Breast cancer and ovarian cancer develop through
multiple molecular pathways guided by genetic and epigenetic clonal
selections. Both cancers have been recognized as a heterogeneous
disease with regard to clinical and biological properties. The
majority of cancer cases are considered sporadic-appearing tumors
in nature because there is no obvious family history, but a number
of families with a known genetic cause or inherited predisposition
to cancer have been identified (1).
Individuals who carry an inherited genetic mutation and epigenetic
aberrations in the tumor suppressor genes have an increased
lifetime risk of developing cancer. Germline mutations in cancer
susceptibility genes cause cancer if the wild-type allele is lost
or inactivated. Breast and ovarian cancers (5–10%) may be
hereditary and occur in cancer prone syndromes (2–7).
Patients predisposed to breast and ovarian cancers are known as the
hereditary breast and ovarian cancer (HBOC) syndrome.
HBOC syndrome is an autosomal dominantly inherited
disease characterized by a young age of onset, more than one
synchronous or metachronous tumor, and a family history of first
and second degree relatives with similar cancers (8). Mainly HBOC syndrome results from
germline mutations in breast cancer genes BRCA1 or BRCA2. Other
genes or low penetrance alleles might be associated with the HBOC
phenotype (9). There is an
increasing understanding that the interrelationship between BRCA
gene cluster and Fanconi anemia (FA), mismatch repair (MMR) and DNA
repair gene status plays a key role in the pathogenesis of cancer
predisposition syndromes. We reviewed the HBOC syndrome and current
knowledge of inherited susceptibility genes.
A computerized literature search was performed to
identify relevant studies reported in the English language. We
searched MEDLINE electronic databases (http://www.ncbi.nlm.nih.gov/sites/entrez) published
between January 1994 and October 2012, combining the keywords
‘hereditary breast and ovarian cancer’, ‘pathogenesis’, ‘BRCA’ and
‘DNA repair’. Various combinations of the terms were used,
depending on the database searched. Each gene was also linked to
NCBI Entrez Gene pages (http://www.ncbi.nlm.nih.gov/sites/entrez).
Additionally, references in each article were searched to identify
potentially missed studies.
The breast cancer-associated genes BRCA1 on
chromosome 17q and BRCA2 on chromosome 13q are the most well-known
breast cancer susceptibility genes (4,5). The
HBOC syndrome is linked to the BRCA1 gene and, to a lesser extent,
to the BRCA2 gene. Germline mutations in these genes account for
2–5% (up to 10%) of all breast cancers and all ovarian cancers.
Mutations were present with a frequency of more than 10% in the
high risk populations, including patients with a family history of
breast or first-degree ovarian cancer, those with bilateral breast
cancer, multiple organ cancer including younger breast cancer
patients (aged <35 years). Mutations in the BRCA1 and BRCA2
genes are responsible for ~60% (up to 85%) of HBOC. Women carrying
a BRCA1 or BRCA2 genetic mutation have 60–80% and 20–40% lifetime
risk of developing breast cancer and ovarian cancer, respectively.
More modest increases in risk for other cancers have been noted:
additional sites included stomach, pancreas, prostate and colon.
The cancer risk ranged from 20 to 60%, with the greatest increases
in cancer risk in stomach and pancreas. BRCA mutations were also
associated with increased risks for leukemia and lymphoma (10).
A recent study showed that breast cancer patients
belonging to a population with a high probability of being BRCA1
carriers showed a better prognosis compared with those with
sporadic breast cancer (11).
Furthermore, BRCA1 and BRCA2-related invasive epithelial ovarian
cancers have a better 5-year overall survival compared with
sporadic ovarian cancers (12). The
5-year overall survival was better in BRCA2 carriers compared to
BRCA1 carriers (36% for non-carriers, 44% for BRCA1 carrier and 52%
for BRCA2 carriers).
The mutations include partial or complete gene
deletions, duplications, large insertions, splice alteration,
frameshifts as well as missense and nonsense mutations. Deletions
or insertions usually lead to abnormal structure and function.
Germline mutations are usually pathogenic point mutations, and are
scattered throughout their coding regions. The potential hot-spot
mutations within BRCA 1 and BRCA2 are uncommon. The previously
described mutations were identified in the Breast Cancer
Information Core website (BIC, http://research.nhgri.nih.gov/bic/). More than 3500
mutations have been reported throughout both genes. Data from
subjects with a variety of ethnic backgrounds altered the overall
odds of BRCA mutation carrier status. The spectrum of mutations is
different depending on the race. The mutations are detected in
10–12% of Ashkenazi Jewish women diagnosed with breast cancer.
Ashkenazi Jewish subjects are observed at increased frequency
compared to other Caucasian, because this population harbors
ancient BRCA1 and BRCA2 mutant alleles. Mutation analysis revealed
the c. 5266dup (until recently referred in the literature as
5382insC), c. 68_69del (185delAG) and 4153delA mutations in BRCA1
and c. 5946del (6174delT) mutation in BRCA2 (13). BRCA gene founder mutations such as
BRCA1c. 5266dup mutation, BRCA2999del5 mutation and BRCA1delexon17
have also been described in other populations, including the
Slavic, Finnish, Icelandic and German populations,
respectively.
The tumor suppressor BRCA1 and BRCA2 genes are
essential components of the double-strand break (DSB) repair by
homologous recombination (HR) system. Targeting tumor suppressor
loss-of-function is possible based on the concept of synthetic
lethality. Thus, the synthetically lethal effect might be observed
in tumors defective in BRCA1 or BRCA2 that are required for
efficient HR, indicating that ovarian cancer patients carrying
germ-line mutations had improved rates of progression-free and
overall survival (12). Chromosomal
rearrangements might be formed as a consequence of these
error-prone DSB repairs and lead to the development of genomic
instability. Large genomic rearrangements have been identified in
HBOC families and account for 8–15% of deleterious BRCA mutations,
but these rearrangements may escape detection (14). The issue is that BRCA genetic
testing done through sequencing will not capture large
rearrangements in these genes.
BRCA1 functions as a tumor suppressor gene, but
paradoxically, BRCA1 knockout mice are embryonically lethal in
homozygous state. Lack of BRCA1 is thought to result in cellular
lethality, suggesting that BRCA1 regulates stem/progenitor cell
proliferation and differentiation. For cell differentiation, BRCA1
regulates apicobasal polarity, together with several genes such as
RHAMM (hyaluronan-mediated motility receptor), AURKA (aurora kinase
A) and TPX2 (microtubule-associated, homolog). Intracellular RHAMM
associates with BRCA1 and BARD1 (BRCA1 associated RING domain 1).
The complex attenuates the mitotic-spindle-promoting activity of
RHAMM that might contribute to tumor progression. BRCA1 also binds
and regulates AURKA, a cell cycle-regulated kinase that appears to
be strongly involved in centrosome regulation. Genetic variants in
the AURKA gene may contribute to breast cancer development
(5). BRCA1 further accumulates TPX2
and is required for mitotic spindle-pole assembly. BRCA-associated
nuclear core complex proteins are required for the functional
integrity of the pathway of not only DNA damage response and
repair, but also cell differentiation
From a clinical point of view, the triple-negative
breast cancer characterized by the absence of estrogen receptor
(ER), progesterone receptor (PgR), and HER2 (also known as ERBB2)
accounts for ~15% of breast cancers and is diagnosed more
frequently in younger women (15).
Compared with the ER/PR-positive tumors, triple-negative breast
cancershowed a greater risk for recurrence and shortened survival.
Triple-negative breast cancer was frequently associated with
mutations in BRCA genes: the incidence was 12.5–20% (16). Women with an early age-of-onset
triple-negative breast cancer are more likely to be associated with
deleterious mutations in BRCA1 and BRCA2 genes (17). Even in non-BRCA gene mutations, a
subset of triple-negative tumors shares multiple clinicopathologic
features and phenotype with BRCA-mutated breast cancers. They
harbor dysfunctional DNA repair mechanisms, but the nature of this
link remains opaque. A subset of HBOC syndrome also contain
mutations in the TP53 gene, and the TP53 loss-of-function tumors
have a low frequency of HER2 expression (18).
The predominant histologic type of ovarian cancers
associated with the HBOC syndrome was high-grade serous carcinomas
of the ovary. There were no significant differences in ovarian
cancer morphology between BRCA1 and BRCA2 carriers (19). Ovarian cancer patients with BRCA
mutations were associated with an increased chemosensitivity and
improved overall survival, but some investigators failed to confirm
improved survival.
Mutations in BRCA1 and BRCA2 do not account for all
cases of HBOC, implicating that the remaining cases can be
attributed to the involvement of other susceptibility genes. Other
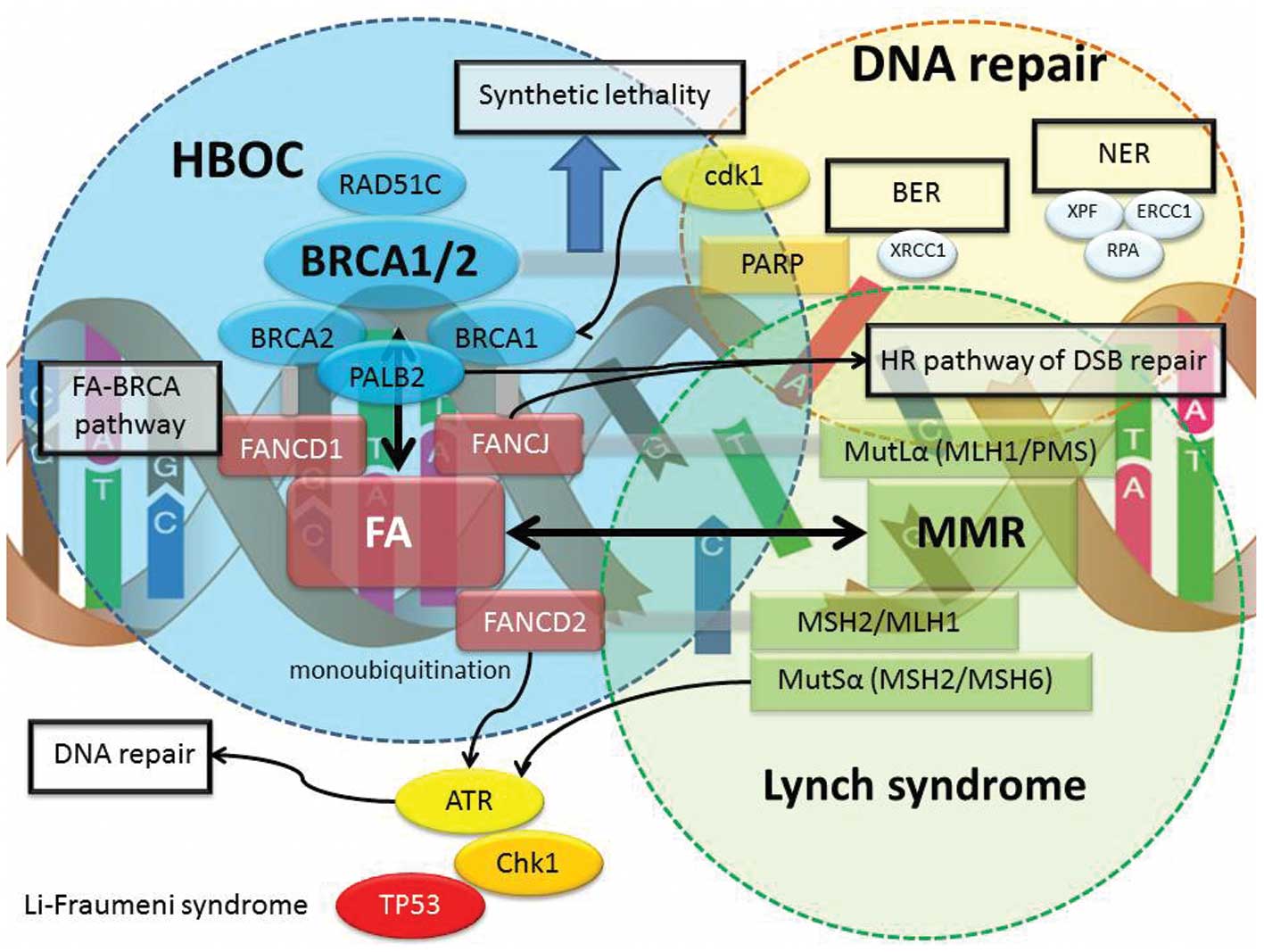
genes, including Fanconi anemia (FA) cluster (FANCD2, FANCA and
FANCC), MMR cluster (MLH1, MSH2, PMS1, PMS2 and MSH6), DNA
checkpoint cluster (ATM, ATR and CHK1/2), and tumor suppressor
cluster (TP53, SKT11 and PTEN) have been associated with increased
risk of breast and ovarian cancer as part of other cancer
syndromes. The contribution of mutations in other genes to the
burden of breast or ovarian cancer is indicated in Table I.
Poly(ADP-ribose) polymerase (PARP) is an enzyme
involved in the recovery of cells from DNA damage and the
regulation of the molecular events such as BER, a key pathway in
the repair of DNA single-strand breaks (SSB). The inhibition of
PARP leads to the induction of synthetic lethality and cell death
by targeting HR-mediated DNA repair deficient tumors (20). Tumors that lack functional BRCA1,
BRCA2, or TP53 are hypersensitive to inhibition of PARP. Several
proteins involved in HR on sensitivity to PARP inhibition may
include BRCA cluster (RAD51C, RAD51D and RAD54), FA cluster
(FANCD2, FANCA and FANCC), Cdk cluster, nucleotide excision repair
(NER) cluster (RPA1 and NBN), DNA repair checkpoint cluster (ATR,
ATM, CHK1 and CHK2) and TP53 cluster. Therefore, therapeutic
approach using PARP inhibitors may be feasible for BRCA
dysregulated tumors and appear promising in a variety of cancer
types, including breast and ovarian cancers. The presence of these
germline mutations and epimutations types might be a hallmark of
BRCAness and a potential biomarker for sensitivity to PARP
inhibition.
There are two types of DNA repair proteins: the
nucleotide excision repair (NER) pathway (ERCC1, excision repair
cross-complementing rodent repair deficiency, complementation group
1) and the base excision repair (BER) pathway (XRCC1, X-ray repair
complementing defective repair in Chinese hamster cells 1)
(23). The NER pathway is
responsible for the removal of bulky DNA lesions. NER is a defense
system against various types of DNA damage and necessary for
maintaining genomic stability. RPA1, replication protein A1, is a
single-stranded DNA binding protein and participates in the
recruitment of the two structure-specific DNA endonucleases, XPG
(xeroderma pigmentosum, also known as ERCC5) and XPF (xeroderma
pigmentosum, complementation group F)-ERCC1 complex, which makes
the 5′ incision in NER. The XPF-ERCC1 complex is essential for
cutting the damaged DNA strand and the DNA repair by the NER
pathway. NBN (nibrin, also known as NBS1) was involved in DNA DSB
repair and DNA damage-induced checkpoint activation as a component
of the MRE11-RAD50-NBS1 complex. Mutations in NBN is thought to be
associated with breast-cancer risk.
Between 50–80% of HBOC syndrome can be explained by
defective germline mutations in BRCA1 and BRCA2 as well as, to a
lesser degree, other genes described above, but for the remaining
families the factors driving susceptibility remain unknown
(24). Approximately one third of
the HBOC families do not have evidence of the germline mutations in
BRCA1 and BRCA2. The loss of BRCA function might be due to either
germline/somatic mutation or epigenetic silencing. Since little is
known about the contribution of epimutations to the remaining
BRCA1/2 mutation-negative cases, epigenetic silencing has been
explored in HBOC syndrome. The activities of tumor suppressor genes
and cancer susceptibility genes could be influenced by genetic and
epigenetic alterations. Decreased expression of cancer
susceptibility genes has been observed in sporadic breast and
ovarian cancer where it is often associated with the aberrant
epimutations or hypermethylation of the BRCA1 and BRCA2 genes. The
loss of BRCA1 function due to somatic hypermethylation explained
~10% of sporadic breast cancer cases. A subset of the sporadic
tumor patients demonstrated hypermethylation of BRCA2 and their
interacting protein including HP1γ (heterochromatin protein
1gamma), RAD51C, ATM and PALB2 (25). Based on BRCA1 deletion, TP53
mutations, ER- and PgR-negative status, young age at diagnosis and
high grade tumor, phenotypic features of sporadic breast cancers
resemble BRCA1 mutated cancers termed ‘BRCAness’. Phenotypic
similarities were most closely observed in BRCAness, epigenetic
silencing and deletion of the BRCA1 and BRCA2 genes.
Many genes have been implicated in the DNA damage
response pathways where the BRCA1 and BRCA2 genes are involved.
Genetic susceptibility to cancer is attributed to deleterious
germline mutations in the DNA mismatch repair (MMR) genes. Another
important non-BRCA1/BRCA2 hereditary condition is hereditary
non-polyposis colorectal cancer (HNPCC) syndrome, also known as
‘Lynch syndrome’. Two manifestations of hereditary ovarian cancer
are currently recognized: the HBOC syndrome and the HNPCC syndrome.
Lynch syndrome has been defined clinically and genetically and is
an autosomal-dominant cancer predisposition syndrome that increases
the risk for several forms of malignancy, including colorectal
(lifetime cancer risk, 70–80%), endometrial (50–60%), stomach
cancer (13–19%), ovarian cancer (9–14%), small intestine, liver and
biliary tract, brain, as well as transitional cell carcinoma of the
ureters and renal pelvis. Mutations in four MMR genes [MLH1 (mutL
homolog 1), MSH2 (mutS homolog 2), MSH6 (mutS homolog 6) and PMS2
(postmeiotic segregation increased 2)] are associated with Lynch
syndrome and account for another 10% of hereditary ovarian cancer
(26). The MMR genes encode
proteins involved in the same pathway of DNA mismatch repair. These
genetic defects in the DNA MMR system result in DNA replication
errors, including base substitutions and insertion-deletion loops,
known as microsatellite instability (MSI). MutS α complex (composed
of MSH2 and MSH6) or MutS β complex (MSH2–MSH3) recognizes
single-base mismatches and small insertion-deletion loops, binds to
mismatched DNA, and recruits the MutL α complex (MLH1–PMS2), which
leads to strand discrimination and removal of the errors and
coordinates the remaining steps in MMR (27–29).
Both HNPCC and HBOC associated ovarian cancer
develop along distinct genetic pathways (30). Although some deviating reports
exist, breast cancer incidence has been found to be elevated in
Lynch syndrome patients (31).
Furthermore, germline mutations associated with Lynch syndrome has
been described in 2–3% of patients diagnosed with endometrial
cancer. Among Lynch syndrome-related cancers, endometrial cancer is
riskier than colorectal cancer in terms of estimated lifetime
cumulative risk (32). The overall
5-year survival rate for endometrial (88 vs. 82%) or ovarian cancer
(64 vs. 58%) was not significantly different between patients with
endometrial or ovarian cancer that are associated with Lynch
syndrome and the controls with sporadic cases (33,34).
Although immunohistochemical analysis of tumor tissue proved to be
a good pre-screening test before proceeding to germline mutation
analysis, the discordant results are sometimes observed between
immunohistochemistry and replication error phenotyping.
Notwithstanding this limitation, immunohistochemistry may be
helpful in the evaluation of women with a likely diagnosis of Lynch
syndrome. Immunohistochemistry for DNA MMR showed loss of proteins
in 9% of women with synchronous endometrial and ovarian cancer
(35).
Attention has been paid to the role of modifiable
risk factors like reproductive histories and exogenous hormones.
Potential modifying factors include age of menarche, parity,
breastfeeding and oophorectomy. Oral contraceptives (OCs) have a
significant protective effect on the risk of ovarian cancer by ~50%
in the general population (28).
OCs reduce the risk of ovarian cancer also in BRCA1/BRCA2 mutation
carriers. The effect of parity may be different in BRCA1 and BRCA2
carriers. Parity protects against breast cancer in BRCA1 mutation
carriers. Multiparity may be associated with an increase in risk in
BRCA2 carriers. However, the effect of multiparity on ovarian
cancer risk for BRCA2 mutation carriers has only been investigated
in a small number of studies. Therefore, the association is
controversial. Women who took OCs before the age of 30 years and
long-term user of 5 or more years have been associated with a
slight increase in risk of breast cancer among BRCA1 mutation
carriers (29). Furthermore,
prolonged hormone replacement therapy (HRT) use is an established
risk factor of breast cancer. HRT also affects ovarian cancer risk.
Among a subgroup of patients who had a familial history of breast
cancer, tamoxifen and raloxifene, selective estrogen receptor
modulators, reduced breast cancer risk. Tamoxifen and raloxifene
reduced the risk of invasive breast cancer: compared with placebo,
raloxifene reduced breast cancer risk by 38%, tamoxifen showed a
50% reduction (37). Additionally,
use of PARP inhibitors is a potential synthetic lethal therapeutic
strategy and may be considered as targeted chemoprevention in
patients with specific DNA-repair defects. Other agents under
preclinical and clinical investigation include cyclooxygenase-2
inhibitors, aromatase inhibitors, tyrosine kinase inhibitors, and
difluoromethylornithine (a polyamine inhibitor) (38,39).
Future efficacy studies are expected.
Prophylactic surgeries are appropriate treatment
options for BRCA mutation-associated cancer (4). Prophylactic bilateral mastectomy
reduces the risk of breast cancer in BRCA mutation carriers by 90%
at any age. Prophylactic bilateral salpingo-oophorectomy (BSO)
lowers the ovarian cancer risk by 80%. BSO performed before age 50
years also exhibits a 50% reduction in subsequent breast cancer
risk. In conclusion, prophylactic surgeries lead to a reduction in
breast and ovarian cancer-specific mortality. Knowledge of these
risk factors and prevention strategies will have a great impact on
the management of hereditary breast and ovarian cancers.
Since identification of the mutation screening is
currently labor intensive and expensive, the screening should be
directed to asymptomatic individuals only if they belong to
high-risk families. Various safe and effective screening protocols
have been recommended for early cancer detection and reduction of
cancer risk in clinical practice. Women with HBOC syndrome often
utilize the latest medical advances in increased surveillance,
prevention, early detection, chemoprevention and optimal treatment.
Among BRCA1 and BRCA2 mutation carriers, use of screening
mammography alone led to increased early detection rates of
non-palpable breast cancer, but the rate of interval cancers was
high (7,40). Therefore, the effectiveness of
mammography alone (sensitivity 40%) is questionable for screening
high-risk women. In the high risk group of women, magnetic
resonance imaging (unenhanced MR imaging with combined
diffusion-weighted and T2-weighted images, sensitivity 50%)
surveillance largely out performed mammography. Furthermore,
dynamic contrast-enhanced MRI exhibited highest sensitivity (86%)
(41). MRI is a better screening
method and will detect the majority of breast cancers at an early
stage. The addition of MRI to screening mammography increased
sensitivity (42), supporting the
benefits of breast MRI examination annually (the sensitivity was
80%, the false positive rate was 10%) in BRCA mutation carriers
(43). Alternating MRI and
mammography screening at 6-month intervals might be a clinically
effective approach. The HBOC carriers have two options to reduce
their risk of ovarian cancer: periodic screening and risk-reducing
surgeries (44). Periodic screening
consists of annual or semi-annual pelvic examination with the
longitudinal CA125 blood test plus concurrent transvaginal
ultrasound. Unfortunately, to date, this screening regimen is
ineffective for early detection of ovarian cancer in high-risk
women. Risk-reducing salpingo-oophorectomy represents a potentially
valuable intervention and is the only way for many women at
high-risk by age 40 years, or on completion of childbearing.
Preventive surgery can reduce ovarian cancer risk by 80–90% and
breast cancer risk by 50–60% in BRCA mutation carriers.
HBOC syndrome is the inherited tendency to develop
breast, ovarian and other cancers and believed to be transmitted by
mutations in the specific genes. Clinical characteristics,
including the type of tumor and age at occurrence as well as family
history, predict the prevalence of BRCA germline mutations. A
number of clinicians usually take into account the age of the
youngest breast cancer patient and the number of ovarian cancer
cases in a family as well as pathological diagnosis. Up to 80% of
the HBOC cases are due to mutations in BRCA1 or BRCA2 genes. Both
BRCA1 and BRCA2 mutations are scattered throughout the whole coding
exons.
To maintain and restore the genomic integrity,
normal cells possess DNA repair mechanisms. The structural
modifications, such as DNA base damage, DNA strand break, inter-
and intra-strand crosslinks and DNA-protein crosslinks, are
involved in mutation and cancer. A variety of intelligent
mechanisms can activate DNA repair pathways and cell cycle
checkpoints and recognize and repair SSB or DSB by the master
sensors and regulators of DNA damage response such as BRCA1 and
BRCA2. BRCA1 and BRCA2 genes were recruited to the sites of DNA
damage. BRCA1 associates with several proteins and is an integral
member of the repair of DNA damage by functional HR, NER and
possibly NHEJ. BRCA1 activated by cdk1 physically interacts with
MutL α, through the interaction of the FA-BRCA pathway (45). BRCA2, also known as FANCD1, has a
more specific role in DNA repair and is directly involved in the
mechanism of HR, regulating the activity of RAD51, a gene
implicated in the HR pathway as well as interacting with PALB2, a
gene implicated in the HR repair and checkpoint functions.
Interestingly, the nuclear function of BRCA proteins is tightly
regulated by the FA-BRCA, MMR, BER and NER pathway. However, the
exact mechanisms of the BRCA-associated interaction and
accumulation of other DNA repair proteins are not comprehensively
known. Therefore, germline mutations in other susceptibility genes,
such as FA genes, MMR genes and DNA repair genes, might be the
predisposing factors in HBOC cases. These predisposing genes encode
for upstream and downstream regulators of BRCA gene products and
also may be associated with the BRCA core complex, including
mutations in FANCD1, FANCD2, FANCJ, FANCP, TP53, PTEN, STK11, CDH1,
CHK2, ATM, ATR, MSH1, M SH2, MLH1 and PMS2 genes. Families affected
by other syndromes, such as Lynch syndrome (mutations in MMR
genes), Fanconi anemia, Cowden syndrome (mutations in PTEN),
Li-Fraumeni syndrome (mutations in TP53), xeroderma pigmentosum and
ataxia-telangiectasia, exhibit additional types of cancers outside
the previously defined HBOC cancer spectrum.
In conclusion, genetic or epigenetic
loss-of-function mutations of genes that are known to be involved
in the repair of DNA damage might lead to increased risk of
developing a broad spectrum of breast and ovarian cancers.
Th present review was supported by grant-in-aid for
Scientific Research from the Ministry of Education, Science, and
Culture of Japan to the Department of Obstetrics and Gynecology,
Nara Medical University (to H.K.).
|
1
|
Lynch HT, Lynch J, Conway T, Watson P,
Feunteun J, Lenoir G, Narod S and Fitzgibbons R Jr: Hereditary
breast cancer and family cancer syndromes. World J Surg. 18:21–31.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ford D, Easton DF, Bishop DT, Narod SA and
Goldgar DE: Risks of cancer in BRCA1-mutation carriers. Breast
Cancer Linkage Consortium. Lancet. 343:692–695. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sokolenko AP, Iyevleva AG, Mitiushkina NV,
Suspitsin EN, Preobrazhenskaya EV, Kuligina ESh, Voskresenskiy DA,
Lobeiko OS, Krylova NY, Gorodnova TV, Buslov KG, Bit-Sava EM,
Dolmatov GD, Porhanova NV, Polyakov IS, Abysheva SN, Katanugina AS,
Baholdin DV, Yanus GA, Togo AV, Moiseyenko VM, Maximov SY,
Semiglazov VF and Imyanitov EN: Hereditary breast-ovarian cancer
syndrome in Russia. Acta Nat. 2:31–35. 2010.PubMed/NCBI
|
|
4
|
Smith KL and Isaacs C: BRCA mutation
testing in determining breast cancer therapy. Cancer J. 17:492–499.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ruan Y, Song AP, Wang H, Xie YT, Han JY,
Sajdik C, Tian XX and Fang WG: Genetic polymorphisms in
AURKA and BRCA1 are associated with breast cancer
susceptibility in a Chinese Han population. J Pathol. 225:535–543.
2011.
|
|
6
|
Zhang B, Beeghly-Fadiel A, Long J and
Zheng W: Genetic variants associated with breast-cancer risk:
comprehensive research synopsis, meta-analysis, and epidemiological
evidence. Lancet Oncol. 12:477–488. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Narod SA and Salmena L: BRCA1 and BRCA2
mutations and breast cancer. Discov Med. 12:445–453.
2011.PubMed/NCBI
|
|
8
|
Vasickova P, Machackova E, Lukesova M,
Damborsky J, Horky O, Pavlu H, Kuklova J, Kosinova V, Navratilova M
and Foretova L: High occurrence of BRCA1 intragenic
rearrangements in hereditary breast and ovarian cancer syndrome in
the Czech Republic. BMC Med Genet. 8:322007.
|
|
9
|
Walsh T and King MC: Ten genes for
inherited breast cancer. Cancer Cell. 11:103–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Friedenson B: The BRCA1/2 pathway prevents
hematologic cancers in addition to breast and ovarian cancers. BMC
Cancer. 7:1522007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cortesi L, Masini C, Cirilli C, Medici V,
Marchi I, Cavazzini G, Pasini G, Turchetti D and Federico M:
Favourable ten-year overall survival in a Caucasian population with
high probability of hereditary breast cancer. BMC Cancer.
10:902010.PubMed/NCBI
|
|
12
|
Bolton KL, Chenevix-Trench G, Goh C,
Sadetzki S, Ramus SJ, Karlan BY, Lambrechts D, Despierre E,
Barrowdale D, McGuffog L, Healey S, Easton DF, Sinilnikova O,
Benítez J, García MJ, Neuhausen S, Gail MH, Hartge P, Peock S,
Frost D, Evans DG, Eeles R, Godwin AK, Daly MB, Kwong A, Ma ES,
Lázaro C, Blanco I, Montagna M, D'Andrea E, Nicoletto MO, Johnatty
SE, Kjær SK, Jensen A, Høgdall E, Goode EL, Fridley BL, Loud JT,
Greene MH, Mai PL, Chetrit A, Lubin F, Hirsh-Yechezkel G, Glendon
G, Andrulis IL, Toland AE, Senter L, Gore ME, Gourley C, Michie CO,
Song H, Tyrer J, Whittemore AS, McGuire V, Sieh W, Kristoffersson
U, Olsson H, Borg Å, Levine DA, Steele L, Beattie MS, Chan S,
Nussbaum RL, Moysich KB, Gross J, Cass I, Walsh C, Li AJ, Leuchter
R, Gordon O, Garcia-Closas M, Gayther SA, Chanock SJ, Antoniou AC
and Pharoah PD; EMBRACE; kConFab Investigators; Cancer Genome Atlas
Research Network. Association between BRCA1 and BRCA2
mutations and survival in women with invasive epithelial ovarian
cancer. JAMA. 307:382–390. 2012.
|
|
13
|
Dillenburg CV, Bandeira IC, Tubino TV,
Rossato LG, Dias ES, Bittelbrunn AC and Leistner-Segal S:
Prevalence of 185delAG and 5382insC mutations in BRCA1, and
6174delT in BRCA2 in women of Ashkenazi Jewish origin in
southern Brazil. Genet Mol Biol. 35:599–602. 2012.PubMed/NCBI
|
|
14
|
Ewald IP, Ribeiro PL, Palmero EI, Cossio
SL, Giugliani R and Ashton-Prolla P: Genomic rearrangements in
BRCA1 and BRCA2: a literature review. Genet Mol Biol.
32:437–446. 2009.
|
|
15
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gonzalez-Angulo AM, Timms KM, Liu S, Chen
H, Litton JK, Potter J, Lanchbury JS, Stemke-Hale K, Hennessy BT,
Arun BK, Hortobagyi GN, Do KA, Mills GB and Meric-Bernstam F:
Incidence and outcome of BRCA mutations in unselected
patients with triple receptor-negative breast cancer. Clin Cancer
Res. 17:1082–1089. 2011.
|
|
17
|
Young SR, Pilarski RT, Donenberg T,
Shapiro C, Hammond LS, Miller J, Brooks KA, Cohen S, Tenenholz B,
Desai D, Zandvakili I, Royer R, Li S and Narod SA: The prevalence
of BRCA1 mutations among young women with triple-negative breast
cancer. BMC Cancer. 9:862009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Holstege H, Joosse SA, van Oostrom CT,
Nederlof PM, de Vries A and Jonkers J: High incidence of
protein-truncating TP53 mutations in BRCA1-related breast
cancer. Cancer Res. 69:3625–3633. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yates MS, Meyer LA, Deavers MT, Daniels
MS, Keeler ER, Mok SC, Gershenson DM and Lu KH: Microscopic and
early-stage ovarian cancers in BRCA1/2 mutation carriers:
building a model for early BRCA-associated tumorigenesis. Cancer
Prev Res. 4:463–470. 2011.PubMed/NCBI
|
|
20
|
Tinker AV and Gelmon K: The role of PARP
inhibitors in the treatment of ovarian carcinomas. Curr Pharm Des.
18:3770–3774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaye SB, Lubinski J, Matulonis U, Ang JE,
Gourley C, Karlan BY, Amnon A, Bell-McGuinn KM, Chen LM,
Friedlander M, Safra T, Vergote I, Wickens M, Lowe ES, Carmichael J
and Kaufman B: Phase II, open-label, randomized, multicenter study
comparing the efficacy and safety of olaparib, a poly (ADP-ribose)
polymerase inhibitor, and pegylated liposomal doxorubicin in
patients with BRCA1 or BRCA2 mutations and recurrent ovarian
cancer. J Clin Oncol. 30:372–379. 2012. View Article : Google Scholar
|
|
22
|
Ledermann J, Harter P, Gourley C,
Friedlander M, Vergote I, Rustin G, Scott C, Meier W,
Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C,
Carmichael J and Matulonis U: Olaparib maintenance therapy in
platinum-sensitive relapsed ovarian cancer. N Engl J Med.
366:1382–1392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vaezi A, Feldman CH and Niedernhofer LJ:
ERCC1 and XRCC1 as biomarkers for lung and head and
neck cancer. Pharmgenomics Pers Med. 4:47–63. 2011.
|
|
24
|
Chen Y, Toland AE, McLennan J, Fridlyand
J, Crawford B, Costello JF and Ziegler JL: Lack of germ-line
promoter methylation in BRCA1-negative families with
familial breast cancer. Genet Test. 10:281–284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Potapova A, Hoffman AM, Godwin AK,
Al-Saleem T and Cairns P: Promoter hypermethylation of the
PALB2 susceptibility gene in inherited and sporadic breast
and ovarian cancer. Cancer Res. 68:998–1002. 2008.
|
|
26
|
Lynch HT, Casey MJ, Snyder CL, Bewtra C,
Lynch JF, Butts M and Godwin AK: Hereditary ovarian carcinoma:
heterogeneity, molecular genetics, pathology, and management. Mol
Oncol. 3:97–137. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vogel VG, Costantino JP, Wickerham DL,
Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon
ER Jr, Wade JL III, Robidoux A, Margolese RG, James J, Lippman SM,
Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan
VC and Wolmark N; National Surgical Adjuvant Breast and Bowel
Project (NSABP). Effects of tamoxifen vs raloxifene on the risk of
developing invasive breast cancer and other disease outcomes: the
NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA.
295:2727–2741. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McLaughlin JR, Risch HA, Lubinski J,
Moller P, Ghadirian P, Lynch H, Karlan B, Fishman D, Rosen B,
Neuhausen SL, Offit K, Kauff N, Domchek S, Tung N, Friedman E,
Foulkes W, Sun P and Narod SA: Hereditary Ovarian Cancer Clinical
Study Group: reproductive risk factors for ovarian cancer in
carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet
Oncol. 8:26–34. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Milne RL, Knight JA, John EM, Dite GS,
Balbuena R, Ziogas A, Andrulis IL, West DW, Li FP, Southey MC,
Giles GG, McCredie MR, Hopper JL and Whittemore AS: Oral
contraceptive use and risk of early-onset breast cancer in carriers
and noncarriers of BRCA1 and BRCA2 mutations. Cancer
Epidemiol Biomarkers Prev. 14:350–356. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Domanska K, Malander S, Staaf J, Karlsson
A, Borg A, Jönsson G and Nilbert M: Genetic profiles distinguish
different types of hereditary ovarian cancer. Oncol Rep.
24:885–895. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lotsari JE, Gylling A, Abdel-Rahman WM,
Nieminen TT, Aittomäki K, Friman M, Pitkänen R, Aarnio M, Järvinen
HJ, Mecklin JP, Kuopio T and Peltomäki P: Breast carcinoma and
Lynch syndrome: molecular analysis of tumors arising in mutation
carriers, non-carriers, and sporadic cases. Breast Cancer Res.
14:R902012. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meyer LA, Broaddus RR and Lu KH:
Endometrial cancer and Lynch syndrome: clinical and pathologic
considerations. Cancer Control. 16:14–22. 2009.PubMed/NCBI
|
|
33
|
Masuda K, Banno K, Yanokura M, Kobayashi
Y, Kisu I, Ueki A, Ono A, Nomura H, Hirasawa A, Susumu N and Aoki
D: Carcinoma of the lower uterine segment (LUS):
clinicopathological characteristics and association with lynch
syndrome. Curr Genomics. 12:25–29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Crijnen TE, Janssen-Heijnen ML, Gelderblom
H, Morreau J, Nooij MA, Kenter GG and Vasen HF: Survival of
patients with ovarian cancer due to a mismatch repair defect. Fam
Cancer. 4:301–305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim MK, Song SY, Do IG, Kim SH, Choi CH,
Kim TJ, Lee JW, Bae DS and Kim BG: Synchronous gynecologic
malignancy and preliminary results of Lynch syndrome. J Gynecol
Oncol. 22:233–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Williams SA, Wilson JB, Clark AP,
Mitson-Salazar A, Tomashevski A, Ananth S, Glazer PM, Semmes OJ,
Bale AE, Jones NJ and Kupfer GM: Functional and physical
interaction between the mismatch repair and FA-BRCA pathways. Hum
Mol Genet. 20:4395–4410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vogel VG, Costantino JP, Wickerham DL,
Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon
ER, Wade JL III, Robidoux A, Margolese RG, James J, Runowicz CD,
Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC and
Wolmark N: National Surgical Adjuvant Breast and Bowel Project:
Update of the National Surgical Adjuvant Breast and Bowel Project
Study of tamoxifen and raloxifene (STAR) P-2 trial: preventing
breast cancer. Cancer Prev Res. 3:696–706. 2010. View Article : Google Scholar
|
|
38
|
Burga LN, Hu H, Juvekar A, Tung NM, Troyan
SL, Hofstatter EW and Wulf GM: Loss of BRCA1 leads to an
increase in epidermal growth factor receptor expression in mammary
epithelial cells, and epidermal growth factor receptor inhibition
prevents estrogen receptor-negative cancers in BRCA1-mutant
mice. Breast Cancer Res. 13:R302011.PubMed/NCBI
|
|
39
|
Uray IP and Brown PH: Chemoprevention of
hormone receptor-negative breast cancer: new approaches needed.
Recent Results Cancer Res. 188:147–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saito M, Matsuzaki M, Sakuma T, Katagata
N, Watanabe F, Yamaguchi Y, Schetter AJ, Takenoshita S and Nomizu
T: Clinicopathological study of non-palpable familial breast cancer
detected by screening mammography and diagnosed as DCIS. Breast
Cancer. Aug 9–2012.(Epub ahead of print).
|
|
41
|
Yabuuchi H, Matsuo Y, Sunami S, Kamitani
T, Kawanami S, Setoguchi T, Sakai S, Hatakenaka M, Kubo M, Tokunaga
E, Yamamoto H and Honda H: Detection of non-palpable breast cancer
in asymptomatic women by using unenhanced diffusion-weighted and
T2-weighted MR imaging: comparison with mammography and dynamic
contrast-enhanced MR imaging. Eur Radiol. 21:11–17. 2011.
View Article : Google Scholar
|
|
42
|
Passaperuma K, Warner E, Causer PA, Hill
KA, Messner S, Wong JW, Jong RA, Wright FC, Yaffe MJ, Ramsay EA,
Balasingham S, Verity L, Eisen A, Curpen B, Shumak R, Plewes DB and
Narod SA: Long-term results of screening with magnetic resonance
imaging in women with BRCA mutations. Br J Cancer. 107:24–30. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Warner E, Hill K, Causer P, Plewes D, Jong
R, Yaffe M, Foulkes WD, Ghadirian P, Lynch H, Couch F, Wong J,
Wright F, Sun P and Narod SA: Prospective study of breast cancer
incidence in women with a BRCA1 or BRCA2 mutation
under surveillance with and without magnetic resonance imaging. J
Clin Oncol. 29:1664–1669. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Westin SN, Sun CC, Lu KH, Schmeler KM,
Soliman PT, Lacour RA, Johnson KG, Daniels MS, Arun BK, Peterson SK
and Bodurka DC: Satisfaction with ovarian carcinoma risk-reduction
strategies among women at high risk for breast and ovarian
carcinoma. Cancer. 117:2659–2667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dohrn L, Salles D, Siehler SY, Kaufmann J
and Wiesmüller L: BRCA1-mediated repression of mutagenic
end-joining of DNA double-strand breaks requires complex formation
with BACH1. Biochem J. 441:919–926. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Corso G, Marrelli D, Pascale V, Vindigni C
and Roviello F: Frequency of CDH1 germline mutations in gastric
carcinoma coming from high- and low-risk areas: metanalysis and
systematic review of the literature. BMC Cancer. 12:82012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chun N and Ford JM: Genetic testing by
cancer site: stomach. Cancer J. 18:355–363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lytras A and Tolis G: Reproductive
disturbances in multiple neuroendocrine tumor syndromes. Endocr
Relat Cancer. 16:1125–1138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Melbārde-Gorkuša I, Irmejs A, Bērzin̦a D,
Strumfa I, Abolin̦š A, Gardovskis A, Subatniece S, Trofimovičs G,
Gardovskis J and Miklaševičs E: Challenges in the management of a
patient with Cowden syndrome: case report and literature review.
Hered Cancer Clin Pract. 10:52012.PubMed/NCBI
|
|
50
|
Buller RE, Lallas TA, Shahin MS, Sood AK,
Hatterman-Zogg M, Anderson B, Sorosky JI and Kirby PA: The
p53 mutational spectrum associated with BRCA1 mutant
ovarian cancer. Clin Cancer Res. 7:831–838. 2001.
|
|
51
|
Meng AG and Jiang LL: Induction of G2/M
arrest by pseudolaric acid B is mediated by activation of the ATM
signaling pathway. Acta Pharmacol Sin. 30:442–450. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lindeman GJ, Hiew M, Visvader JE, Leary J,
Field M, Gaff CL, Gardner RJ, Trainor K, Cheetham G, Suthers G and
Kirk J: Frequency of the ATM IVS10-6T>G variant in Australian
multiple-case breast cancer families. Breast Cancer Res.
6:R401–R407. 2004. View
Article : Google Scholar
|
|
53
|
Wang X, Kennedy RD, Ray K, Stuckert P,
Ellenberger T and D'Andrea AD: Chk1-mediated phosphorylation of
FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell
Biol. 27:3098–3108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kuusisto KM, Bebel A, Vihinen M,
Schleutker J and Sallinen SL: Screening for BRCA1,
BRCA2, CHEK2, PALB2, BRIP1,
RAD50, and CDH1 mutations in high-risk Finnish
BRCA1/2-founder mutation-negative breast and/or ovarian
cancer individuals. Breast Cancer Res. 13:R202011.PubMed/NCBI
|
|
55
|
de Garibay GR, Díaz A, Gaviña B, Romero A,
Garre P, Vega A, Blanco A, Tosar A, Díez O, Pérez-Segura P,
Díaz-Rubio E, Caldés T and de la Hoya M: Low prevalence of SLX4
loss-of-function mutations in non-BRCA1/2 breast and/or ovarian
cancer families. Eur J Hum Genet. Dec 5–2012.(Epub ahead of
print).
|
|
56
|
Seal S, Thompson D, Renwick A, Elliott A,
Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K,
North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR and
Rahman N; Breast Cancer Susceptibility Collaboration (UK).
Truncating mutations in the Fanconi anemia J gene BRIP1 are
low-penetrance breast cancer susceptibility alleles. Nat Genet.
38:1239–1241. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sy SM, Huen MS and Chen J: PALB2 is an
integral component of the BRCA complex required for homologous
recombination repair. Proc Natl Acad Sci USA. 106:7155–7160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Willers H, Kachnic LA, Luo CM, Li L,
Purschke M, Borgmann K, Held KD and Powell SN: Biomarkers and
mechanisms of FANCD2 function. J Biomed Biotechnol.
2008:8215292008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Johnson N, Li YC, Walton ZE, Cheng KA, Li
D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD, Newell DR,
D'Andrea AD, Curtin NJ, Wong KK and Shapiro GI: Compromised CDK1
activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat
Med. 17:875–882. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Clague J, Wilhoite G, Adamson A, Bailis A,
Weitzel JN and Neuhausen SL: RAD51C germline mutations in
breast and ovarian cancer cases from high-risk families. PLoS One.
6:e256322011. View Article : Google Scholar
|
|
61
|
Hsu HM, Wang HC, Chen ST, Hsu GC, Shen CY
and Yu JC: Breast cancer risk is associated with the genes encoding
the DNA double-strand break repair Mre11/Rad50/Nbs1 complex. Cancer
Epidemiol Biomarkers Prev. 16:2024–2032. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Meindl A, Hellebrand H, Wiek C, Erven V,
Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L,
Schaal H, Ramser J, Honisch E, Kubisch C, Wichmann HE, Kast K,
Deissler H, Engel C, Muller-Myhsok B, Neveling K, Kiechle M, Mathew
CG, Schindler D, Schmutzler RK and Hanenberg H: Germline mutations
in breast and ovarian cancer pedigrees establish RAD51C as a human
cancer susceptibility gene. Nat Genet. 42:410–414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pelttari LM, Heikkinen T, Thompson D,
Kallioniemi A, Schleutker J, Holli K, Blomqvist C, Aittomäki K,
Bützow R and Nevanlinna H: RAD51C is a susceptibility gene
for ovarian cancer. Hum Mol Genet. 20:3278–3288. 2011. View Article : Google Scholar
|
|
64
|
Esteban Cardeñosa E, de Juan Jiménez I,
Palanca Suela S, Chirivella González I, Segura Huerta A, Santaballa
Beltran A, Casals El Busto M, Barragán González E, Fuster Lluch O,
Bermúdez Edo J and Bolufer Gilabert P: Low penetrance alleles as
risk modifiers in familial and sporadic breast cancer. Fam Cancer.
11:629–636. 2012.PubMed/NCBI
|
|
65
|
Jakubowska A, Gronwald J, Menkiszak J,
Górski B, Huzarski T, Byrski T, Edler L, Lubiński J, Scott RJ and
Hamann U: The VEGF_936_C>T 3′UTR polymorphism reduces
BRCA1-associated breast cancer risk in Polish women. Cancer
Lett. 262:71–76. 2008.
|
|
66
|
Runnebaum IB, Wang-Gohrke S, Vesprini D,
Kreienberg R, Lynch H, Moslehi R, Ghadirian P, Weber B, Godwin AK,
Risch H, Garber J, Lerman C, Olopade OI, Foulkes WD, Karlan B,
Warner E, Rosen B, Rebbeck T, Tonin P, Dubé MP, Kieback DG and
Narod SA: Progesterone receptor variant increases ovarian cancer
risk in BRCA1 and BRCA2 mutation carriers who were never exposed to
oral contraceptives. Pharmacogenetics. 11:635–638. 2001. View Article : Google Scholar
|
|
67
|
Ratner E, Lu L, Boeke M, Barnett R, Nallur
S, Chin LJ, Pelletier C, Blitzblau R, Tassi R, Paranjape T, Hui P,
Godwin AK, Yu H, Risch H, Rutherford T, Schwartz P, Santin A,
Matloff E, Zelterman D, Slack FJ and Weidhaas JB: A
KRAS-variant in ovarian cancer acts as a genetic marker of
cancer risk. Cancer Res. 70:6509–6515. 2010.
|
|
68
|
Ripperger T, Gadzicki D, Meindl A and
Schlegelberger B: Breast cancer susceptibility: current knowledge
and implications for genetic counselling. Eur J Hum Genet.
17:722–731. 2009. View Article : Google Scholar : PubMed/NCBI
|