Introduction
Silent mating-type information regulation 2
homologue 1 (SIRT1) is a protomember of the sirtuin family
(SIRT1–7) that is involved in a variety of physiological processes
such as genomic stability, metabolism, neurogenesis and cell
survival due to its ability to deacetylate both histone and
numerous non-histone substrates such as gene silencing, metabolism,
neuroprotection, and genomic stability (1,2). SIRT1
acts as a deacetylase not only on histone but also by deacetylating
cell cycle regulators such as p53 (3), p73 (4)
and Rb (5). With regard to the
growth of cancer cells, it has been reported that SIRT1 regulates
cell proliferation, survival, and death, and plays a pivotal role
in tumorigenesis and longevity.
SIRT1 prompts cell proliferation and plays an
important role in the drug resistance of tumor cells, especially
during chemotherapy. In humans, several types of cancer, including
prostate (6,7), breast cancer (8,9), colon
(10,11), glioblastoma (12), lymphoma (13) and acute myeloid leukemia (14) have been demonstrated to have a
significantly increased expression of SIRT1. Furthermore, a recent
study showed that SIRT1 regulates the DNA damage signaling pathway
through deacetylation of NBS1 to promote cellular survival
(15). It was reported that SIRT1
upregulates the expression of multidrug resistance 1 (MDR1) in
cancer, and this may cause cancer resistance to chemotherapy
(16). SIRT1-deficient cells
exhibited p53 hyperacetylation following DNA damage and increased
ionizing radiation-induced thymocyte apoptosis in vivo along
with downregulation of MDR1 (16–19).
This evidence indicates the oncogenic role of SIRT1.
By contrast, several studies have shown the tumor
suppressor role of SIRT1. The reduced transformation capability of
c-myc can be compromised in the presence of SIRT1 (20). Ectopic induction of SIRT1 in a
β-catenin-driven mouse model of colon cancer significantly reduced
colon cancer formation, growth, and animal morbidity (21). Moreover, the mouse embryonic
fibroblasts (MEFs) derived from SIRT1−/− mice are prone
to spontaneous immortalization, suggesting that SIRT1 behaves as a
growth-suppressive gene (22).
Analysis of SIRT1 mutant mice revealed that SIRT1−/−
MEFs displayed chromosome aneuploidy and repair defects, and that
SIRT1 played an important role in the maintenance of the
heterochromatin structure through deacetylation of histones in
vivo(23). Collectively, these
previous findings indicated that SIRT1 may possess both pro- and
anti-proliferative functions, depending on the cell types. In spite
of the controversial role of SIRT1 in tumorigenesis (24,25),
it is evident that SIRT1 is significantly involved in the process
of tumorigenesis.
Since SIRT1 is involved in both the death and
survival of cells, we proposed that SIRT1 may be a critical
determinant of cell fate during tumorigenesis. To test this
hypothesis, we investigated the expression of SIRT1 in tumor breast
tissues and matched normal breast tissues of Taiwanese women, and
used breast cancer cell lines to examine the role of SIRT1 in
breast tumorigenesis.
Materials and methods
Patients
A total of 27 breast cancer patients were included
in this study between 2007 and 2008. Tumor tissues and the paired
normal breast tissues were immediately frozen after collection. The
cases were diagnosed as infiltrating ductal carcinoma (IDC; n=27)
according to the World Health Organization classification system
(26). Clinical data were collected
retrospectively. All patients provided informed consent and the
study was approved by the Institutional Review Board of Changhua
Christian Hospital.
Antibodies and reagents
The antibodies used for western blotting were:
anti-SIRT1 (ab53517; Abcam, Cambridge, UK), anti-β-actin (A-5441)
and anti-Bcl-2 (B-3170) (both from Sigma-Aldrich Co., MI, USA). The
primary antibodies used for immunohistochemical analysis were:
anti-SIRT1 (ab7343; Abcam) and anti-Ki67 (RM-9106-S1; Lab Vision,
Fremont, CA, USA). Chemicals and reagents were purchased from
Sigma-Aldrich Co., unless otherwise indicated.
Immunohistochemical analysis
The 5-μm sections of formalin-fixed,
paraffin-embedded tissues were deparaffinized, rehydrated and the
antigen retrieved. The tissue sections were incubated with hydrogen
peroxide for 5 min at room temperature to quench the endogenous
peroxidase. After blocking in normal goat serum, the tissue
sections were incubated for 1 h at room temperature with the
primary antibodies anti-SIRT1 (1:100 dilution) and anti-Ki67 (1:50
dilution), respectively. The slides were then washed with 0.5%
Triton X-100 in PBS and incubated with HRP-conjugated secondary
antibody for 1 h (Vector Laboratories, Burlingame CA, USA). Nuclei
were counterstained with hematoxylin. As a negative control, normal
goat serum was substituted for the primary antibody. All slides
were examined by two independent investigators who were blind to
the clinical data. To evaluate the expression of SIRT1 and Ki67 in
tumor breast tissue and matched normal breast tissue, the staining
intensity was scored from 0 to 3. The ratio of the positively
stained area to the total section area was assessed.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was isolated from breast tissue using
TRIzol Reagent and cDNA was synthesized by MMLV Reverse
transcriptase (both from Invitrogen, Carlsbad, CA, USA). The primer
sequences used for PCR are shown in Table I. Synthesized cDNA was amplified and
the PCR product was then visualized on 1% agarose gel. The
intensity of the PCR product was determined by densitometry using
the Gel Image Analysis System (Alpha Innotech Co., San Leandro, CA,
USA).
 | Table IOligonucleotide sequences for
RT-PCR. |
Table I
Oligonucleotide sequences for
RT-PCR.
| Primer | Primer sequence |
|---|
| SIRT1 | F:
5′-CATCTCTCTGTCACAAATTCATAGCC-3′
R: 5′-GTAATTTCTGAAAGCTTTACAGGGTT-3′ |
| GAPDH | F:
5′-GAGTCAACGGATTTGGTCGT-3′
R: 5′-GGTGCTAAGCAGTTGGTGGT-3′ |
Cell culture
The human breast cancer cell lines and MCF-7 and
MDA-MB-231 cell lines were grown in Dulbecco's modified Eagle's
medium: Nutrient Mixture F-12 (DMEM/F12) supplemented with 10%
fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 100
μg/ml streptomycin (Gibco, Invitrogen Ltd., Carlsbad, CA, USA) in a
humidified atmosphere containing 5% CO2 at 37°C.
Cell proliferation assay
The cytotoxicity effect of sirtinol on MCF-7 and
MDA-MB-231 cells was determined by trypan blue dye exclusion and
Alamar-blue assays (AbD Serotec, Kidlington, UK). Briefly, the
cells were plated at a density of 2×105 cells in 6-well
plates with an increasing dose of sirtinol (10–50 μM in DMSO) or
vehicle alone. Following incubation for 24 or 48 h, cells were
collected and an aliquot of cell suspension was mixed with an equal
volume of trypan blue, and cells were then counted under the
microscope. The cell viability assay was performed following the
instructions of the Alamar blue assay kit. In brief, cells were
plated at a density of 2×103 cells in 200 μl of complete
medium in a 96-well plate and treated with increasing doses of
sirtinol or vehicle alone. Each treatment was performed in
triplicate. The cells were incubated for 24 or 48 h at 37°C in 5%
CO2. Five hours before the end of the culture, the
Alamar blue reagent (20 μl in phenol red free RPMI-1640 medium) was
added into each well and incubated up to 24 or 48 h. Absorbance of
590 nm wavelength emissions was recorded.
Flow cytometry analysis
Cell cycle analysis was performed with flow
cytometry. Cells were grown at a density of 5×105 cells
in 6-well culture plates, treated with different concentrations of
sirtinol for 24 and 48 h, and maintained at 37°C in a 5%
CO2 incubator. Following treatment, the cells were
gently trypsinized, added to the culture medium, and pelleted by
centrifugation. The cell pellet was resuspended in 1 ml PBS, washed
at 1,500 × g for 30 min at 4°C, and fixed in ethanol (75% v/v). The
cells were then washed with PBS and labeled with propidium
iodide/RNase A solution. Labeled cells were analyzed by flow
cytometry (Epics XL-MCL Flow Cytometry; Beckman Coulter, Inc.,
Fullerton, CA, USA) and the analyses were performed using WinMDI
2.9 software.
Western blot analysis
The cells were harvested at 24 and 48 h following
sirtinol treatment, as described above, and washed with PBS,
respectively. The cells were lysed with ice-cold RIPA lysis buffer
(1X TBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS,
0.004% sodium azide with protease inhibitor) and harvested. The
lysate was centrifuged at 14,000 × g for 30 min at 4°C, and the
supernatant was collected and stored at −20°C until analysis. The
protein concentration was determined using the bicinchoninic acid
protein assay (Pierce, Rockford, IL, USA) according to the
manufacturer's instructions. Fifty micrograms of protein were
separated by SDS-PAGE and then transferred onto a PVDF membrane
(0.4 μm; Millipore, Billerica, MA, USA). Non-specific interactions
were blocked by incubating the membrane with 5% non-fat dry milk in
0.5% PBST for 1 h at room temperature. The membrane was then
blotted with anti-SIRT1, Bcl-2, or β-actin antibodies at 4°C
overnight. HRP-conjugated secondary antibodies were added for 1 h,
and the immunoreactive signals were then detected using the
SuperSignal West Pico Chemiluminescent Substrate (Pierce). The
level of protein expression was analyzed using the Gel Image
Analysis System (Alpha Innotech Co.).
Statistical analysis
Statistical analyses were performed using the
Student's t-test. A P-value of <0.05 was considered to indicate
a statistically significant difference. All data are shown as the
means ± SEM. Statistical calculations were performed using SPSS
software (SPSS, Chicago, IL, USA).
Results
Increased gene expression of SIRT1 in
breast cancer
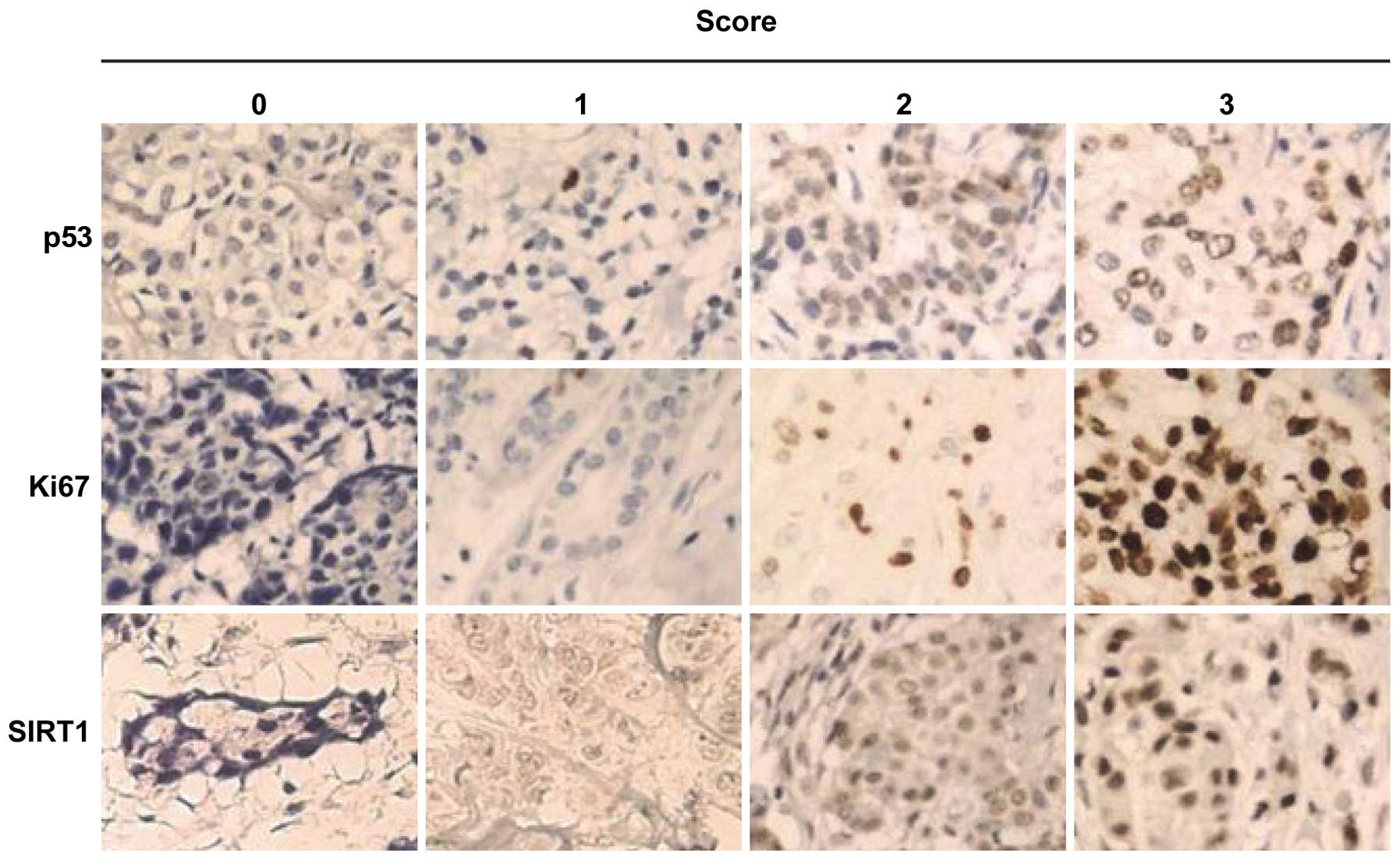
The results of immunohistochemical analysis revealed
that SIRT1, Ki67 and p53 were localized in tumor and in normal
cells with varying intensities (Fig.
1). The expression of SIRT1 and Ki67 was significantly higher
in tumor tissue than in normal tissue (P<0.001 and P<0.005
for SIRT1 and Ki67, respectively). Twenty-three (82.1%) tumor cases
had a higher level of SIRT1 and Ki67 proteins. Furthermore, the
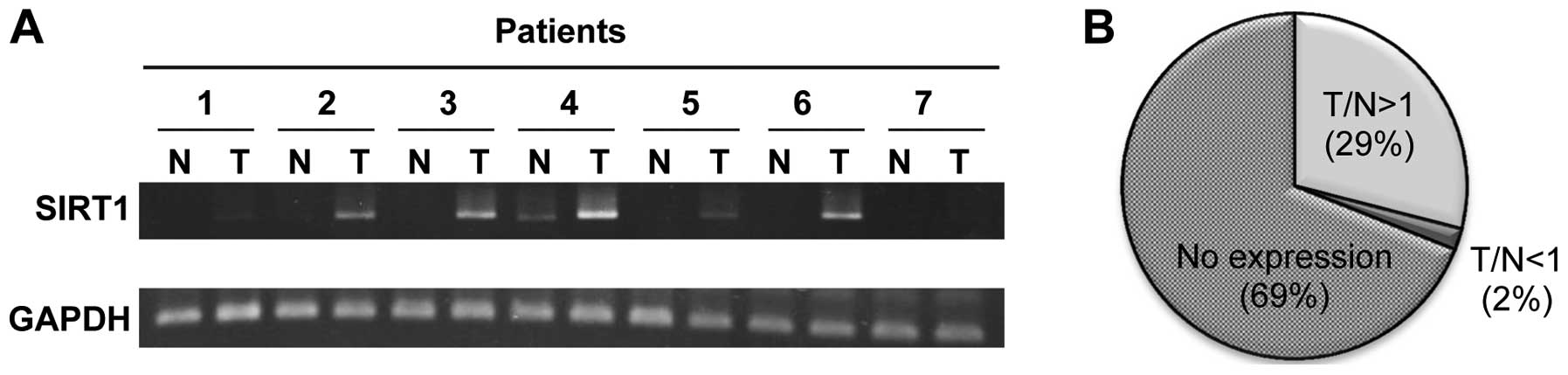
RT-PCR results showed that the mRNA level of SIRT1 was
overexpressed in breast tumor tissues (29%), compared to the paired
normal tissues (Fig. 2).
Sirtinol induces cell growth inhibition
through cell cycle arrest in the G0/G1 phase in breast cancer
cells
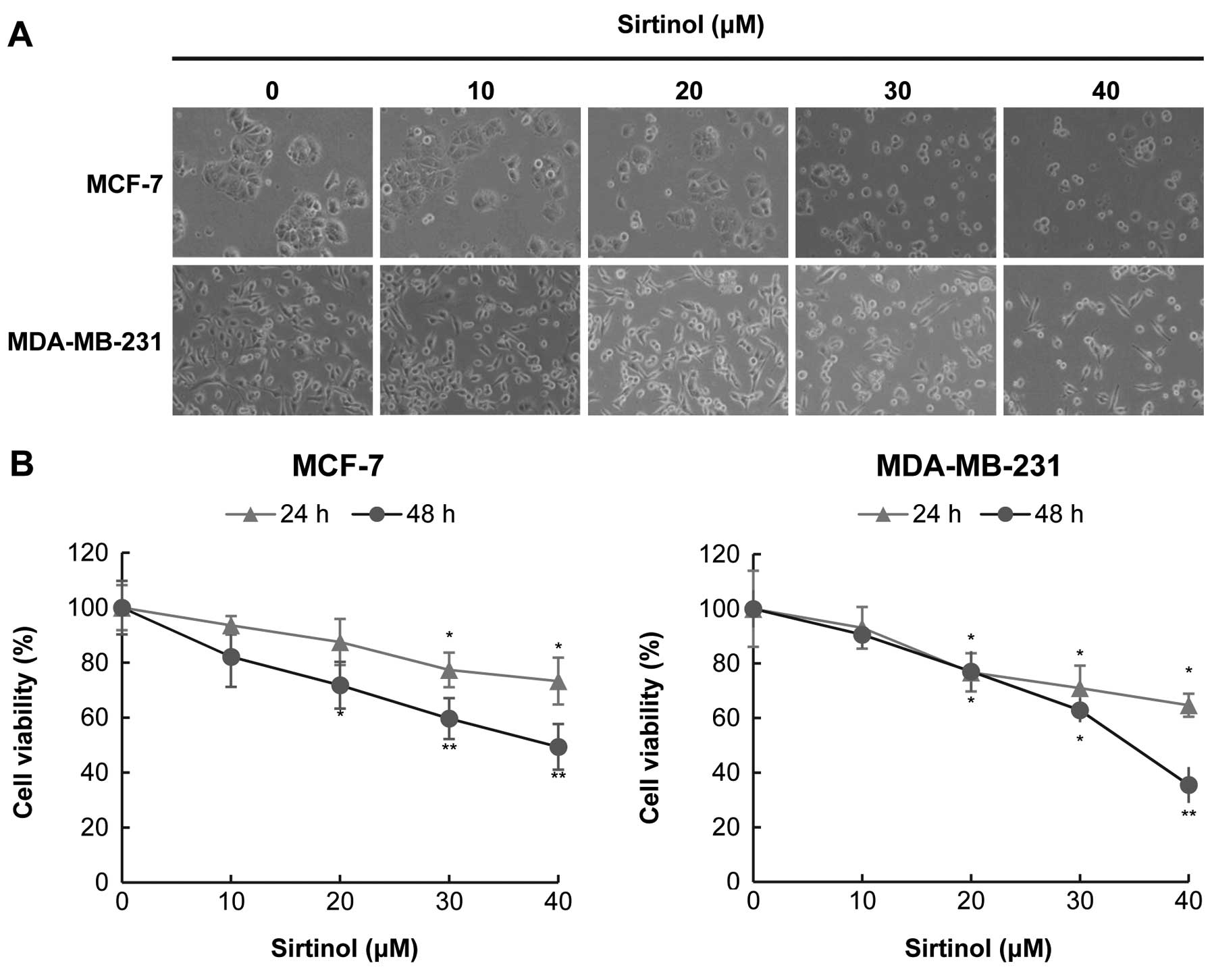
When cells were treated with sirtinol at higher
concentrations (30–40 μM), cell shrinkage was observed (Fig. 3A). The cell viability was reduced by
sirtinol in a time- and dose-dependent manner in both MCF-7 and
MDA-MB-23 cells (Fig. 3B). Flow
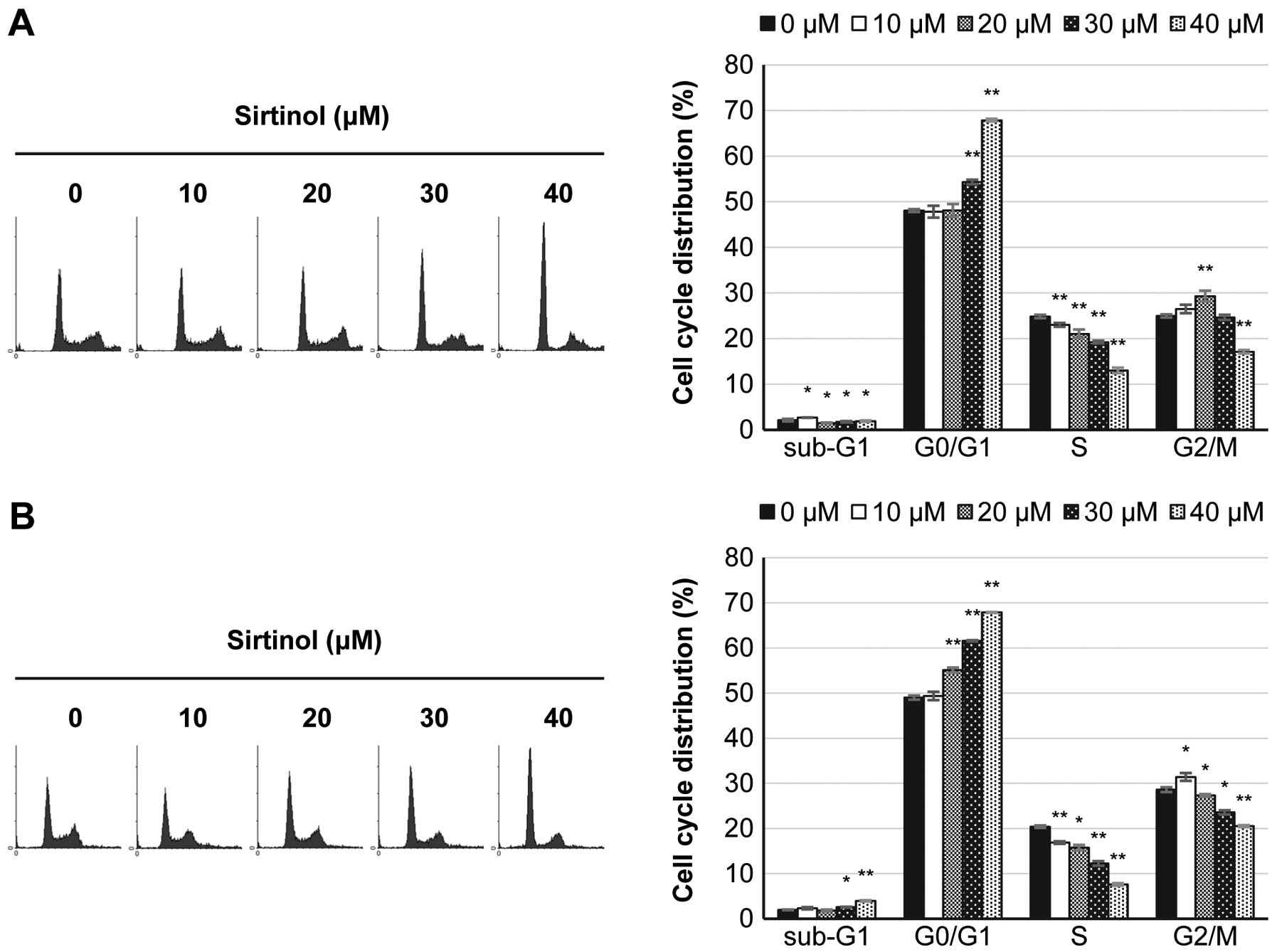
cytometry analysis was used to examine the effects of sirtinol on
the cell cycle progression of breast cancer cells. As shown in
Fig. 4, sirtinol induced a cell
cycle arrest in the G0/G1 phase in both cell lines. The proportion
of the sub-G1 population also increased slightly in MDA-MB-231
cells, but not in MCF-7 cells.
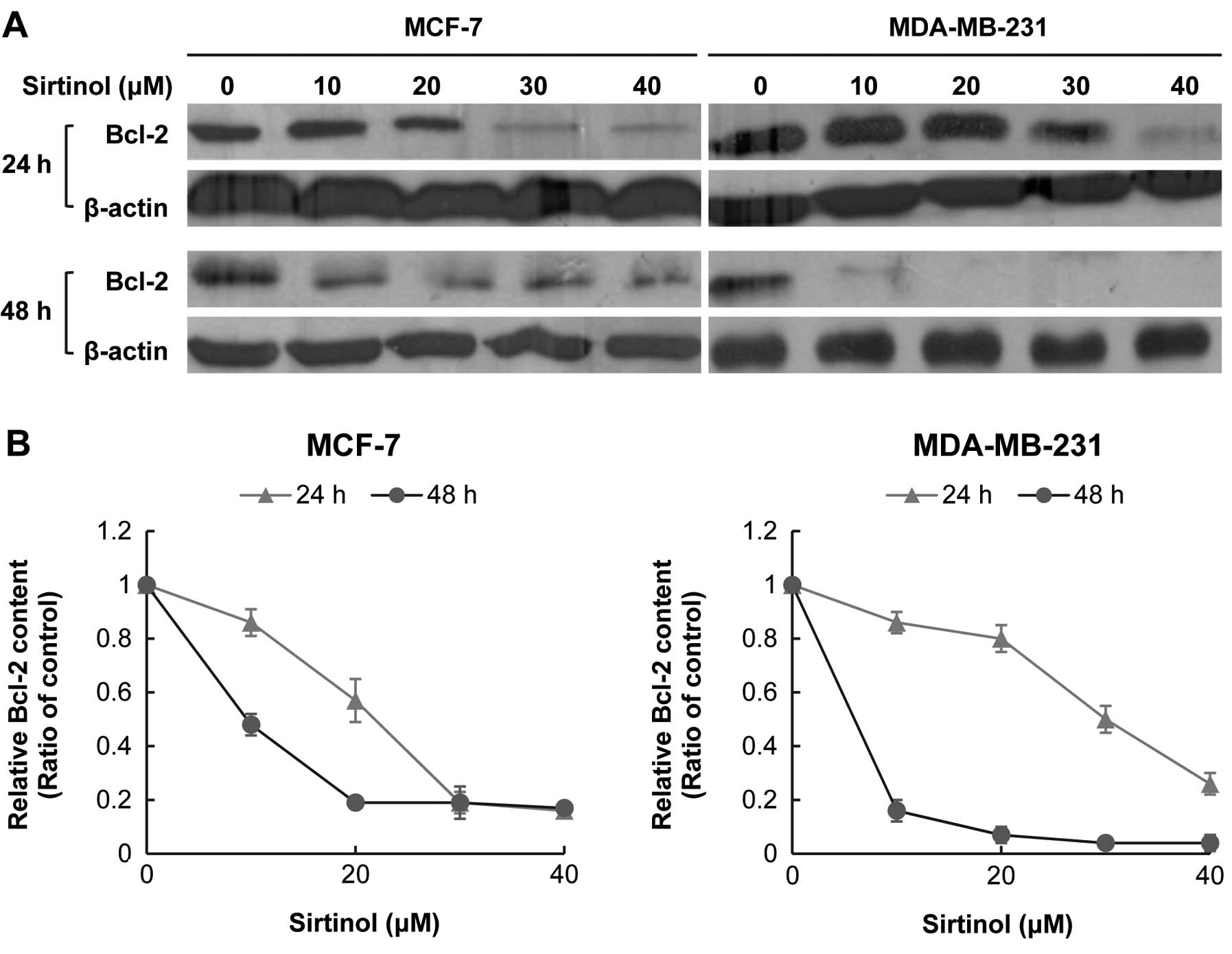
Sirtinol attenuates the abundance of
pro-survival Bcl-2 protein
To characterize the molecular mechanisms underlying
these different behaviors, we analyzed the protein expression of
Bcl-2 by western blotting. The Bcl-2 protein level in MCF-7 and
MDA-MB-231 cells was significantly decreased with sirtinol
treatment, particularly with 48-h treatment (Fig. 5).
Discussion
The expression of SIRT1 in breast cancer tissues is
controversial. Kuzmichev et al(8) and Sung et al(27) demonstrated an overexpression of the
SIRT1, while Wang et al(23)
reported a decreased expression of the SIRT1 protein in breast
tumor tissues. This study showed that there was an extremely high
frequency of overexpressed SIRT1 in breast tumor tissues compared
to their paired normal tissues (92.59%, n=27) (Fig. 1). Notably, the immuhistochemistry
results indicated that there is a positive and high correlation of
Ki67 and SIRT1 expression (Fig.
1A). It was also found that breast cancer tissue with less
proliferative potential (Ki67 scores 0 and 1) exerts a lower
expression level of SIRT1, suggesting that SIRT1 could be a
predictive marker for the prognosis of breast cancer. Therefore, we
proposed that an overexpression of SIRT1 may be involved in breast
tumorigenesis, particularly in breast tumor promotion.
In general, estrogen receptor (ER)-negative breast
cancer is often more aggressive and drug-resistant than ER-positive
breast cancer. To examine the roles of SIRT1 in breast cancer
cells, two human breast cancer cell lines, MCF-7 (ER-positive) and
MDA-MB-231 (ER-negative), were examined. We demonstrated that
sirtinol effectively induced proliferation inhibition in both MCF-7
and MDA-MB-231 cells, indicating the promising therapeutic strategy
of targeting SIRT1 for breast cancer without having to consider the
ER status.
Furthermore, inhibition of SIRT1 causes an
accumulation of G0/G1 phase and a decrease in both S and G2/M
phases (Fig. 3), suggesting the
role of SIRT1 in cell cycle progression. Furthermore, we explored
whether sirtinol inhibits cell proliferation through the apoptosis
pathway in MCF-7 and MDA-MB-231 cells. Several studies reported
that knockdown of SIRT1 or inhibition of SIRT1 activity, such as
sirtinol, splitomycin, and cambinol (28–30),
causes an apoptotic death of cancer cells.
Among the proteins involved in the control of the
apoptotic pathway, Bcl-2, a pro-survival protein, has been
demonstrated to protect cells from apoptosis (31). Bcl-2 inhibits the mitochondrial
pathways of apoptosis through local effects at the mitochondrial
and endoplasmic reticulum membranes (32). Furthermore, Bcl-2 was reported to be
overexpressed in most breast cancer cells (33,34).
Therefore, downregulation of Bcl-2 is a potential chemotherapeutic
strategy (35). Our data showed
that sirtinol promoted apoptosis by decreasing the Bcl-2 expression
in both MCF-7 and MDA-MB-231 cells.
The results suggest that inhibition of SIRT1 has
therapeutic potential as an antitumor strategy. The present study
confirmed the correlation between SIRT1 and the prognosis of breast
cancer patients. Markedly, the blockade of SIRT activity causes an
anti-breast cancer cell effect through downregulating the Bcl-2
protein. Therefore, the specific targeting of the SIRT1 protein may
be a beneficial chemotherapeutic strategy, particularly for
ER-negative breast cancer.
Acknowledgements
This study was supported by a grant from and the
Changhua Christian Hospital-Kaohsiung Medical University joint
grant (97-CCH-KMU-014), Taiwan. Editorial support was provided by
Ms. Yu-Fen Wang, M.S.
References
|
1
|
Blander G and Guarente L: The Sir2 family
of protein deacetylases. Annu Rev Biochem. 73:417–435. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Michan S and Sinclair D: Sirtuins in
mammals: insights into their biological function. Biochem J.
404:1–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaziri H, Dessain SK, Ng Eaton E, et al:
hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell.
107:149–159. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dai JM, Wang ZY, Sun DC, Lin RX and Wang
SQ: SIRT1 interacts with p73 and suppresses p73-dependent
transcriptional activity. J Cell Physiol. 210:161–166. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wong S and Weber JD: Deacetylation of the
retinoblastoma tumour suppressor protein by SIRT1. Biochem J.
407:451–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huffman DM, Grizzle WE, Bamman MM, et al:
SIRT1 is significantly elevated in mouse and human prostate cancer.
Cancer Res. 67:6612–6618. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jung-Hynes B and Ahmad N: Role of p53 in
the anti-proliferative effects of Sirt1 inhibition in prostate
cancer cells. Cell Cycle. 8:1478–1483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuzmichev A, Margueron R, Vaquero A, et
al: Composition and histone substrates of polycomb repressive group
complexes change during cellular differentiation. Proc Natl Acad
Sci USA. 102:1859–1864. 2005. View Article : Google Scholar
|
|
9
|
Zhang Y, Zhang M, Dong H, et al:
Deacetylation of cortactin by SIRT1 promotes cell migration.
Oncogene. 28:445–460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stünkel W, Peh BK, Tan YC, et al: Function
of the SIRT1 protein deacetylase in cancer. Biotechnol J.
2:1360–1368. 2007.
|
|
11
|
Kabra N, Li Z, Chen L, et al: SirT1 is an
inhibitor of proliferation and tumor formation in colon cancer. J
Biol Chem. 284:18210–18217. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu G, Yuan X, Zeng Z, et al: Analysis of
gene expression and chemoresistance of CD133+ cancer
stem cells in glioblastoma. Mol Cancer. 5:672006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jang KY, Hwang SH, Kwon KS, et al: SIRT1
expression is associated with poor prognosis of diffuse large
B-cell lymphoma. Am J Surg Pathol. 32:1523–1531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bradbury CA, Khanim FL, Hayden R, et al:
Histone deacetylases in acute myeloid leukaemia show a distinctive
pattern of expression that changes selectively in response to
deacetylase inhibitors. Leukemia. 19:1751–1759. 2005. View Article : Google Scholar
|
|
15
|
Yuan Z, Zhang X, Sengupta N, Lane WS and
Seto E: SIRT1 regulates the function of the Nijmegen breakage
syndrome protein. Mol Cell. 27:149–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chu F, Chou PM, Zheng X, Mirkin BL and
Rebbaa A: Control of multidrug resistance gene mdr1 and cancer
resistance to chemotherapy by the longevity gene sirt1. Cancer Res.
65:10183–10187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng HL, Mostoslavsky R, Saito S, et al:
Developmental defects and p53 hyperacetylation in Sir2 homolog
(SIRT1)-deficient mice. Proc Natl Acad Sci USA. 100:10794–10799.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matsushita N, Takami Y, Kimura M, et al:
Role of NAD-dependent deacetylases SIRT1 and SIRT2 in radiation and
cisplatin-induced cell death in vertebrate cells. Genes Cells.
10:321–332. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kojima K, Ohhashi R, Fujita Y, et al: A
role for SIRT1 in cell growth and chemoresistance in prostate
cancer PC3 and DU145 cells. Biochem Biophys Res Commun.
373:423–428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yuan J, Minter-Dykhouse K and Lou Z: A
c-Myc-SIRT1 feedback loop regulates cell growth and transformation.
J Cell Biol. 185:203–211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Firestein R, Blander G, Michan S, et al:
The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon
cancer growth. PLoS One. 3:e20202008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chua KF, Mostoslavsky R, Lombard DB, et
al: Mammalian SIRT1 limits replicative life span in response to
chronic genotoxic stress. Cell Metab. 2:67–76. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang RH, Sengupta K, Li C, et al: Impaired
DNA damage response, genome instability, and tumorigenesis in SIRT1
mutant mice. Cancer Cell. 14:312–323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lim CS: SIRT1: tumor promoter or tumor
suppressor? Med Hypotheses. 67:341–344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li K and Luo J: The role of SIRT1 in
tumorigenesis. N Am J Med Sci. 4:104–106. 2011. View Article : Google Scholar
|
|
26
|
Tavassoli FA and Devilee P; World Health
Organization Classification of Tumours. Pathology and Genetics of
Tumours of the Breast and Female Genital Organs. IARC Press; Lyon:
2003
|
|
27
|
Sung JY, Kim R, Kim JE and Lee J: Balance
between SIRT1 and DBC1 expression is lost in breast cancer. Cancer
Sci. 101:1738–1744. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heltweg B, Gatbonton T, Schuler AD, et al:
Antitumor activity of a small-molecule inhibitor of human silent
information regulator 2 enzymes. Cancer Res. 66:4368–4377. 2006.
View Article : Google Scholar
|
|
29
|
Ota H, Tokunaga E, Chang K, et al: Sirt1
inhibitor, Sirtinol, induces senescence-like growth arrest with
attenuated Ras-MAPK signaling in human cancer cells. Oncogene.
25:176–185. 2006.PubMed/NCBI
|
|
30
|
Ford J, Jiang M and Milner J:
Cancer-specific functions of SIRT1 enable human epithelial cancer
cell growth and survival. Cancer Res. 65:10457–10463. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reed JC: Bcl-2-family proteins and
hematologic malignancies: history and future prospects. Blood.
111:3322–3330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hockenbery DM: Targeting mitochondria for
cancer therapy. Environ Mol Mutagen. 51:476–489. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amundson SA, Myers TG, Scudiero D, Kitada
S, Reed JC and Fornace AJ Jr: An informatics approach identifying
markers of chemosensitivity in human cancer cell lines. Cancer Res.
60:6101–6110. 2000.PubMed/NCBI
|
|
34
|
Zhang M, Guo R, Zhai Y and Yang D: LIGHT
sensitizes IFNgamma-mediated apoptosis of MDA-MB-231 breast cancer
cells leading to down-regulation of anti-apoptosis Bcl-2 family
members. Cancer Lett. 195:201–210. 2003. View Article : Google Scholar
|
|
35
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|