Introduction
The ERas gene, which can support embryonic
stem (ES) cell tumor-like growth (1–4), was
first identified in murine ES cells by Takahashi et
al(1). It encodes a protein of
227 amino acids with 43, 46 and 47% identity to conventional
Ras oncogenes H-ras, K-ras and N-ras,
respectively, defined as a new member of the Ras family.
Unlike other Ras family members, the ERas product is
a constitutively active protein without any mutation (1), while conventional ras oncogenes
acquire activation of carcinogenesis by point mutation of several
amino acids, which include Gly12, Ala59 or Glu63 (5,6). The
ERas protein contains amino acid residues identical to those
present in active mutants of the conventional Ras oncogenes
K-ras, N-ras and H-ras (7). The conventional Ras members
mainly function through activating either phosphatidylinositol-3-OH
kinase (PI3K) or Raf pathway (8,9), while
ERas interacts with PI3K (10) but
not with Raf (11,12). The human ERas gene was
initially erroneously recognized as a processed pseudogene
HRasp (Ha-Ras2) (13,14)
with deficiency or meaningless mutation, but it was more recently
described as a gene potentially encoding a functional human ERas
protein (15–18). Therefore, a few studies on human
ERas have previously been reported.
In 2002, Bjorklund et al(19) found that mouse ES cells developed
into teratomas when transplanted into nude mice. In 2003, Takahashi
et al(1) found that
ERas is key in the tumor-like growth properties of ES cells.
In 2009, Kaizaki et al(17)
discovered ERas was actively expressed in gastric cancer
(GC) and was closely related to its oncogenesis. These results
indicate that the expression of ERas may be associated with
cell proliferation and transformation.
However, in 2010, Kubota et al(20) detected 142 clinical samples of GC,
which showed that ERas expression was strongly associated
with liver and lymph node metastases. Meanwhile, there was no
significant correlation between ERas expression and
histological differentiation. Overexpression of ERas in GC
cell lines promoted colony formation while it showed no significant
effect on cell proliferation. These results indicated that
ERas was only associated with the metastasis of GC. However,
clonality is an indicator of proliferation capacity. Hence,
considering the multiaspect influences of oncogenesis, we decided
to knock down the ERas gene in order to study the impactions
on proliferation and clonality of GC cells.
In addition, Kameda and Thomson (2) used RT-PCR to analyze expression of the
ERas gene in human ES cells in 2005, and they could not
detect a full-length ERas coding transcript. Instead, a
truncated noncoding transcript was found, which was caused by a
premature polyadenylation signal predicted through sequence
analysis and confirmed by 3′RACE analysis. Except for the premature
PloyA locus, humans and chimpanzees have
typical-Alu-s-retrotransposon insertions, which also influence the
expression of ERas at this specific locus. Moreover, the
lack of ERas expression in human ES cells indicates that the
oncogenesis is very different from that of murines (21). These findings indicate that further
studies should be performed to ascertain whether or not a
full-length ERas coding transcript is present in human GC
cells.
In this study, 8 cell strains from different
sources, GC lymph node metastasis, GC liver metastasis, GC ascites
and GC tissues, with different differentiation degrees, were chosen
to determine whether a full-length ERas mRNA exists and to
elucidate the difference of degree of ERas expression among
these GC strains by RT-PCR, real-time PCR and western blotting.
Furthermore, we confirmed the effect of ERas knockdown on
cell proliferation, metastasis and clonality in ERas highly
expressed GC strains.
Materials and methods
Cell culture and cell lines
The cell lines GES-1, MKN-28, MKN-45, BGC-823,
NCL-N87, SNU-16, SGC-7901 and AGS were cultured in RPMI-1640,
supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT,
USA). They were cultured in an atmosphere of 5% CO2 at
37°C. BGC-823, NCL-N87 and AGS cells were obtained from Shanghai
Institute of Cell Bank (Shanghai, China); GES-1, MKN-28, MKN-45,
SNU-16 and SGC-7901 were purchased from the American Type Culture
Collection (ATCC, Rockville, MD, USA).
Amplification of the ERas full-length
transcripts and sequencing analysis
Total cellular RNA was extracted from each cell line
with TRIzol® (Invitrogen Life Technologies, Carlsbad,
CA, USA), and cDNA was synthesized with Reverse Transcription kit
(Promega Corp., Madison, WI, USA), and both were performed
according to the manufacturer’s protocol. The cDNA was synthesized
by PCR with PrimeSTAR HS DNA polymerase (Takara Bio, Shiga, Japan).
The primers used for the full-length ERas coding sequence
were: forward, 5′-atggagctgccaacaaagcctggca-3′ and reverse,
5′-ttcaggccacagagcagccacagt-3′, which gave an amplified fragment of
702 bp. The reaction conditions were: 94°C for 15 sec, 62°C for 45
sec and 72°C for 30 sec, repeated for 35 cycles. The products were
separated by 1.2% agarose gel electrophoresis, and the 702 bp bands
were recycled using aqua-Spin gel extraction mini kit (Watson
Biotechnologies, Inc., Shanghai, China). The identification of PCR
products was confirmed by sequencing analysis (Songon Biotech Co.,
Shanghai, China).
Real-time quantitative PCR
The real-time quantitative PCR analyses were
performed in triplicate using SYBR® Premix Ex Taq™II kit
(Takara Bio). The GAPDH gene was chosen as an endogenous
control. The primers used for ERas were: forward,
5′-cacatggagcccttccttc-3′ and reverse, 5′-tgtccagggtcaactccttc-3′;
the primers used for GAPDH were: forward,
5′-ggacctgacctgccgtctag-3′ and reverse, 5′-gtagcccaggatgcccttga-3′;
the PCR conditions were as follows: 94°C for 15 sec, 58°C for 45
sec, 72°C for 20 sec, repeated for 35 cycles. Amplified products
were separated by 0.9% agarose gel electrophoresis.
Western blot analysis
Cells were lysed in a lysis buffer containing 2%
sodium dodecyl sulfate (SDS) and 0.125 M Tris-HCl (pH 6.8) on ice
for 30 min, followed by high-speed centrifugation, and the
supernatant protein was finally collected. SDS-PAGE was performed
using 10% polyacrylamide gels. PAGE separated proteins were
electrophoretically transferred onto nitrocellulose membranes. The
membrane filters were blocked with 5% powdered milk in TBST (0.1%
Tween-20) for 2 h and then incubated in rabbit ERas antibody
(Abgent, Suzhou, China) diluted 1:100 in TBST at 4°C overnight, and
finally incubated with HRP anti-rabbit secondary antibody (Kangchen
Biotech, Shanghai, China) diluted 1:2,000 for 1 h at room
temperature. Antigens on the membrane were detected with enhanced
chemiluminescense detection reagents (Roche, Basel,
Switzerland).
Small interfering RNA transfection
Two ERas stealth siRNA, no. 30 forward,
GCAACUAGCUUUGAGGGAC(dTdT) and reverse, GUCCCUCAAAGCUAGUUGC(dTdT);
no. 32 forward, GUAACAUGGGAGUGCCUAA(dTdT) and reverse,
UUAGGCACUCCCAUGUUAC(dTdT); one high GC% negative control siRNA
forward, CCUACGCCACCAAUUUC GU(dTdT) and reverse,
ACGAAAUUGGUGGCGUAGG (dTdT) were designed and synthesized (Bioneer,
Daejon, Korea). siRNA was mixed with Lipofectamine™ 2000
(Invitrogen Life Technologies) in an OptiMEM serum-free medium
(HyClone) for 30 min at room temperature and then added to each
24-well plate containing MKN-28 or BGC-823 cells. Cells were
maintained in a humidified 5% CO2 incubator at 37°C for
6 h with the old medium being replaced by a fresh medium. After 24
h of transfection, cells were harvested for cell proliferation,
migration and colony formation assays.
CCK-8 assay
siRNA-transfected MKN-28 and BGC-823 cells were
seeded into 96-well plates at a density of 3×103
cells/well and maintained in culture medium for 5 days. Each well
set five duplicates. We measured cell growth using cholecystokinin
(CCK) assay by Cell Counting Kit (Dojindo, Tokyo, Japan) according
to the manufacturer’s instructions.
Cell migration assay
For wound-healing experiments, MKN-28 and BGC-823
cells transfected with siRNA were cultured to 80% confluence after
being seeded into 6-well plates, then scraped using a p10 tip (time
0), and suspended cells were washed with PBS three times. Cells
were incubated for another 4 days and images were captured by
microscope (Zeiss, Oberkochen, German) at the same time every day.
Migration distance was measured from images (5 fields) at each
indicated time point.
Transwell assay of MKN-28 and BGC-823 cells was
assessed using 6.5 mm diameter inserts (Corning Costar Corp.,
Corning, NY, USA). A total of 3×104 cells were suspended
in 100 μl serum-free RPMI-1640 medium and loaded into upper wells;
lower chambers were filled with 600 μl of complete medium
(RPMI-1640 supplemented with 10% FBS). Migration chambers were
incubated in a humidified 5% CO2 incubator at 37°C for
24 and 48 h. Cells were then fixed with 600 μl of paraformaldehyde
for 20 min. The inner surfaces of the upper chambers were wiped
using cotton swabs to remove non-migrated cells in the migration
assay. The chambers were then washed with PBS and stained with 500
μl crystal violet for 20 min at room temperature. Stained cells
were counted using the ImageJ software, and 5 random fields were
counted (Zeiss).
Colony formation assay
A total of 500 siRNA-transfected MKN-28 and BGC-823
cells were seeded in 6-well plates and incubated for 14 days
respectively, with the medium replaced every 4 days. On the 15th
day, the cells were stained with crystal violet for 20 min and
washed with tap water for 10 min. For each dish, colonies in five
random fields were counted using the ImageJ software.
Statistical analysis
Each measurement was performed in triplicate.
Original real-time PCR data, CCK-8 data, migration/invasion data
and colony formation data were recorded as continuous variables and
analyzed using Student’s t-test or linear polynomial ANOVA with LSD
post hoc examination. All statistical analyses were performed using
SPSS 16.0 software. P-values <0.05 were considered to indicate
statistically significant differences.
Results
ERas expressed in GC cells
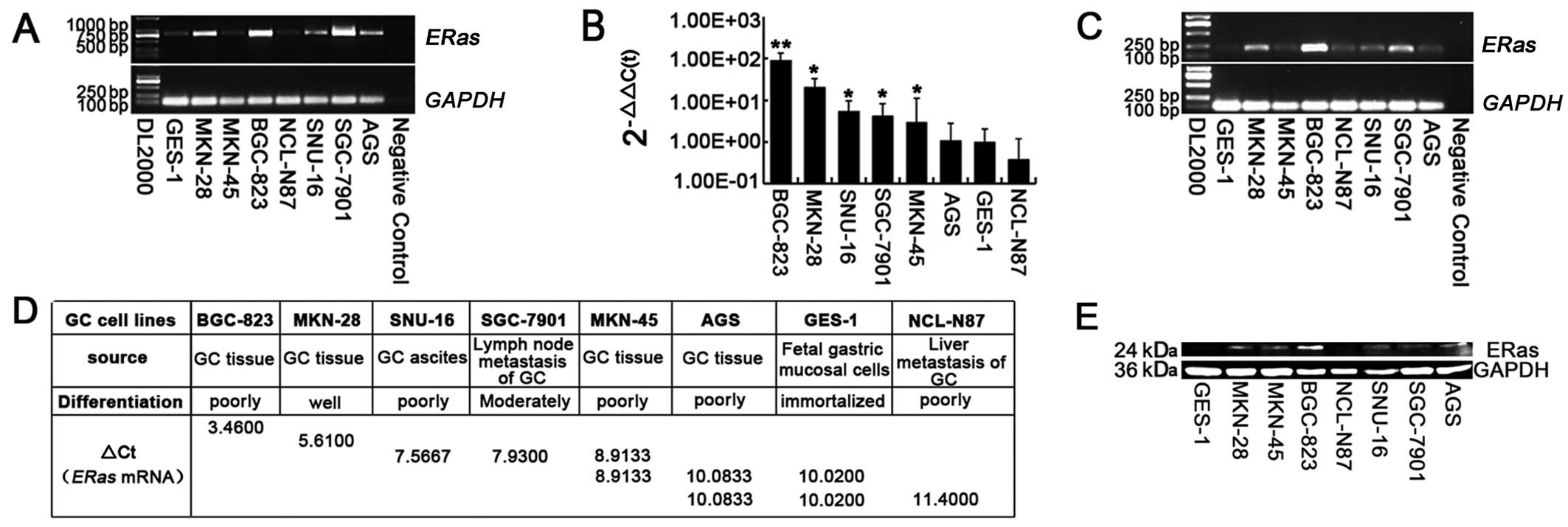
The full-length ERas mRNA transcript was
detected in all seven GC cell lines and GES-1 gastric mucosa cell
line by RT-PCR. The forward and reverse primers were located in
open reading frame (ORF) 1–25 and 680–702 bp separately, which give
rise to an amplified fragment of 702 bp (Fig. 1A). To assess whether ERas
expression was different with mutant H-ras, K-ras and
N-ras, mutation analysis was performed by sequencing
analysis, revealing no mutation of ERas in all the gastric
cell lines that we selected (data not shown).
The expression levels of ERas mRNA were also
determined by real-time PCR. All eight cell lines were divided into
five groups by one-way ANOVA. The GC cell line BGC-823 expressed
most highly (P<0.01), MKN-28 expressed highly (P<0.05),
SNU-16, SGC-7901, MKN-45 expressed moderately (P<0.05), while
AGS, NCL-N87 expressed poorly, similar to the normal gastric
mucosal cell line, GES-1 (P>0.05) (Fig. 1B-D). ERas protein expression was
also confirmed by western blotting (Fig. 1E).
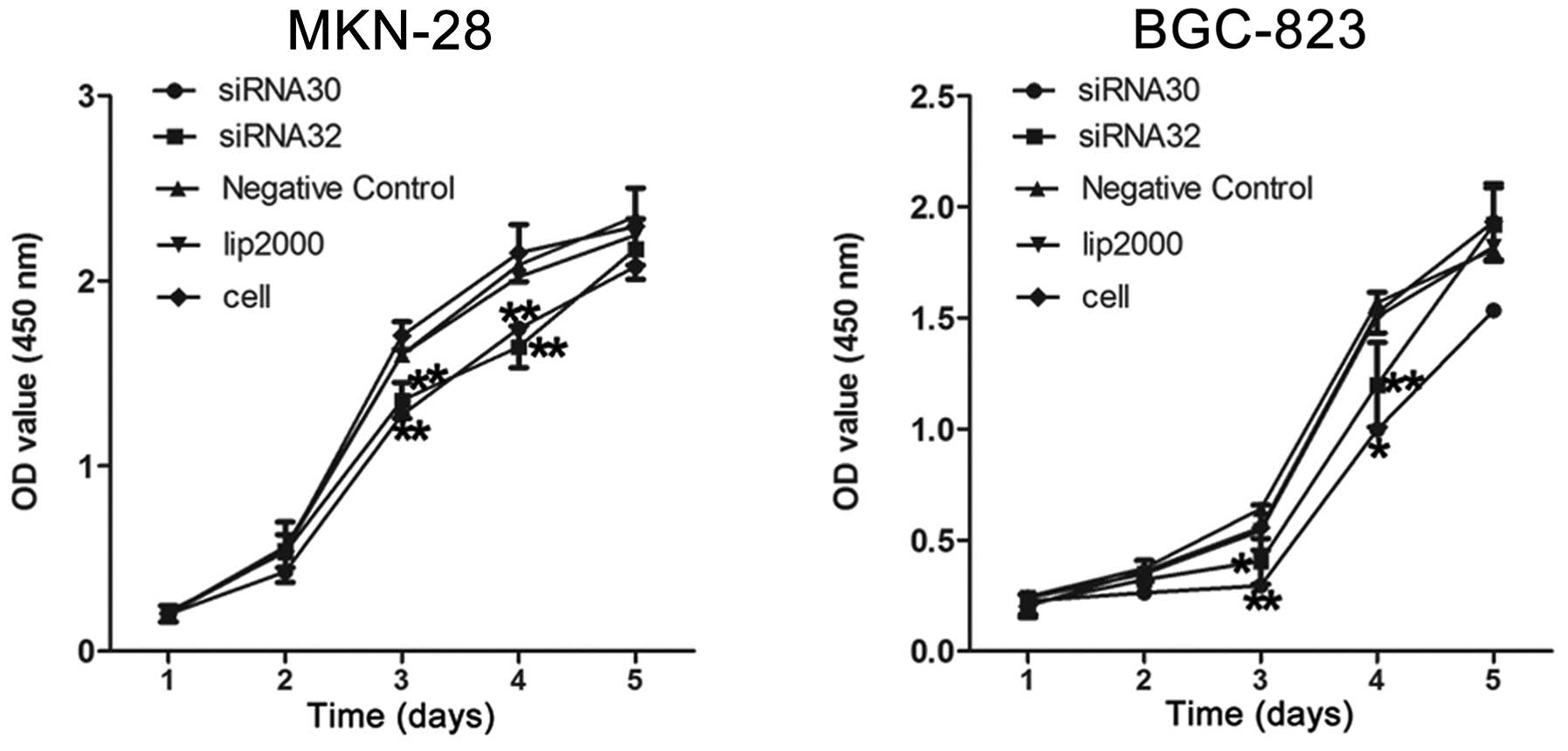
ERas increases GC cell proliferation
To examine the role of ERas in cell proliferation,
we measured BGC-823, MKN-28 cell growth by CCK-8 assay after
transfecting with ERas siRNA30 and siRNA32.
We observed a significant decrease in cell
proliferation when the expression of ERas was knocked down
in MKN-28 and BGC-823 cells (Fig.
2).
On the third day, the OD value of MKN-28 cells
decreased from 1.60±0.05 to 1.28±0.10 when transfected with siRNA30
(P<0.01), and it decreased to 1.35±0.10 when transfected with
siRNA32 (P<0.01). Meanwhile, the OD value of BGC-823 cells
decreased from 0.64±0.18 to 0.30±0.08 and 0.40±0.10 when
transfected with siRNA30 (P<0.01) and siRNA32 (P<0.05)
individually.
On the fourth day, the OD value decreased from
2.09±0.09 to 1.74±0.10 and 1.64±0.11 individually in MKN-28 cells
when transfected with siRNA30 (P<0.01) and siRNA32 (P<0.01),
respectively, while it decreased from 1.57±0.07 to 1.00±0.46
(P<0.05) and 1.20±0.19 (P<0.01) in BGC-823 cells. These data
indicate that the proliferation of these GC cell lines is
significantly promoted by the ERas gene.
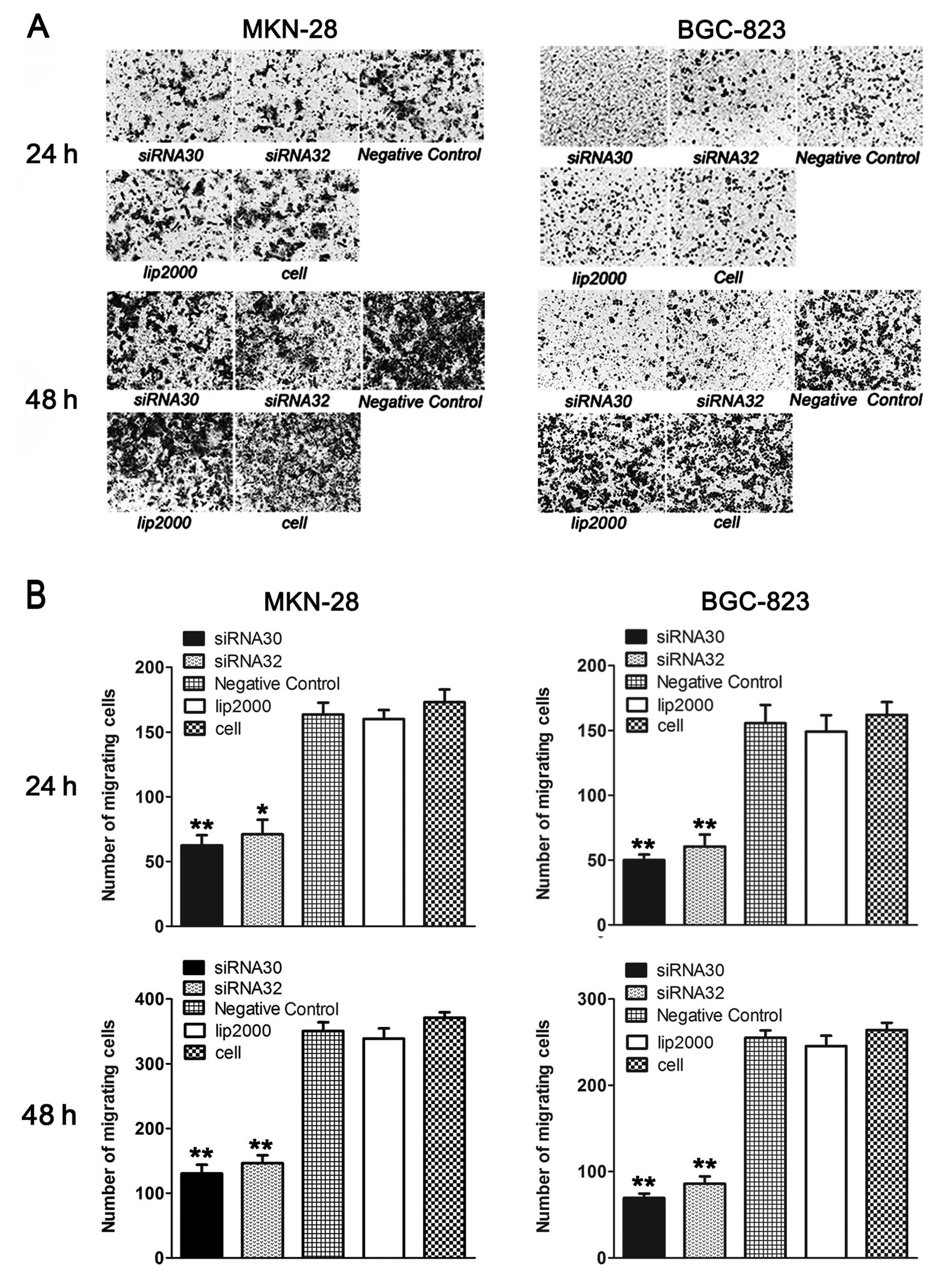
ERas promotes GC cell migration
We also confirmed the effect of ERas on migration in
MKN-28 and BGC-823 cells by Transwell and wound-healing assay.
As shown in Fig. 3A and
B, knockdown of ERas by siRNA significantly impaired the
ability of MKN-28 and BGC-823 cells to migrate through the
membranes. Twenty-four hours later, the number of migratory cells
decreased from 163.5±9.19 to 62.50±7.78 (P<0.05) and 71±11.31
(P<0.01) when transfected with siRNA30 and siRNA32 in MKN-28
cells, while it decreased from 155.50±9.19 to 50±4.24 (P<0.01)
and 60.5±9.19 (P<0.05) in BGC-823 cells. Forty-eight hours
later, the number of migratory MKN-28 cells decreased from
350.5±13.44 to 62.50±7.78 (P<0.01) and 71±11.31 (P<0.01)
respectively, while the number of migratory BGC-823 cells decreased
from 255±8.49 to 69.5±4.95 (P<0.01) and 86±8.49 (P<0.01).
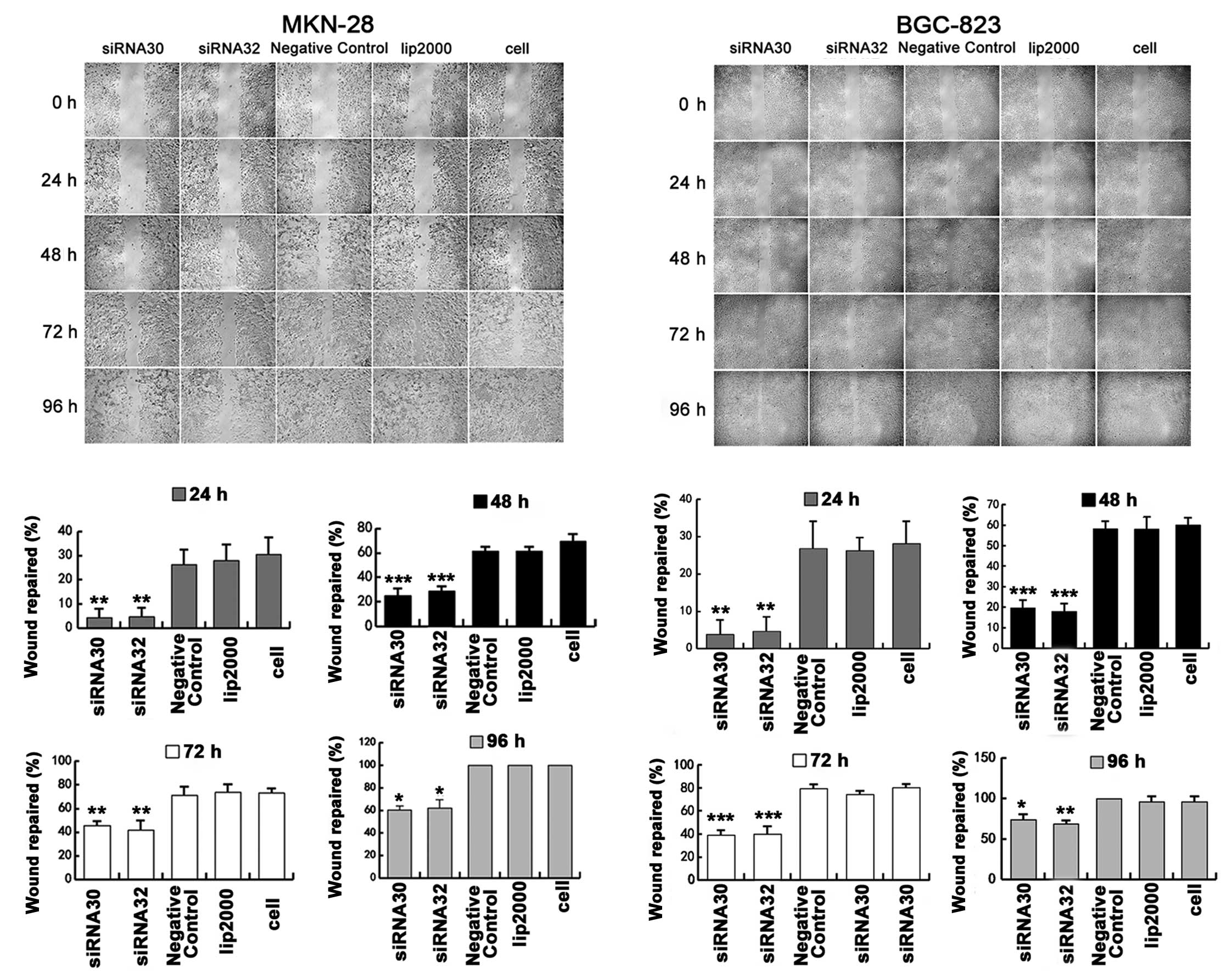
Then, we further analyzed migration ability by using
wound-healing for 4 days. As shown in Fig. 4, the speed of wound repair was
markedly slower when ERas was silenced by siRNA in MKN-28
and BGC-823 cells at 24, 48, 72 and 96 h. The statistical
significance was most notable at 48 h, when the wound repair
percentage decreased from 61.35±3.54% to 25±6.25% (P<0.001) and
28.89±3.85% (P<0.001) after ERas was knocked down by
siRNA30 and siRNA32 respectively in MKN-28 cells, while the
percentage decreased from 58.13±3.61% to 19.61±3.78% (P<0.001)
and 18±3.85% (P<0.001) respectively in BGC-823 cells.
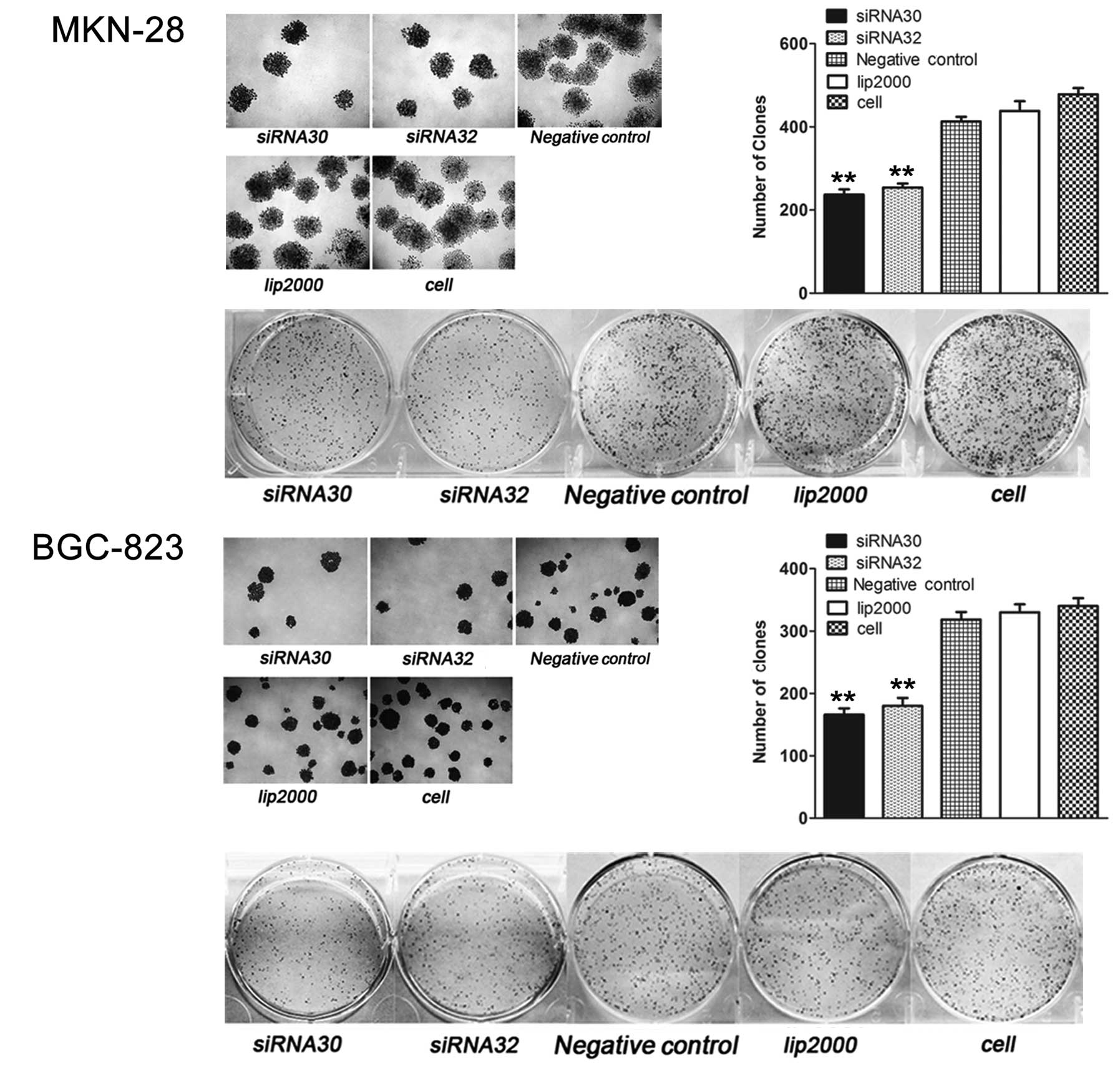
ERas promotes GC cell colony
formation
The colony formation was assessed two weeks later.
As shown in Fig. 5, ERas
knockdown by siRNA30 or siRNA32 reduced the number of colonies from
413±11.31 to 237±12.73 (P<0.01) and 254±9.90 (P<0.01) in
MKN-28 cells, while the number of colonies in BGC-823 cells was
reduced from 318.5±12.02 to 166±9.90 (P<0.01) and 180±12.73
(P<0.01).
Discussion
The ERas gene is strongly expressed in
BGC-823 and MKN-28 cell strains among the eight gastric cell lines
(Fig. 1B-D). To examine the effect
of ERas on GC cell proliferation, these two highly expressing
endogenous ERas cell strains were investigated after
treatment with ERas siRNA by CCK-8 assay for 5 days.
Knockdown of ERas inhibited the proliferation ability
markedly compared to control in BGC-823 and MKN-28 cells on the
third and fourth day (Fig. 2).
Kubota et al(20) concluded
that ERas could not promote the proliferation in GCIY cells
transfected with an ERas-overexpressing vector by MTS assays
for 6 days, whereas it could enhance colony formation as the cell
clonality experiment results showed. They came to the same result
that there was no relationship between proliferation and the
ERas gene in GCIY and NUGC-4 cells with ERas knocked
down by stealth siRNA using MTS assays for 2 days. However, whether
or not knockdown of ERas inhibits cell colony formation was
not further established. GCIY cell strain was used in both
ERas overexpression and knockdown experiment. However, the
expression level of ERas in this strain was relatively low,
indicating it was not suitable for the knockdown experiment. On the
other hand, the observation period in their study was relatively
short. These reasons may lead to the disparity. Meanwhile, the
results of Transwell test, scratch test and colony formation test
(22,23) proved that ERas is able to increase
GC cell migration and colony formation which were also two aspects
that could reflect cell proliferation. Hence, there is sufficient
evidence to prove that ERas enhances GC cell proliferation.
Furthermore, ERas was expressed most highly
in poorly differentiated BGC-823 cells and well differentiated
MKN-28 cells, less highly in poorly differentiated SNU-16 cells,
moderately differentiated SGC-7901 cells and poorly differentiated
MKN-45 cells, almost silently in poorly differentiated AGC cells,
immortal GES-1 cells and poorly differentiated NCL-N87 cells
(Fig. 1A-C). From these date, the
conclusion that expression of ERas is not related to
histological differentiation is the same as that of Kubota et
al. However, from analyzing the following seven cell strains,
SGC-7901 from GC lymph metastasis, NCL-N87 from GC liver
metastasis, SNU-16 from GC ascites, GES-1 from fetal gastric mucosa
and others from GC tissues, the connection between ERas and
metastasis of gastric lymph and liver is not so strong, which is
different from the result of Kubota et al. Therefore, to
fully understand the connection between ERas and gastric lymph
metastasis and liver metastasis, further research is required.
The full-length transcript of ERas (2,20) in
those 7 GC cell strains were examined (Fig. 1A), suggesting that activated ERas is
present universally in GC cells. Since a full-length transcript
cannot be found in human ES cells, there should be some common
activation factors which inhibit the prematuration of early
polyadenylation signal and the insertion of Alu-S transposons to
activate ERas expression.
To date, there are few studies on the relationship
between ERas and cancer, and the function of ERas has yet to be
determined. This study used CCK-8, Transwell, scratch test and
clonality test to successfully prove that ERas has the ability to
enhance GC cell proliferation, metastasis and clonality. Compared
to Kubota et al(20), this
study proved that the activation of the ERas gene in GC
cells is common and the function of ERas in the processes of GC
cell development and metastasis is important; there must be a
significance of clarifying the correlative key signal pathways and
the cytokines to activate the pathways.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (30672440). We thank Yufei Wang, Qiang
Fu, Ronghua Wang, Ran Hu for their technical assistance. We also
gratefully acknowledge the entire staff of the Experimental Center
of Shanghai Jiaotong University Affiliated First People’s
Hospital.
Abbreviations:
|
ES
|
embryonic stem
|
|
PI3K
|
phosphatidylinositol-3-OH kinase
|
|
GC
|
gastric carcinoma
|
References
|
1
|
Takahashi K, Mitsui K and Yamanaka S: Role
of ERas in promoting tumour-like properties in mouse embryonic stem
cells. Nature. 423:541–545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kameda T and Thomson JA: Human ERas gene
has an upstream premature polyadenylation signal that results in a
truncated, noncoding transcript. Stem Cells. 23:1535–1540. 2005.
View Article : Google Scholar
|
|
3
|
Takahashi K, Murakami M and Yamanaka S:
Role of the phosphoinositide 3-kinase pathway in mouse embryonic
stem (ES) cells. Biochem Soc Trans. 33:1522–1525. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takahashi K, Nakagawa M, Young SG and
Yamanaka S: Differential membrane localization of ERas and Rheb,
two Ras-related proteins involved in the phosphatidylinositol
3-kinase/mTOR pathway. J Biol Chem. 280:32768–32774. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fasano O, Aldrich T, Tamanoi F, et al:
Analysis of the transforming potential of the human H-ras gene by
random mutagenesis. Proc Natl Acad Sci USA. 81:4008–4012. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schubbert S, Shannon K and Bollag G:
Hyperactive Ras in developmental disorders and cancer. Nat Rev
Cancer. 7:295–308. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seeburg PH, Colby WW, Capon DJ, et al:
Biological properties of human c-Ha-ras1 genes mutated at codon 12.
Nature. 312:71–75. 1984. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cass LA and Meinkoth JL: Ras signaling
through PI3K confers hormone-independent proliferation that is
compatible with differentiation. Oncogene. 19:924–932. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deora AA, Hajjar DP and Lander HM:
Recruitment and activation of Raf-1 kinase by nitric
oxide-activated Ras. Biochemistry. 39:9901–9908. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rodriguez-Viciana P, Warne PH, Dhand R, et
al: Phosphatidylinositol-3-OH kinase as a direct target of Ras.
Nature. 370:527–532. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moodie SA, Willumsen BM, Weber MJ and
Wolfman A: Complexes of Ras.GTP with Raf-1 and mitogen-activated
protein kinase kinase. Science. 260:1658–1661. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang XF, Settleman J, Kyriakis JM, et al:
Normal and oncogenic p21ras proteins bind to the amino-terminal
regulatory domain of c-Raf-1. Nature. 364:308–313. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang EH, Gonda MA, Ellis RW, et al: Human
genome contains four genes homologous to transforming genes of
Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci USA.
79:4848–4852. 1982. View Article : Google Scholar
|
|
14
|
Miyoshi J, Kagimoto M, Soeda E and Sakaki
Y: The human c-Ha-ras2 is a processed pseudogene inactivated by
numerous base substitutions. Nucleic Acids Res. 12:1821–1828. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yasuda K, Yashiro M, Sawada T, et al: ERas
oncogene expression and epigenetic regulation by histone
acetylation in human cancer cells. Anticancer Res. 27:4071–4075.
2007.PubMed/NCBI
|
|
16
|
Yashiro M, Yasuda K, Nishii T, et al:
Epigenetic regulation of the embryonic oncogene ERas in
gastric cancer cells. Int J Oncol. 35:997–1003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaizaki R, Yashiro M, Shinto O, et al:
Expression of ERas oncogene in gastric carcinoma. Anticancer Res.
29:2189–2193. 2009.PubMed/NCBI
|
|
18
|
Kubota E, Kataoka H, Tanaka M, et al: ERas
enhances resistance to CPT-11 in gastric cancer. Anticancer Res.
31:3353–3360. 2011.PubMed/NCBI
|
|
19
|
Bjorklund LM, Sánchez-Pernaute R, Chung S,
et al: Embryonic stem cells develop into functional dopaminergic
neurons after transplantation in a Parkinson rat model. Proc Natl
Acad Sci USA. 99:2344–2349. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kubota E, Kataoka H, Aoyama M, et al: Role
of ES cell-expressed Ras (ERas) in tumorigenicity of gastric
cancer. Am J Pathol. 177:955–963. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rao M: Conserved and divergent paths that
regulate self-renewal in mouse and human embryonic stem cells. Dev
Biol. 275:269–286. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hsu RJ, Ho JY, Cha TL, et al: WNT10A plays
an oncogenic role in renal cell carcinoma by activating
WNT/β-catenin pathway. PLoS One. 7:e476492012.PubMed/NCBI
|
|
23
|
Li WG, Yuan YZ, Qiao MM and Zhang YP: High
dose glargine alters the expression profiles of microRNAs in
pancreatic cancer cells. World J Gastroenterol. 18:2630–2639. 2012.
View Article : Google Scholar : PubMed/NCBI
|