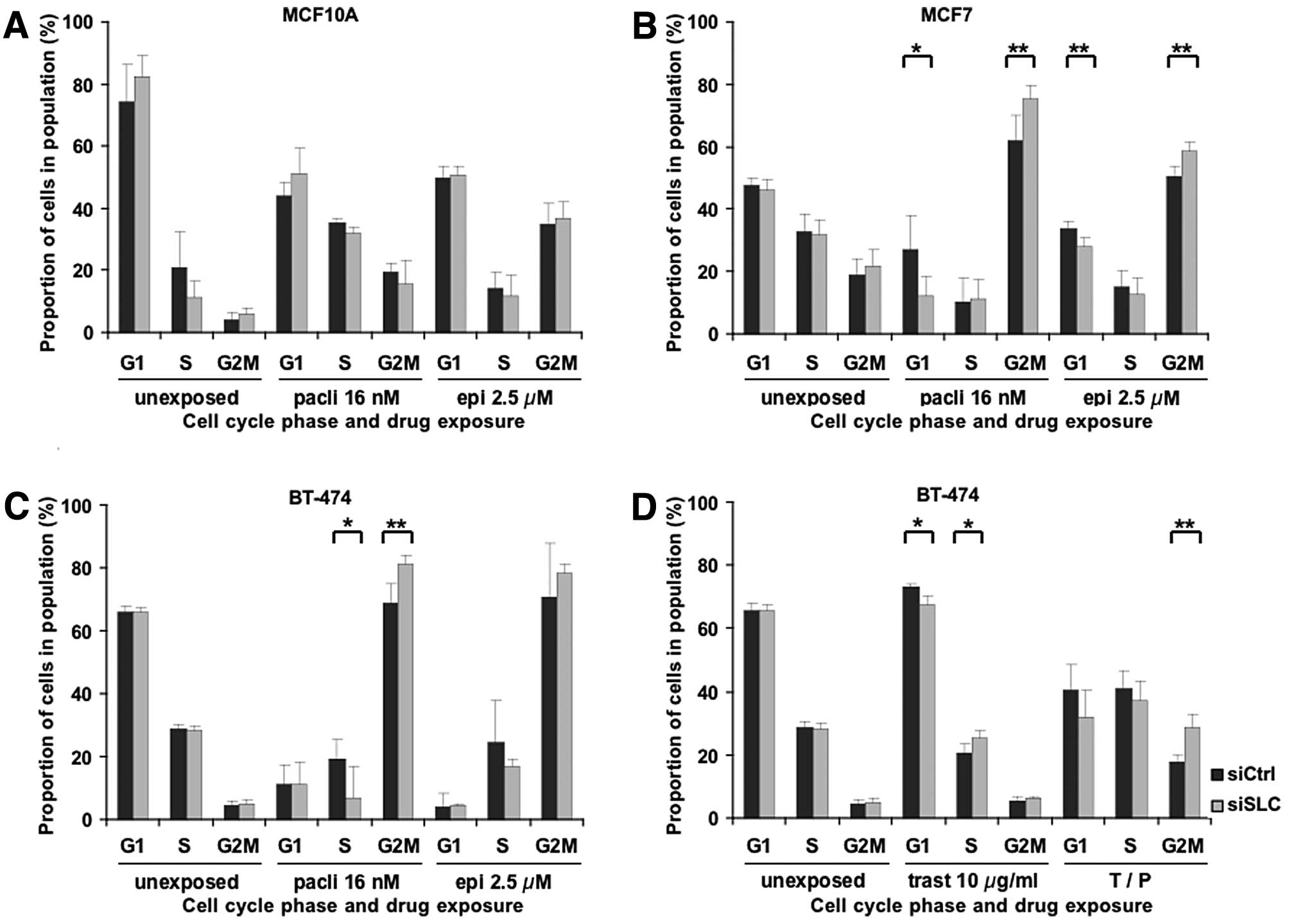
Paclitaxel, epirubicin and trastuzumab are all known
to induce cell cycle arrest, thus leading to cell death or
inhibition of proliferation. To evaluate a possible influence of
SLC25A43 KD on the cell cycle, we analysed the cell cycle
phase distribution in surviving cells following drug exposure.
Paclitaxel blocks the cells in the late G2 and M
phases of the cell cycle (5), and,
as shown in Fig. 2A-C, exposure to
paclitaxel induced a G2/M block in all three cell lines. Notably,
SLC25A43 KD resulted in a significantly higher G2/M block at
16 nM paclitaxel in MCF7 and BT-474 compared to control. This,
however, was not seen in MCF10A cells. The significant difference
in G2/M distribution remained in MCF7 (P<0.05) and BT-474
(P<0.05) cells at the higher concentration of paclitaxel.
Epirubicin induces permanent double strand breaks
leading to G2/M block (7,8). Similar to paclitaxel, epirubicin also
induced G2/M block in the cell lines upon exposure (Fig. 2A-C). However, only MCF7 cells showed
a significant change in cell cycle phase distribution between
siCtrl and SLC25A43 KD at the lower concentration (2.5 μM)
of epirubicin. Exposure of the cell lines to 10 μM of epirubicin
resulted in less G2/M block compared to 2.5 μM, but there was a
significantly higher fraction of cells in G2/M phase in
SLC25A43 KD compared to control in both MCF7 (P<0.05) and
BT-474 (P<0.05) cells.
Next, we examined the effects of trastuzumab on the
cell cycle phase distribution after KD of SLC25A43.
Trastuzumab is known to exhibit growth arrest in the G1 phase,
partly due to inhibition of proteins involved in regulating the
cell cycle (11–15). This was also confirmed in the BT-474
cells (Fig. 2D). Furthermore, KD of
SLC25A43 leads to a significantly decreased G1 block at both
10 and 100 μg/ml of trastuzumab (P<0.05). This result suggests
that KD of SLC25A43 causes a reduced sensitivity to the
inhibitory mechanisms of trastuzumab, which correlates with the
results regarding the reduced inhibition of proliferation in
SLC25A43 KD cells compared to control, following trastuzumab
treatment (Fig. 1D). Exposure to
trastuzumab in combination with paclitaxel resulted in reduced G2/M
block compared to paclitaxel alone, however, the significant effect
of SLC25A43 KD on the G2/M block remained (Fig. 2C and D). These results demonstrate
that KD of SLC25A43 in combination with drug exposure alters
the cell cycle phase distribution in the breast cancer cell
lines.
Ki-67 expression is altered by paclitaxel
and epirubicin exposure
Loss of growth control, including aberrations in the
mechanisms regulating the integrity of cell cycle progression, is a
hallmark of cancer (27).
Determining the expression of cell proliferation markers in tumour
has therefore become increasingly important in the clinic (31,32).
One marker of interest is Ki-67, a protein present in all phases of
the cell cycle except in the G0 and the early G1 phase (33). In light of this and regarding the
findings that KD of SLC25A43 alters the cell cycle
distribution upon drug exposure, we further investigated the
effects of SLC25A43 KD on the Ki-67 expression.
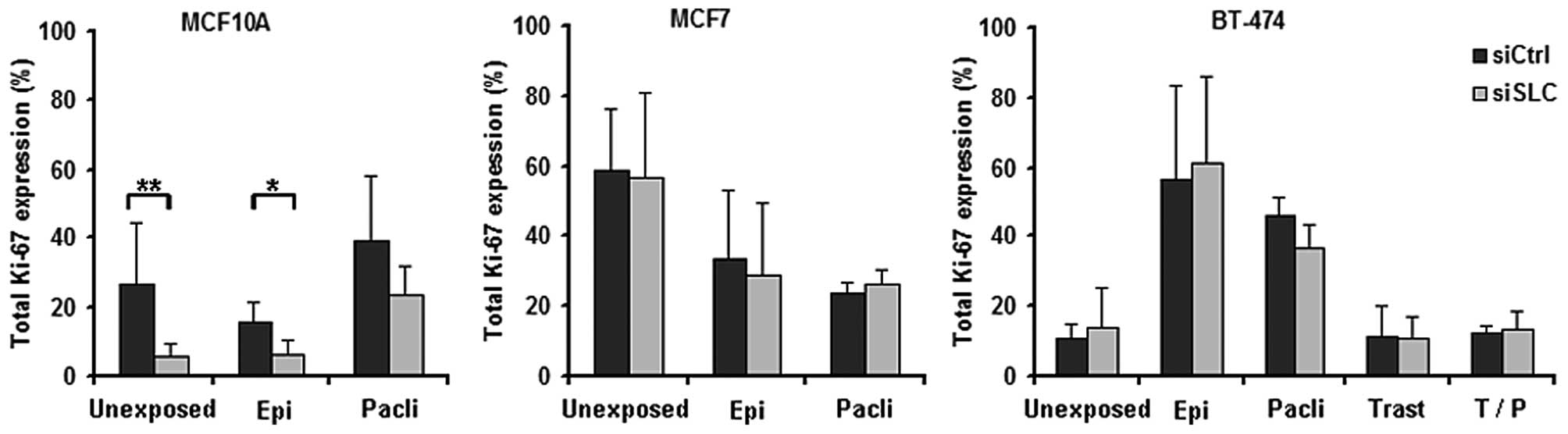
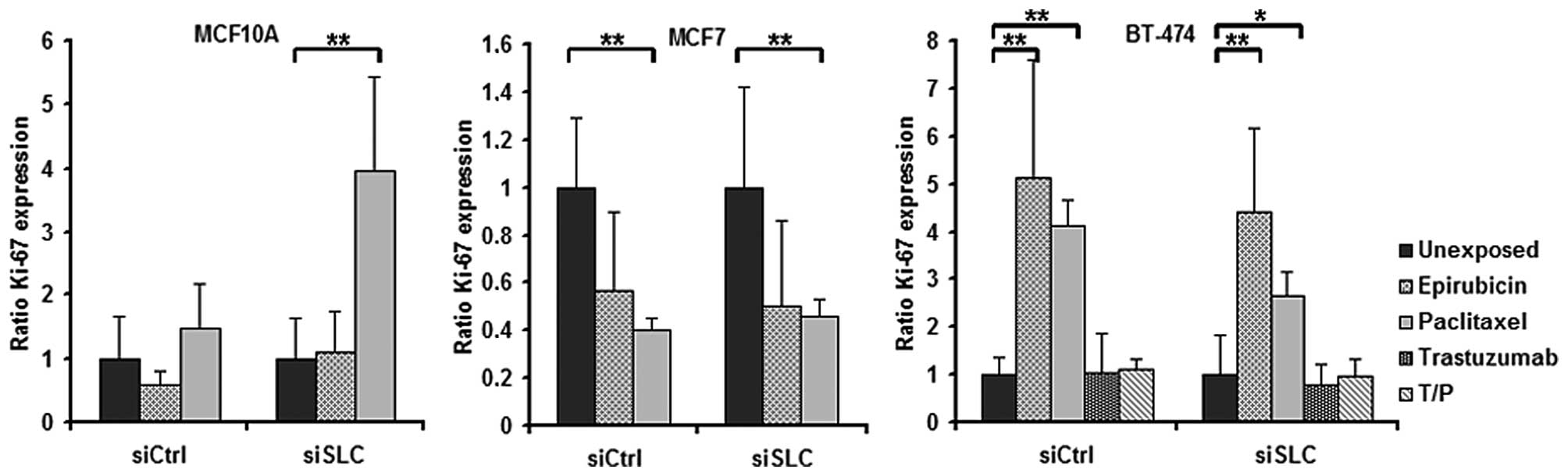
In MCF10A cells, KD of SLC25A43 led to a
decreased total percentage of Ki-67-positive cells compared to
siCtrl both in unexposed- and in epirubicin-exposed cells (Fig. 3). When exposing the cell lines to
paclitaxel, the fraction of Ki-67-positive cells was found not to
be altered by KD of SLC25A43 when compared to siCtrl.
However, paclitaxel was shown to influence the Ki-67 expression in
all three cell lines (Fig. 4). In
addition, exposure to epirubicin resulted in an increased fraction
of Ki-67-positive BT-474 cells (Fig.
4).
As the mitochondrion has been demonstrated to impact
the cell cycle regulation (20–26),
we investigated if KD of SLC25A43 in the three cell lines,
when exposed to drugs, would contribute to changes in cell cycle
regulation by analysis of p21 positivity. In our study,
SLC25A43 KD and drug exposure did not influence the p21
positivity in any of the three cell lines (data not shown).
Discussion
The theory that mitochondria are important players
of apoptosis, cell proliferation and cell cycle regulation
(20–26) has expanded the field of cancer
research, and it is now well established that mitochondrial
dysfunction plays a key role in tumour development and response to
therapy (27).
We previously demonstrated a common loss of the
mitochondrial gene SLC25A43 in HER2-positive breast cancer
(28). We further showed that
HER2-positive breast tumours with a lower expression of SLC25A43
also have a lower S phase fraction. These previous findings led to
the hypothesis that an altered mitochondrial function through a
reduced expression of SLC25A43 may alter the efficacy of antitumour
drugs in vitro.
Using siRNA, we investigated the effects of
SLC25A43 knockdown (KD) on drug cytotoxicity in immortalised
mammary epithelial cells, HER2-negative, and HER2-positive breast
cancer cells after exposure to different drugs. In our study, KD of
SLC25A43 did not alter the viability following epirubicin
exposure in any of the cell lines. This suggests that the mechanism
of action of epirubicin cytotoxicity is, at least in part,
independent of altered mitochondrial function due to
SLC25A43 KD. Paclitaxel exerts is cytotoxic effects through
other mechanisms, involving microtubule assembly, cell cycle arrest
and the mitochondrial apoptotic pathway (34–36).
Deregulations in this pathway and other mitochondrial functions
have been suggested to induce paclitaxel resistance (36–38).
In our experiments, exposure of the HER2-negative MCF7 cells and
the HER2-positive BT-474 cells to paclitaxel after KD increased the
viability of the cells compared to control. This change in
viability was accompanied by an increased G2/M block in the exposed
KD cells, indicating that further blocking of the cells in G2/M may
be beneficial for cell survival. Tan et al(39) demonstrated that delaying cell entry
into M phase conferred survival advantages following paclitaxel
exposure in breast cancer cells. BT-474 cells were further
subjected to exposure to trastuzumab. KD of SLC25A43
followed by exposure to trastuzumab resulted in decreased
inhibitory effect on proliferation of the antibody and reduced the
G0/G1 arrest. Trastuzumab is known to increase the association
between p27Kip1 and CDK2 complexes resulting in induced
G1 cell cycle arrest (11–15,40).
Altered expression of these proteins has been connected with
trastuzumab resistance (41). Our
findings show that KD of SLC25A43 reduces the G0/G1 arrest,
possibly leading to the reduced inhibitory effect of trastuzumab in
the HER2-positive BT-474 cells. When combining trastuzumab and
paclitaxel to BT-474 cells, all survival and proliferative
advantages due to SLC25A43 KD previously observed are
eradicated. This demonstrates the beneficial effect of dual
exposure with cytostatic drugs and target therapies after
SLC25A43 KD.
Ki-67 is widely used as a proliferation marker in
the clinic and it has also been shown to be a prognostic marker
(31); however, studies
investigating the predictive value of Ki-67 for chemotherapy
present inconclusive data (42–45).
When investigating the Ki-67 expression in the cell lines after
drug exposure, SLC25A43 KD was shown to influence the
expression in the non-malignant MCF10A cells only after epirubicin
exposure. However, both epirubicin and paclitaxel were found to
alter the Ki-67 expression in the two breast cancer cell lines when
compared to unexposed cells. MCF7 cells showed a decreased Ki-67
positivity while BT-474 cells showed an increased Ki-67 positivity
following paclitaxel exposure. These findings may not be a direct
result of the drug exposure; instead, it is more likely to be a
secondary finding following the cell cycle arrest. It has
previously been described in a clinical study that the Ki-67
expression was altered after chemotherapy, compared to before
treatment, and a decreased expression was associated with reduced
disease-free survival in rectal carcinoma (46). Also, p21 expression has been shown
to be altered after treatment (46,47).
In our study, however, we were not able to assess any differences
in p21 expression due to drug exposure. The clinical importance of
an altered Ki-67 and/or p21 expression after chemotherapy for
patient outcome remains to be further elucidated.
Collectively, we have demonstrated that SLC25A43
alters the efficacy of paclitaxel through increased viability and
increased G2/M arrest. A reduced expression of SLC25A43 also
diminishes the antiproliferative effect of the target therapy
trastuzumab. Our findings support the role of altered mitochondrial
function in cancer and drug resistance.
Acknowledgements
We thank Edwin Reizer for the laboratory assistance
and Professor Olle Stål for helpful discussions concerning the
manuscript. This study was supported by grants from the Lion Cancer
Foundation Sweden and the Research Committee at Örebro County
Council.
References
|
1
|
Fossati R, Confalonieri C, Torri V, et al:
Cytotoxic and hormonal treatment for metastatic breast cancer: a
systematic review of published randomized trials involving 31,510
women. J Clin Oncol. 16:3439–3460. 1998.PubMed/NCBI
|
|
2
|
Abal M, Andreu JM and Barasoain I:
Taxanes: microtubule and centrosome targets, and cell cycle
dependent mechanisms of action. Curr Cancer Drug Targets.
3:193–203. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Minotti G, Menna P, Salvatorelli E, Cairo
G and Gianni L: Anthracyclines: molecular advances and
pharmacologic developments in antitumor activity and
cardiotoxicity. Pharmacol Rev. 56:185–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schiff PB, Fant J and Horwitz SB:
Promotion of microtubule assembly in vitro by taxol. Nature.
277:665–667. 1979. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Frankel A, Buckman R and Kerbel RS:
Abrogation of taxol-induced G2-M arrest and apoptosis in human
ovarian cancer cells grown as multicellular tumor spheroids. Cancer
Res. 57:2388–2393. 1997.PubMed/NCBI
|
|
6
|
Sparano JA: Taxanes for breast cancer: an
evidence-based review of randomized phase II and phase III trials.
Clin Breast Cancer. 1:32–42. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cersosimo RJ and Hong WK: Epirubicin: a
review of the pharmacology, clinical activity, and adverse effects
of an adriamycin analogue. J Clin Oncol. 4:425–439. 1986.PubMed/NCBI
|
|
8
|
Knoop AS, Knudsen H, Balslev E, et al:
Retrospective analysis of topoisomerase IIa amplifications and
deletions as predictive markers in primary breast cancer patients
randomly assigned to cyclophosphamide, methotrexate, and
fluorouracil or cyclophosphamide, epirubicin, and fluorouracil:
Danish Breast Cancer Cooperative Group. J Clin Oncol. 23:7483–7490.
2005.
|
|
9
|
Early Breast Cancer Trialists’
Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal
therapy for early breast cancer on recurrence and 15-year survival:
an overview of the randomised trials. Lancet. 365:1687–1717.
2005.PubMed/NCBI
|
|
10
|
Levine MN, Bramwell VH, Pritchard KI, et
al: Randomized trial of intensive cyclophosphamide, epirubicin, and
fluorouracil chemotherapy compared with cyclophosphamide,
methotrexate, and fluorouracil in premenopausal women with
node-positive breast cancer. National Cancer Institute of Canada
Clinical Trials Group. J Clin Oncol. 16:2651–2658. 1998.
|
|
11
|
Baselga J, Albanell J, Molina MA and
Arribas J: Mechanism of action of trastuzumab and scientific
update. Semin Oncol. 28:4–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sliwkowski MX, Lofgren JA, Lewis GD,
Hotaling TE, Fendly BM and Fox JA: Nonclinical studies addressing
the mechanism of action of trastuzumab (Herceptin). Semin Oncol.
26:60–70. 1999.PubMed/NCBI
|
|
13
|
Mirza AM, Gysin S, Malek N, Nakayama K,
Roberts JM and McMahon M: Cooperative regulation of the cell
division cycle by the protein kinases RAF and AKT. Mol Cell Biol.
24:10868–10881. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mullany LK, Nelsen CJ, Hanse EA, et al:
Akt-mediated liver growth promotes induction of cyclin E through a
novel translational mechanism and a p21-mediated cell cycle arrest.
J Biol Chem. 282:21244–21252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yakes FM, Chinratanalab W, Ritter CA, King
W, Seelig S and Arteaga CL: Herceptin-induced inhibition of
phosphatidylinositol-3 kinase and Akt is required for
antibody-mediated effects on p27, cyclin D1, and antitumor action.
Cancer Res. 62:4132–4141. 2002.PubMed/NCBI
|
|
16
|
Montemurro F and Aglietta M: Incorporating
trastuzumab into the neoadjuvant treatment of HER2-overexpressing
breast cancer. Clin Breast Cancer. 6:77–80. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Montemurro F, Valabrega G and Aglietta M:
Trastuzumab-based combination therapy for breast cancer. Expert
Opin Pharmacother. 5:81–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Buzdar AU, Ibrahim NK, Francis D, et al:
Significantly higher pathologic complete remission rate after
neoadjuvant therapy with trastuzumab, paclitaxel, and epirubicin
chemotherapy: results of a randomized trial in human epidermal
growth factor receptor 2-positive operable breast cancer. J Clin
Oncol. 23:3676–3685. 2005. View Article : Google Scholar
|
|
19
|
Piccart-Gebhart MJ, Procter M,
Leyland-Jones B, et al: Trastuzumab after adjuvant chemotherapy in
HER2-positive breast cancer. N Engl J Med. 353:1659–1672. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Finkel T and Hwang PM: The Krebs cycle
meets the cell cycle: mitochondria and the G1-S transition. Proc
Natl Acad Sci USA. 106:11825–11826. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Detmer SA and Chan DC: Functions and
dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol.
8:870–879. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schieke SM, McCoy JP Jr and Finkel T:
Coordination of mitochondrial bioenergetics with G1 phase cell
cycle progression. Cell Cycle. 7:1782–1787. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mandal S, Guptan P, Owusu-Ansah E and
Banerjee U: Mitochondrial regulation of cell cycle progression
during development as revealed by the tenured mutation in
Drosophila. Dev Cell. 9:843–854. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jones RG, Plas DR, Kubek S, et al:
AMP-activated protein kinase induces a p53-dependent metabolic
checkpoint. Mol Cell. 18:283–293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gwinn DM, Shackelford DB, Egan DF, et al:
AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Owusu-Ansah E, Yavari A, Mandal S and
Banerjee U: Distinct mitochondrial retrograde signals control the
G1-S cell cycle checkpoint. Nat Genet. 40:356–361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tina E, Malakkaran Lindqvist B, Gabrielson
M, Lubovac Z, Wegman P and Wingren S: The mitochondrial transporter
SLC25A43 is frequently deleted and may influence cell proliferation
in HER2-positive breast tumors. BMC Cancer. 12:3502012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tina E, Prenkert M, Hoglund M, Paul C and
Tidefelt U: Topoisomerase IIα expression in acute myeloid leukaemia
cells that survive after exposure to daunorubicin or ara-C. Oncol
Rep. 22:1527–1531. 2009.
|
|
30
|
Lantz E, Cunningham I and Higa GM:
Targeting HER2 in breast cancer: overview of long-term experience.
Int J Womens Health. 1:155–171. 2010.PubMed/NCBI
|
|
31
|
Goldhirsch A, Ingle JN, Gelber RD, Coates
AS, Thurlimann B and Senn HJ: Thresholds for therapies: highlights
of the St Gallen International Expert Consensus on the primary
therapy of early breast cancer 2009. Ann Oncol. 20:1319–1329. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Butt AJ, Caldon CE, McNeil CM, Swarbrick
A, Musgrove EA and Sutherland RL: Cell cycle machinery: links with
genesis and treatment of breast cancer. Adv Exp Med Biol.
630:189–205. 2008.PubMed/NCBI
|
|
33
|
Gerdes J, Lemke H, Baisch H, Wacker HH,
Schwab U and Stein H: Cell cycle analysis of a cell
proliferation-associated human nuclear antigen defined by the
monoclonal antibody Ki-67. J Immunol. 133:1710–1715.
1984.PubMed/NCBI
|
|
34
|
Tudor G, Aguilera A, Halverson DO, Laing
ND and Sausville EA: Susceptibility to drug-induced apoptosis
correlates with differential modulation of Bad, Bcl-2 and Bcl-xL
protein levels. Cell Death Differ. 7:574–586. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sunters A, Madureira PA, Pomeranz KM, et
al: Paclitaxel-induced nuclear translocation of FOXO3a in breast
cancer cells is mediated by c-Jun NH2-terminal kinase and Akt.
Cancer Res. 66:212–220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kutuk O and Letai A: Alteration of the
mitochondrial apoptotic pathway is key to acquired paclitaxel
resistance and can be reversed by ABT-737. Cancer Res.
68:7985–7994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou M, Zhao Y, Ding Y, et al: Warburg
effect in chemosensitivity: targeting lactate dehydrogenase-A
re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer.
9:332010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gogvadze V, Orrenius S and Zhivotovsky B:
Mitochondria as targets for chemotherapy. Apoptosis. 14:624–640.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tan M, Jing T, Lan KH, et al:
Phosphorylation on tyrosine-15 of p34(Cdc2) by ErbB2 inhibits
p34(Cdc2) activation and is involved in resistance to taxol-induced
apoptosis. Mol Cell. 9:993–1004. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lane HA, Motoyama AB, Beuvink I and Hynes
NE: Modulation of p27/Cdk2 complex formation through 4D5-mediated
inhibition of HER2 receptor signaling. Ann Oncol. 12(Suppl 1):
S21–S22. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nahta R, Takahashi T, Ueno NT, Hung MC and
Esteva FJ: P27(kip1) down-regulation is associated with trastuzumab
resistance in breast cancer cells. Cancer Res. 64:3981–3986. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chang J, Ormerod M, Powles TJ, Allred DC,
Ashley SE and Dowsett M: Apoptosis and proliferation as predictors
of chemotherapy response in patients with breast carcinoma. Cancer.
89:2145–2152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
MacGrogan G, Mauriac L, Durand M, et al:
Primary chemotherapy in breast invasive carcinoma: predictive value
of the immunohistochemical detection of hormonal receptors, p53,
c-erbB-2, MiB1, pS2 and GST pi. Br J Cancer. 74:1458–1465. 1996.
View Article : Google Scholar
|
|
44
|
Colleoni M, Viale G, Zahrieh D, et al:
Chemotherapy is more effective in patients with breast cancer not
expressing steroid hormone receptors: a study of preoperative
treatment. Clin Cancer Res. 10:6622–6628. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rozan S, Vincent-Salomon A, Zafrani B, et
al: No significant predictive value of c-erbB-2 or p53 expression
regarding sensitivity to primary chemotherapy or radiotherapy in
breast cancer. Int J Cancer. 79:27–33. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rau B, Sturm I, Lage H, et al: Dynamic
expression profile of p21WAF1/CIP1 and Ki-67 predicts survival in
rectal carcinoma treated with preoperative radiochemotherapy. J
Clin Oncol. 21:3391–3401. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pohl G, Rudas M, Taucher S, et al:
Expression of cell cycle regulatory proteins in breast carcinomas
before and after preoperative chemotherapy. Breast Cancer Res
Treat. 78:97–103. 2003. View Article : Google Scholar : PubMed/NCBI
|