Introduction
Gallic acid (GA; 3,4,5-trihydroxyl-benzoic acid) is
a polyhydroxylphenolic compound, which is broadly disseminated in a
variety of plants, fruits and foods (1). It is easily absorbed in humans;
micromolar concentrations of free and glucuronidated forms of GA
and its major metabolite 4-O-methylgallic acid have been detected
in human blood plasma following the ingestion of GA-rich food
(2). Diverse biological activities
of GA have been reported, including anti-bacterial (3), anti-viral (4) and anti-inflammatory (5). The main focus among the properties of
GA is connected to its antitumoral action. In fact, anticancer
activity of GA has been reported in various cancer cells such as
leukemia (6), prostate cancer
(7,8), lung cancer (9,10),
gastric, colon, breast, cervical and esophageal cancer (11). Apoptosis induced by GA is associated
with oxidative stresses derived from reactive oxygen species (ROS),
mitochondrial dysfunction and an increase in intracellular
Ca2+ level (6,12). GA has both pro- and anti-oxidant
properties depending on the concentrations of iron or hydrogen
peroxide (H2O2) in medium and plasma
(13,14).
The major ROS include H2O2,
superoxide anion (O2•-) and hydroxyl radical
(•OH). O2•- is metabolized to
H2O2 by superoxide dismutases (15). H2O2 is further
detoxified to O2 and H2O by catalase or
glutathione (GSH) (16). They
affect the activity of mitogen-activated protein kinases (MAPKs),
which are involved in crucial signaling pathways in cell
proliferation, differentiation and cell death in response to a
variety of stimuli (17,18). MAPKs can sense the cellular redox
status and are common targets for ROS. There are currently three
well known MAPKs: the extracellular signal regulated kinase
(ERK1/2), the c-Jun N-terminal kinase/stress-activated protein
kinase (JNK/SAPK) and the p38 (17). Each MAP kinase pathway has
comparatively different upstream activators and specific substrates
(19). In general, JNK and p38 are
activated by ROS or a mild oxidative shift, initiating procedures
related to apoptosis (20,21). ROS also induce ERK phosphorylation
and activate the ERK pathway (22).
In most cases, ERK signaling has pro-survival and proliferative
roles rather than pro-apoptotic effects (23).
Lung cancer is a major cause of cancer-related
mortality in developed countries. The carcinogenesis of lung cancer
is associated with excessive inflammation mediated by ROS of
airborne and bloodborne origin. Various novel remedial strategies
are still under consideration since the clinical use of
conventional drugs is limited due to intrinsic or acquired
resistance and toxicity (24). We
recently reported that GA inhibits the growth of Calu-6 and A549
lung cancer cells (25,26). In addition, MEK inhibitor PD98059
attenuates growth inhibition and death in GA-treated Calu-6 cells
(27). Since different and opposite
effects of MAPKs by a variety of ROS can occur even in the same
type of cells, the relationship between ROS and MAPKs requires
further elucidation regarding signalings related to cell survival
and cell death. In the present study, we investigated the effects
of MAPK inhibitors on cell growth, death, ROS and GSH levels in
GA-treated A549 lung cancer cells.
Materials and methods
Cell culture
The human pulmonary adenocarcinoma A549 cell line
was obtained from the American Type Culture Collection (ATCC,
Manassas, VA, USA) and maintained in a humidified incubator
containing 5% CO2 at 37°C. A549 cells were cultured in
RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 1%
penicillin-streptomycin (Gibco- BRL, Grand Island, NY, USA).
Reagents
GA was purchased from Sigma-Aldrich Chemical Co.
(St. Louis, MO, USA) and was dissolved in ethanol at 200 mM as a
stock solution. MEK inhibitor (PD98059), JNK inhibitor (SP600125)
and p38 inhibitor (SB203580) were obtained from Calbiochem (San
Diego, CA, USA) and were dissolved in DMSO at 10 mM as a stock
solution. Cells were pretreated with each MAPK inhibitor for 30 min
prior to GA treatment. Based on a previous experiment (28), 10 μM of each MAPK inhibitor was used
as an optimal dose in this experiment. Ethanol (0.2%) and DMSO
(0.3%) were used as a control vehicle.
Cell growth assay
The effect of drugs on A549 cell growth was
determined by trypan blue exclusion cell counting or measuring
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
dye absorbance of living cells as previously described (29). In brief, 2×105 cells/well
were seeded in 24-well plates (Nunc, Roskilde, Denmark) for cell
counting and 5×104 cells/well were seeded in 96-well
microtiter plates for an MTT assay. Following exposure to the
indicated dose of GA and/or each MAPK inhibitor for 24 h, 20 μl of
MTT (Sigma) solution (2 mg/ml in PBS) were added to each well of
96-well plates. The plates were incubated for an additional 4 h at
37°C. Medium in plates was withdrawn using pipetting and 200 μl
DMSO was added to each well to solubilize the formazan crystals.
Optical density was measured at 570 nm using a microplate reader
(Spectra MAX 340, Molecular Devices Co., Sunnyvale, CA, USA).
Sub-G1 DNA content analysis
Sub-G1 DNA content cells were determined by
propidium iodide (PI; Ex/Em=488 nm/617 nm, Sigma-Aldrich) staining
as previously described (30). In
brief, 1×106 cells in 60-mm culture dishes (Nunc) were
incubated with GA and/or each MAPK inhibitor for 24 h. Cells were
then washed with PBS and fixed in 70% ethanol. Cells were washed
again with PBS, then incubated with PI (10 μg/ml) with simultaneous
RNase treatment at 37°C for 30 min. Cell DNA content was measured
using a FACStar flow cytometer (Becton Dickinson, San Jose, CA,
USA).
Annexin V staining for cell death
detection
Apoptosis was determined by staining cells with
Annexin V-fluorescein isothiocyanate (FITC; Ex/Em=488 nm/519 nm,
Invitrogen Corp., Camarillo, CA, USA) as previously described
(30,31). In brief, 1×106 cells in
60-mm culture dishes (Nunc) were incubated with GA and/or each MAPK
inhibitor for 24 h. Cells were washed twice with cold PBS and then
resuspended in 500 μl of binding buffer (10 mM HEPES/NaOH pH 7.4,
140 mM NaCl, 2.5 mM CaCl2) at a concentration of
1×106 cells/ml. Annexin V-FITC (5 μl) was then added to
these cells, which were analyzed with a FACStar flow cytometer
(Becton-Dickinson).
Measurement of mitochondrial membrane
potential (MMP; ΔΨm)
MMP (ΔΨm) levels were measured by a
rhodamine 123 fluorescent dye (Ex/Em=485 nm/535 nm, Sigma-Aldrich
Chemical Co.) as previously described (31,32).
In brief, 1×106 cells in 60-mm culture dishes (Nunc)
were incubated with GA and/or each MAPK inhibitor for 24 h. Cells
were washed twice with PBS and incubated with the rhodamine 123
(0.1 μg/ml) at 37°C for 30 min. Rhodamine 123 staining intensity
was determined by flow cytometry. Rhodamine 123 negative cells
indicated the loss of MMP (ΔΨm) in A549 cells. MMP
(ΔΨm) levels in cells except MMP (ΔΨm) loss
cells were expressed as mean fluorescence intensity (MFI), which
was calculated by CellQuest software.
Detection of intracellular ROS
levels
Intracellular ROS such as
H2O2, •OH and ONOO•
were detected by means of an oxidation-sensitive fluorescent probe
dye, 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCFDA; Ex/Em=495 nm/529 nm, Invitrogen Molecular
Probes, Eugene, OR, USA) (33).
H2DCFDA is poorly selective for
O2•−. By contrast, dihydroethidium (DHE;
Ex/Em=518 nm/605 nm, Invitrogen Molecular Probes) is highly
selective for O2•− among ROS. In brief,
1×106 cells in 60-mm culture dishes (Nunc) were
incubated with GA and/or each MAPK inhibitor for 24 h. Cells were
then washed in PBS and incubated with 20 μM H2DCFDA or
DHE at 37°C for 30 min. DCF and DHE fluorescences were detected
using a FACStar flow cytometer (Becton-Dickinson). ROS and
O2•− levels were expressed as MFI, which was
calculated by CellQuest software.
Detection of the intracellular GSH
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em=522 nm/595
nm; Invitrogen Molecular Probes) as previously described (34,35).
In brief, 1×106 cells in 60-mm culture dishes (Nunc)
were incubated with GA and/or each MAPK inhibitor for 24 h. Cells
were then washed with PBS and incubated with 5 μM CMFDA at 37°C for
30 min. CMF fluorescence intensity was determined using a FACStar
flow cytometer (Becton-Dickinson). Negative CMF staining
(GSH-depleted) cells were expressed as the percentage of (−) CMF
cells. CMF levels in cells except GSH-depleted cells were expressed
as MFI, which was calculated by CellQuest software.
Statistical analysis
The results represent at least three independent
experiments (means ± SD). The data were analyzed using Instat
software (GraphPad Prism 4, San Diego, CA, USA). One-way analysis
of variance (ANOVA) with post hoc analysis using Tukey’s multiple
comparison test was used for parametric data. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of MAPK inhibitors on cell growth
in GA-treated A549 cells
We examined the effects of MAPK inhibitors on the
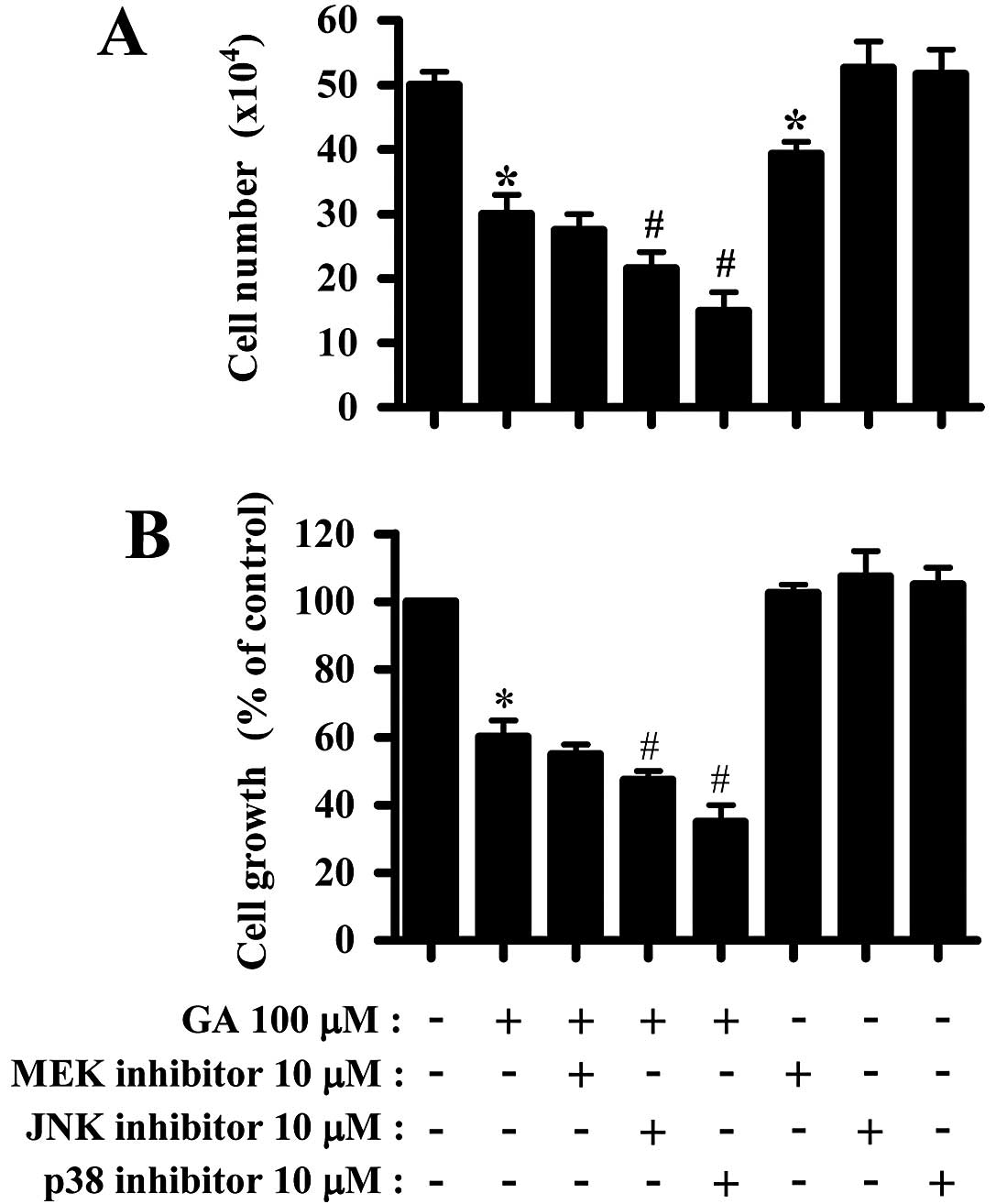
growth of GA-treated A549 cells. GA dose-dependently inhibited A549
cell growth with an IC50 of ~150 μM at 24 h (25). Treatment with 100 μM GA used in this
study inhibited the growth of A549 cells ~40% based on trypan blue
cell counting and MTT assays at 24 h (Fig. 1A and B). It has been demonstrated
that high dose usages of MAPK inhibitors may decrease their
specificity (36). In fact, 20 μM
concentration of each MAPK inhibitor significantly induced cell
growth inhibition and death in A549 cells (data not shown).
Therefore, the concentration of 10 μM each MAPK inhibitor was used
in the current experiments to evade the non-specific inhibition of
other kinases. While MEK inhibitor slightly enhanced the growth
inhibition by GA, JNK and p38 inhibitors significantly enhanced the
growth inhibition (Fig. 1A and B).
Only MEK inhibitor decreased cell number in A549 control cells
(Fig. 1A). The 10 μM MAPK
inhibitors did not affect A549 control cell growth (Fig. 1B).
Effects of MAPK inhibitors on cell death
and MMP (ΔΨm) in GA-treated A549 cells
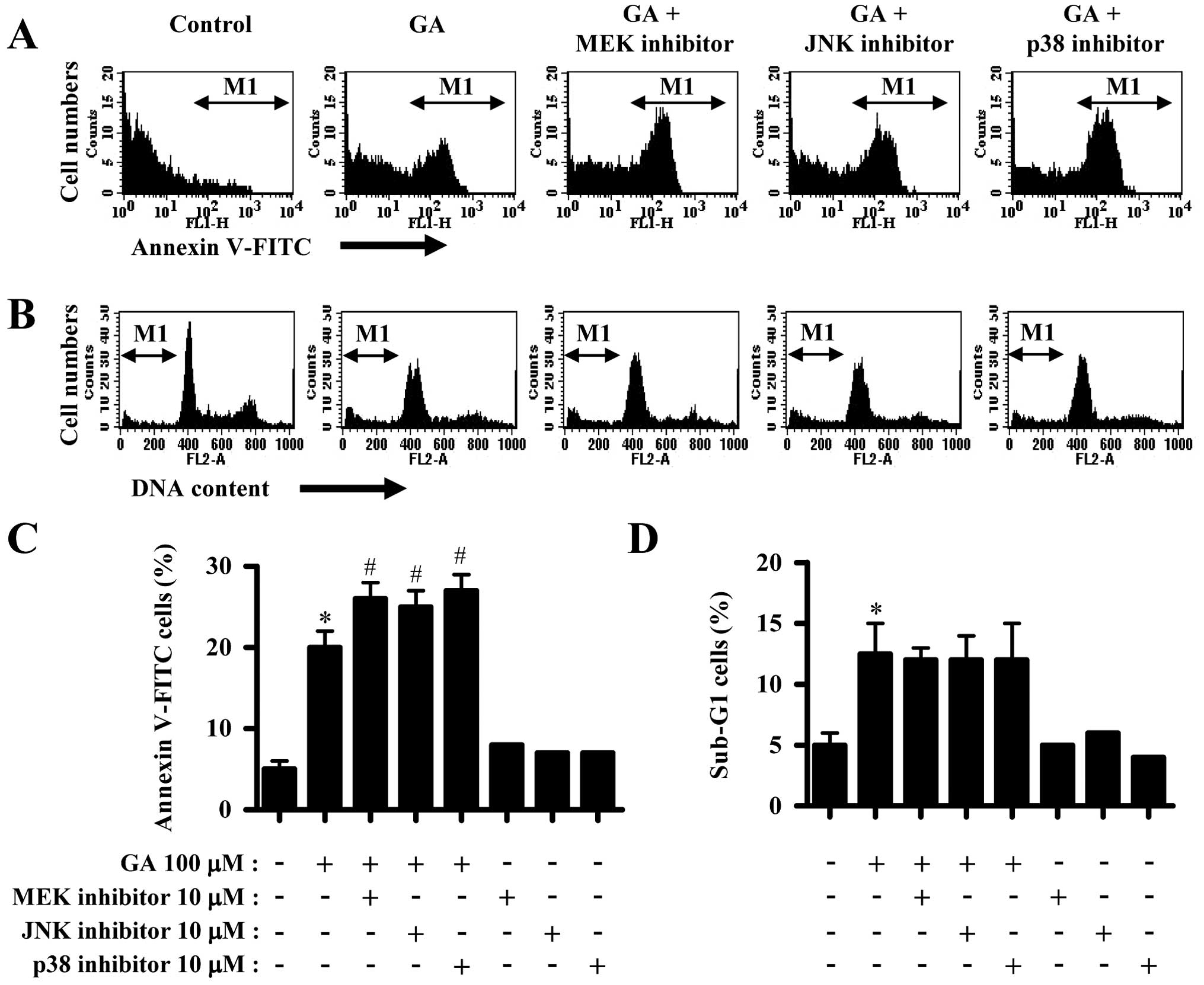
GA significantly induced cell death in A549 cells,
as evidenced by trypan blue positive staining (Fig. 1A) and Annexin V staining cells
(Fig. 2A and C). However, GA
slightly increased the number of sub-G1 DNA content cells (Fig. 2B and D). All the MAPK inhibitors
increased the number of Annexin V staining cells in GA-treated A549
cells (Fig. 2A and C). However,
none of the MAPK inhibitors altered the number of sub-G1 cells
(Fig. 2B and D) and none of the
MAPK inhibitors affected the number of Annexin V staining and
sub-G1 cells in A549 control cells (Fig. 2C and D).
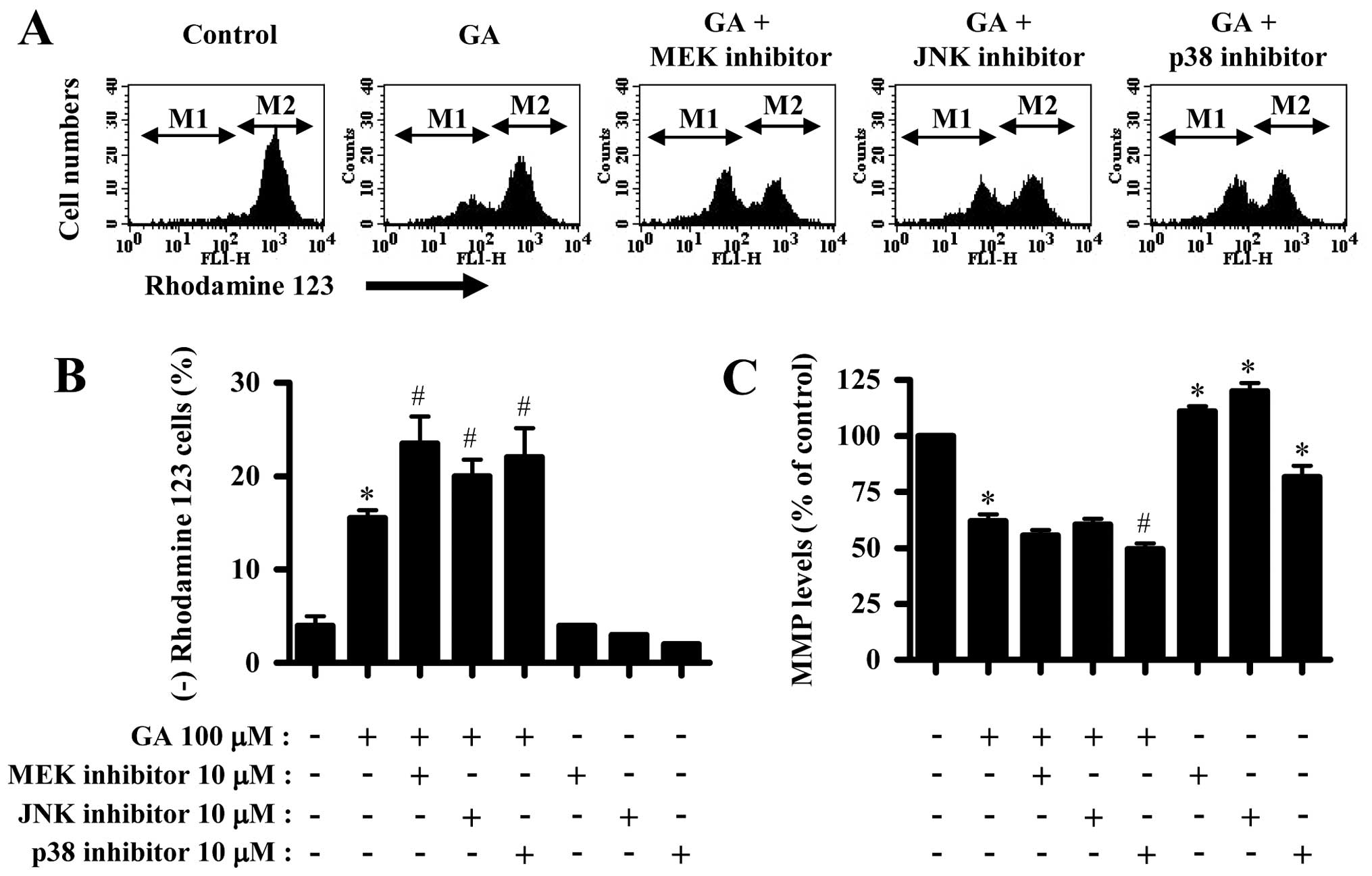
In addition, GA significantly triggered the loss of
MMP (ΔΨm) in A549 cells (Fig. 3A and B). All the MAPK inhibitors
intensified the MMP (ΔΨm) loss in GA-treated A549 cells
(Fig. 3A and B). In relation to MMP
(ΔΨm) levels, GA reduced MMP (ΔΨm) levels in
A549 cells (Fig. 3A and C). Only
p38 inhibitor significantly decreased MMP (ΔΨm) levels
in GA-treated A549 cells (Fig. 3A and
C). Although MEK or JNK inhibitor alone increased MMP
(ΔΨm) levels in A549 control cells, p38 inhibitor
reduced the MMP (ΔΨm) levels (Fig. 3A and C).
Effects of MAPK inhibitors on ROS and GSH
levels in GA-treated A549 cells
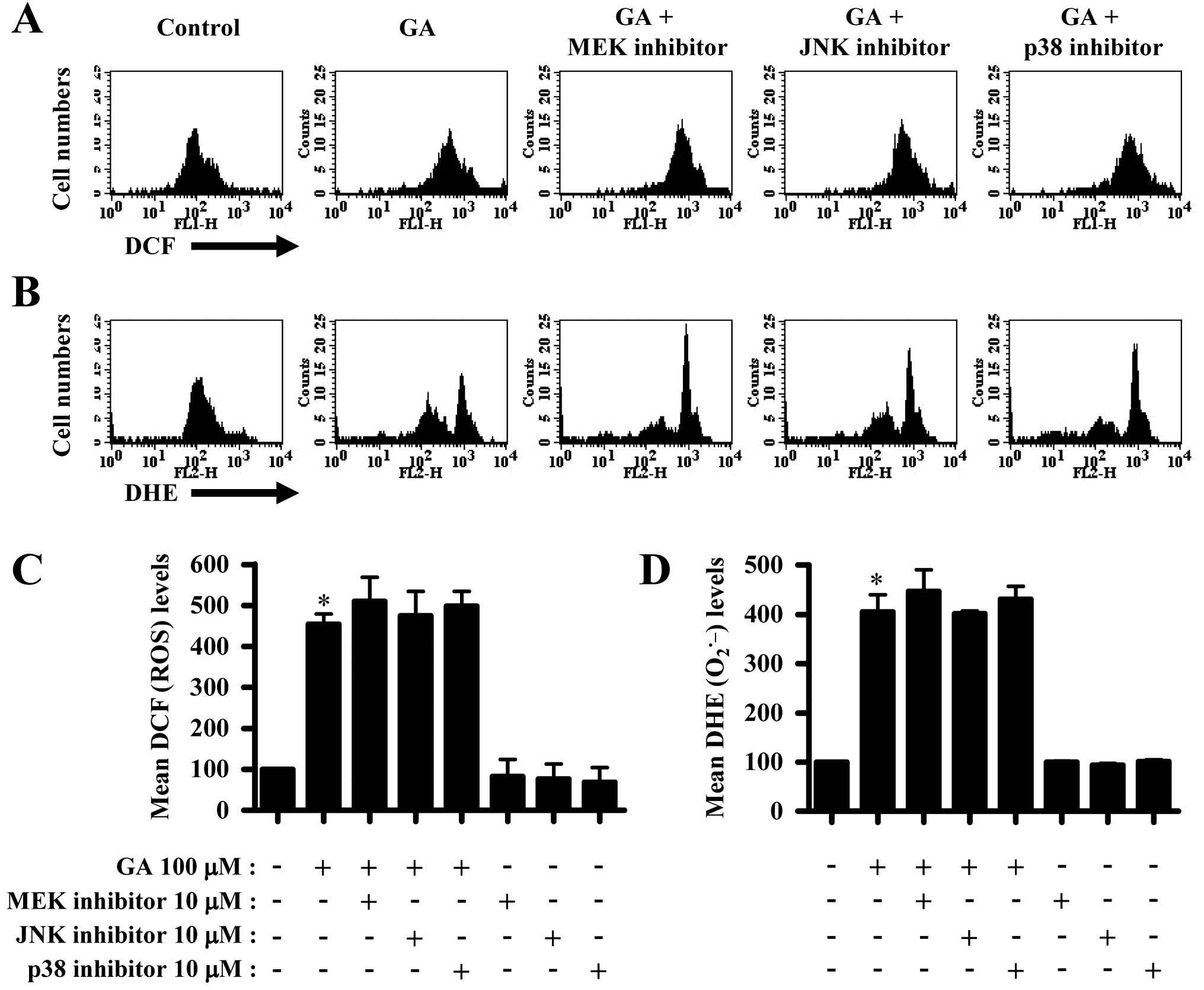
Next, we determined whether intracellular ROS and
GSH levels in GA-treated A549 cells were altered by treatment with
each MAPK inhibitor. ROS (DCF) level such as
H2O2 was increased in GA-treated A549 cells
(Fig. 4A and C). The MAPK
inhibitors did not significantly change ROS level in GA-treated
A549 cells (Fig. 4A and C). Red
fluorescence derived from DHE reflecting intracellular
O2•− level was also increased in A549 cells
(Fig. 4B and D). None of the MAPK
inhibitors altered O2•− levels in GA-treated
A549 cells (Fig. 4B and D). The
MAPK inhibitors did not alter ROS level in A549 control cells
(Fig. 4C and D).
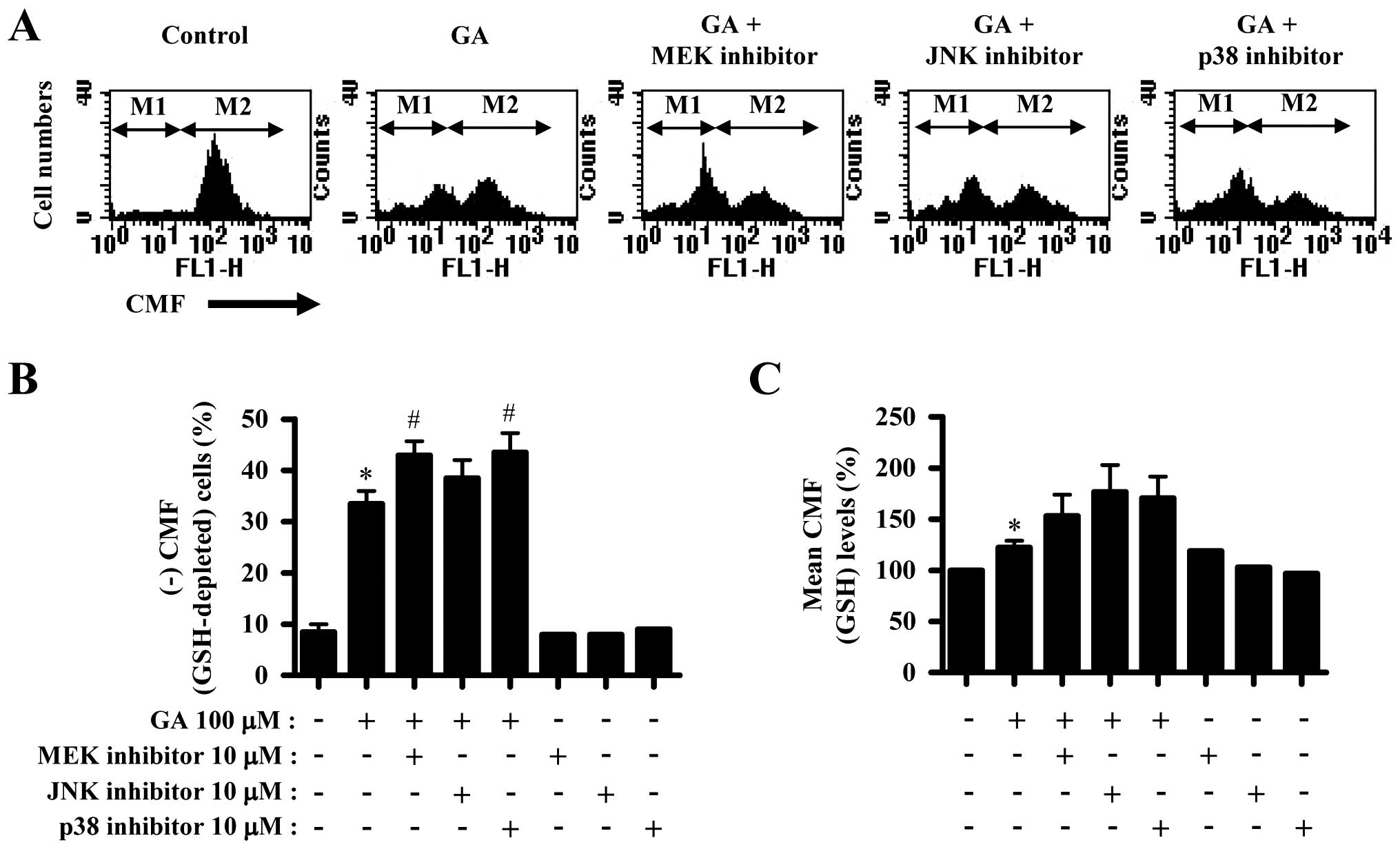
GA increased the number of GSH-depleted cells in
A549 cells (Fig. 5A and B). MEK and
p38 inhibitors significantly increased GSH-depleted cell number in
GA-treated A549 cells and JNK inhibitor showed a light effect on
that (Fig. 5A and B). In addition,
GA increased GSH level in A549 cells (Fig. 5A and C). All the MAPK inhibitors
seemed to intensify the increased GSH level by GA (Fig. 5A and C).
Discussion
Since GA inhibited the growth of A549 cells and
induced their death, we focused on elucidating the toxicological
effect of GA on cell growth and death in A549 cells pretreated with
MAPK inhibitors in relation to ROS and GSH. GA increased Annexin
V-FITC positive cells in A549 cells, which indirectly indicated
that GA-induced A549 cell death occurred via apoptosis. However, GA
did not increase sub-G1 cell number as much as Annexin V positive
cell number by it. In addition, 200 μM GA did not increase sub-G1
cell number compared with the number in 100 μM GA-treated cells
(data not shown). Therefore, GA seemed to induce growth inhibition
in A549 cells via necrosis as well as apoptosis.
ERK activation has a pro-survival function rather
than pro-apoptotic effects (23).
Similarly, MEK inhibitor, which presumably decreased ERK activity,
slightly enhanced the growth inhibition by GA and significantly
intensified cell death by it. In addition, MEK inhibitor alone
significantly reduced the number of A549 control cells. Therefore,
this result indirectly suggested that the inhibition of ERK
signaling by MEK inhibitor plays a pro-apoptotic role in GA-treated
A549 cells and has an anti-growth function in A549 control cells.
However, MEK inhibitor attenuates growth inhibition and death in
GA-treated Calu-6 cells (27),
indicating that MEK inhibitor differently affects cell growth and
death in GA-treated A549 and Calu-6 lung cancer cells. Moreover,
MEK inhibitor did not affect cell growth inhibition and death in
GA-treated human pulmonary fibroblast cells (37). Therefore, the targeted therapy
related to ERK signaling should be carefully considered in lung
cancer treatment. Considerable evidence demonstrates that JNK or
p38 signaling is related to cell death (20,21).
In fact, GA induces PC12 rat pheochromocytoma cell death through
the activation of JNK and its inhibitor protects PC12 cells against
GA-induced cell death (38). In
addition, p38 inhibitor prevented anisomycin-induced macrophage
death (39) and its inhibitor
decreased the death of pyrogallol-induced calf pulmonary artery
endothelial cells (28). Moreover,
JNK and p38 inhibitors do not significantly alter cell death in
GA-treated human pulmonary fibroblast cells and Calu-6 cells
(27,37). However, results of the present study
demonstrated that JNK or p38 inhibitor enhanced growth inhibition
and death in GA-treated A549 cells. It is also demonstrated that
JNK and p38 inhibitors enhance cell growth inhibition and death in
GA-treated HeLa cells (40).
Therefore, the inhibition of JNK or p38 signaling by each inhibitor
seemed to be a pro-apoptotic function in GA-treated A549 cells.
Collectively, anti- or pro-apoptotic effects of JNK or p38
inhibitor depend on cell types or co-treated agents. Markedly, none
of the MAPK inhibitors increased the number of sub-G1 cells in
GA-treated A549 cells. These results indicated that the inhibition
of each MAPK signaling by its inhibitor was involved in the
enhancement of apoptosis rather than necrosis.
Cell death induced by GA is associated with
mitochondrial dysfunction (12).
Accordingly, GA induced the loss of MMP (ΔΨm) in A549
cells and reduced MMP (ΔΨm) levels. All the MAPK
inhibitors intensified MMP (ΔΨm) loss in GA-treated A549
cells. Therefore, the cell death by GA and/or MAPK inhibitors
seemed to be tightly connected with the loss of MMP
(ΔΨm). In addition, p38 inhibitor significantly reduced
MMP (ΔΨm) levels in GA-treated A549 cells. This result
likely clarifies the strong growth inhibition of A549 cells by
co-treatment with GA and p38 inhibitor since MTT reduction is
considered to be an indirect measurement of mitochondrial activity
(41). However, since all the MAPK
inhibitors affected the basal MMP (ΔΨm) level in A549
control cells without changes in cell growth, the changes in MMP
(ΔΨm) levels are not fully associated with those of MTT
reduction in cells. Nevertheless, the basal activity of MAPK
signalings seemed to be involved in the maintenance of MMP
(ΔΨm) in A549 control cells.
Increasing evidence suggests that apoptosis induced
by GA is associated with oxidative stresses derived from ROS
(6,42). Similarly, ROS levels including
O2•− were significantly increased in
GA-treated A549 cells. The MAPK inhibitors did not significantly
alter ROS levels in GA-treated A549 cells. These data suggest that
changes in ROS levels by MAPK inhibitors in GA-treated A549 cells
are not closely related to cell death. GSH is a main non-protein
antioxidant and eliminates the O2•− by
providing electrons for enzymes such as GSH peroxidase, which
reduce H2O2 to H2O. It has been
reported that the intracellular GSH content has a decisive effect
on anticancer drug-induced apoptosis, indicating that apoptotic
effects are inversely comparative to GSH content (34,43,44).
Similarly, GA increased the number of GSH-depleted cells in A549
cells. MEK and p38 inhibitors significantly enhanced GSH depletion
in GA-treated A549 cells, and JNK inhibitor slightly increased the
number. These results might be correlated with the results derived
from Annexin V and MMP (ΔΨm) assays. These results
support the hypothesis that the intracellular GSH content has a
decisive effect on cell death (32,34,45,46).
It is of note that CMF (GSH) level in A549 cells was increased by
GA. The increased GSH level was likely to occur against the
increasing ROS level by GA. It may be that some cells which could
not defend oxidative stress resulting from GA treatment underwent
cell death pathway. All the MAPK inhibitors enhanced GSH level in
GA-treated A549 cells. These results suggest that MAPK inhibitors
are involved in the upregulation of GSH levels in GA-treated A549
cells, consequently affecting ROS levels and the proportions of
GSH-depleted cells. The mechanism underlying the effect of MAPKs on
intracellular GSH level in cells requires further
clarification.
In conclusion, GA induced growth inhibition and
death in A549 cells, which was accompanied by intracellular ROS
increase and GSH depletion. All the MAPK inhibitors enhanced growth
inhibition and death in GA-treated A549 cells, which were related
to GSH depletion rather than ROS level.
Acknowledgements
The present study was supported by a grant from the
Ministry of Science and Technology (MoST)/Korea Science and
Engineering Foundation (KOSEF) through the Diabetes Research Center
at Chonbuk National University (2012-0009323) and the National
Research Foundation of Korea Grant funded by the Korean Government
(MEST) (2010-0021808).
Abbreviations:
|
GA
|
gallic acid
|
|
ROS
|
reactive oxygen species
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
MAP kinase or ERK kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
SOD
|
superoxide dismutase
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
FBS
|
fetal bovine serum
|
|
FITC
|
fluorescein isothiocyanate
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
GSH
|
glutathione
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
PI
|
propidium iodide
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
References
|
1
|
Niemetz R and Gross GG: Enzymology of
gallotannin and ellagitannin biosynthesis. Phytochemistry.
66:2001–2011. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shahrzad S, Aoyagi K, Winter A, Koyama A
and Bitsch I: Pharmacokinetics of gallic acid and its relative
bioavailability from tea in healthy humans. J Nutr. 131:1207–1210.
2001.PubMed/NCBI
|
|
3
|
Kang MS, Oh JS, Kang IC, Hong SJ and Choi
CH: Inhibitory effect of methyl gallate and gallic acid on oral
bacteria. J Microbiol. 46:744–750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kratz JM, Andrighetti-Frohner CR, Leal PC,
Nunes RJ, Yunes RA, Trybala E, Bergstrom T, Barardi CR and Simoes
CM: Evaluation of anti-HSV-2 activity of gallic acid and pentyl
gallate. Biol Pharm Bull. 31:903–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim SH, Jun CD, Suk K, Choi BJ, Lim H,
Park S, Lee SH, Shin HY, Kim DK and Shin TY: Gallic acid inhibits
histamine release and pro-inflammatory cytokine production in mast
cells. Toxicol Sci. 91:123–131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inoue M, Sakaguchi N, Isuzugawa K, Tani H
and Ogihara Y: Role of reactive oxygen species in gallic
acid-induced apoptosis. Biol Pharm Bull. 23:1153–1157. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaur M, Velmurugan B, Rajamanickam S,
Agarwal R and Agarwal C: Gallic acid, an active constituent of
grape seed extract, exhibits anti-proliferative, pro-apoptotic and
anti-tumorigenic effects against prostate carcinoma xenograft
growth in nude mice. Pharm Res. 26:2133–2140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Veluri R, Singh RP, Liu Z, Thompson JA,
Agarwal R and Agarwal C: Fractionation of grape seed extract and
identification of gallic acid as one of the major active
constituents causing growth inhibition and apoptotic death of DU145
human prostate carcinoma cells. Carcinogenesis. 27:1445–1453. 2006.
View Article : Google Scholar
|
|
9
|
Kawada M, Ohno Y, Ri Y, Ikoma T, Yuugetu
H, Asai T, Watanabe M, Yasuda N, Akao S, Takemura G, et al:
Anti-tumor effect of gallic acid on LL-2 lung cancer cells
transplanted in mice. Anticancer Drugs. 12:847–852. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohno Y, Fukuda K, Takemura G, Toyota M,
Watanabe M, Yasuda N, Xinbin Q, Maruyama R, Akao S, Gotou K,
Fujiwara T and Fujiwara H: Induction of apoptosis by gallic acid in
lung cancer cells. Anticancer Drugs. 10:845–851. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Faried A, Kurnia D, Faried LS, Usman N,
Miyazaki T, Kato H and Kuwano H: Anticancer effects of gallic acid
isolated from Indonesian herbal medicine, Phaleria
macrocarpa (Scheff.) Boerl, on human cancer cell lines. Int J
Oncol. 30:605–613. 2007.PubMed/NCBI
|
|
12
|
Chen HM, Wu YC, Chia YC, Chang FR, Hsu HK,
Hsieh YC, Chen CC and Yuan SS: Gallic acid, a major component of
Toona sinensis leaf extracts, contains a ROS-mediated
anti-cancer activity in human prostate cancer cells. Cancer Lett.
286:161–171. 2009.PubMed/NCBI
|
|
13
|
Strlic M, Radovic T, Kolar J and Pihlar B:
Anti- and prooxidative properties of gallic acid in fenton-type
systems. J Agric Food Chem. 50:6313–6317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sakagami H and Satoh K: Prooxidant action
of two antioxidants: ascorbic acid and gallic acid. Anticancer Res.
17:221–224. 1997.PubMed/NCBI
|
|
15
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: a comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wilcox CS: Reactive oxygen species: roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Genestra M: Oxyl radicals, redox-sensitive
signalling cascades and antioxidants. Cell Signal. 19:1807–1819.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Blenis J: Signal transduction via the MAP
kinases: proceed at your own RSK. Proc Natl Acad Sci USA.
90:5889–5892. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kusuhara M, Takahashi E, Peterson TE, Abe
J, Ishida M, Han J, Ulevitch R and Berk BC: p38 Kinase is a
negative regulator of angiotensin II signal transduction in
vascular smooth muscle cells: effects on
Na+/H+ exchange and ERK1/2. Circ Res.
83:824–831. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsin YH, Chen CF, Huang S, Shih TS, Lai PS
and Chueh PJ: The apoptotic effect of nanosilver is mediated by a
ROS- and JNK-dependent mechanism involving the mitochondrial
pathway in NIH3T3 cells. Toxicol Lett. 179:130–139. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guyton KZ, Liu Y, Gorospe M, Xu Q and
Holbrook NJ: Activation of mitogen-activated protein kinase by
H2O2. Role in cell survival following oxidant
injury. J Biol Chem. 271:4138–4142. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Petty RD, Nicolson MC, Kerr KM,
Collie-Duguid E and Murray GI: Gene expression profiling in
non-small cell lung cancer: from molecular mechanisms to clinical
application. Clin Cancer Res. 10:3237–3248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
You BR, Kim SZ, Kim SH and Park WH: Gallic
acid-induced lung cancer cell death is accompanied by ROS increase
and glutathione depletion. Mol Cell Biochem. 357:295–303. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
You BR and Park WH: Gallic acid-induced
lung cancer cell death is related to glutathione depletion as well
as reactive oxygen species increase. Toxicol In Vitro.
24:1356–1362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han YH, Moon HJ, You BR, Yang YM, Kim SZ,
Kim SH and Park WH: The MEK inhibitor PD98059 attenuates growth
inhibition and death in gallic acid-treated Calu-6 lung cancer
cells by preventing glutathione depletion. Mol Med Rep. 3:519–524.
2010.PubMed/NCBI
|
|
28
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: JNK and p38 inhibitors increase and decrease
apoptosis, respectively, in pyrogallol-treated calf pulmonary
arterial endothelial cells. Int J Mol Med. 24:717–722.
2009.PubMed/NCBI
|
|
29
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Effects of carbonyl cyanide p-(trifluoromethoxy)
phenylhydrazone on the growth inhibition in human pulmonary
adenocarcinoma Calu-6 cells. Toxicology. 265:101–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han YH, Kim SZ, Kim SH and Park WH:
Apoptosis in pyrogallol-treated Calu-6 cells is correlated with the
changes of intracellular GSH levels rather than ROS levels. Lung
Cancer. 59:301–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han YH, Moon HJ, You BR and Park WH: The
effect of MG132, a proteasome inhibitor on HeLa cells in relation
to cell growth, reactive oxygen species and GSH. Oncol Rep.
22:215–221. 2009.PubMed/NCBI
|
|
32
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*−) generator induces apoptosis via the
depletion of intracellular GSH contents in Calu-6 cells. Lung
Cancer. 63:201–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han YH, Kim SH, Kim SZ and Park WH:
Caspase inhibitor decreases apoptosis in pyrogallol-treated lung
cancer Calu-6 cells via the prevention of GSH depletion. Int J
Oncol. 33:1099–1105. 2008.PubMed/NCBI
|
|
34
|
Han YH, Kim SZ, Kim SH and Park WH:
Intracellular GSH level is a factor in As4.1 juxtaglomerular cell
death by arsenic trioxide. J Cell Biochem. 104:995–1009. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bain J, McLauchlan H, Elliott M and Cohen
P: The specificities of protein kinase inhibitors: an update.
Biochem J. 371:199–204. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park WH: MAPK inhibitors differentially
affect gallic acid-induced human pulmonary fibroblast cell growth
inhibition. Mol Med Rep. 4:193–204. 2011.PubMed/NCBI
|
|
38
|
Kang MK, Kang NJ, Jang YJ, Lee KW and Lee
HJ: Gallic acid induces neuronal cell death through activation of
c-Jun N-terminal kinase and downregulation of Bcl-2. Ann NY Acad
Sci. 1171:514–520. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Croons V, Martinet W, Herman AG,
Timmermans JP and De Meyer GR: The protein synthesis inhibitor
anisomycin induces macrophage apoptosis in rabbit atherosclerotic
plaques through p38 mitogen-activated protein kinase. J Pharmacol
Exp Ther. 329:856–864. 2009. View Article : Google Scholar
|
|
40
|
You BR and Park WH: The effects of
mitogen-activated protein kinase inhibitors or small interfering
RNAs on gallic acid-induced HeLa cell death in relation to reactive
oxygen species and glutathione. J Agric Food Chem. 59:763–771.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marshall NJ, Goodwin CJ and Holt SJ: A
critical assessment of the use of microculture tetrazolium assays
to measure cell growth and function. Growth Regul. 5:69–84.
1995.PubMed/NCBI
|
|
42
|
Serrano A, Palacios C, Roy G, Cespon C,
Villar ML, Nocito M and Gonzalez-Porque P: Derivatives of gallic
acid induce apoptosis in tumoral cell lines and inhibit lymphocyte
proliferation. Arch Biochem Biophys. 350:49–54. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Han YH, Kim SZ, Kim SH and Park WH:
Enhancement of arsenic trioxide-induced apoptosis in HeLa cells by
diethyldithiocarbamate or buthionine sulfoximine. Int J Oncol.
33:205–213. 2008.PubMed/NCBI
|
|
44
|
Han YH, Kim SZ, Kim SH and Park WH:
Suppression of arsenic trioxide-induced apoptosis in HeLa cells by
N-acetylcysteine. Mol Cells. 26:18–25. 2008.PubMed/NCBI
|
|
45
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
46
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: The effects of buthionine sulfoximine,
diethyldithiocarbamate or 3-amino-1,2,4-triazole on propyl
gallate-treated HeLa cells in relation to cell growth, reactive
oxygen species and glutathione. Int J Mol Med. 24:261–268.
2009.
|