Introduction
According to the 2011 annual report on the global
burden of cancer, 1,600,000 new cases of lung cancer were noted in
2008, which accounted for 13% of the total cancer cases worldwide,
and there were 1,400,000 lung cancer-related deaths during the same
period, which accounted for 18% of all cancer-related mortality
making lung cancer the leading cause of cancer-related death
worldwide (1). Lung cancer has a
very poor prognosis compared with other cancer types, indicating
that there is an urgent need for new therapies for non-small cell
lung cancer (NSCLC) by elucidating the mechanisms of its growth and
progression.
The single-pass heterodimeric transmembrane receptor
protein, Notch1, localizes on the cell surface and mediates
cell-to-cell interactions. When the extracellular domain of Notch1
dissociates from the heterodimerization domain through interaction
with ligand cells, the Notch1 receptor undergoes sequential
proteolytic cleavage and the Notch1 intracellular domain (N1ICD) is
released and translocates to the nucleus where it activates
transcription of target genes (2).
During the neoplastic process, the tumor microenvironment becomes
more solid and hypoxic resulting in intensified interactions
between cells, which eventually generate conditions with amplified
Notch1 signaling (3,4).
Although the oncogenic role of Notch1 has been
established in T-acute lymphoblastic leukemia, its roles in other
neoplastic diseases are controversial (5). In T-cells, N1ICD induces the
expression of cyclin D3, CDK4 and CDK6, leading to G1-S cell cycle
phase progression (6). Other
authors have reported that Notch1 signaling inhibits B lymphocyte
growth (7), small cell lung cancer
(8) and hepatocellular carcinoma
(9). Since deregulated Notch1
signaling is common in NSCLC (10,11)
and several γ-secretase inhibitors (GSIs) are under development for
various clinical applications, clarification of its role is
required.
Among the intercellular junctional complexes which
maintain epithelial integrity and polarity, adherens junctions are
made up of homophillic interactions between E-cadherin molecules
and α-catenin, β-catenin and δ1-catenin (p-120), which are
connected to the actin cytoskeletal network. Interaction between
E-cadherin and β-catenin stabilizes cell-to-cell contacts and
maintains adherens junctions, and is important for cell-to-cell
interaction-mediated signaling (12). Destabilization of adherens junctions
facilitates endocytosis of E-cadherin and changes in the levels of
β-catenin in various cellular compartments. Disruption of
E-cadherin/β-catenin complexes also decreases intercellular
adhesion and further increases cell migration and invasiveness
(13,14). Physical interaction of Notch1 with
β-catenin through the RAM domain of Notch1 suggests another role of
Notch1 in maintaining the stability of adherens junctions (15). By elucidating the function of Notch1
in adherens junctions, the context-dependent role of Notch1 may
become clear.
In the present study, we investigated the role of
Notch1 in adherens junction stability and NSCLC cell growth.
Overexpression of N1ICD decreased E-cadherin through upregulation
of the snail family of transcription repressors and it resulted in
the alteration of total and active β-catenin expression. We found
that Notch1 and β-catenin interact independently to E-cadherin
expression and that Notch1 deregulates Wnt/β-catenin signaling. Our
results suggest that Notch1 is an important component involved in
the physical and signaling homeostasis of adherens junctions.
Materials and methods
Plasmids, antibodies and cell cycle
analysis
A549 cells were purchased from ATCC (Manassas, VA,
USA). H460, H596 and H1650 cells were obtained from the Korean Cell
Line Bank (Seoul, Korea). pMSCV-myc-N1ICD and -control vectors were
obtained from Dr G. Jung (Seoul National University) and transduced
as previously described (5).
pCS2-ICV-6mt and pCS2-ΔEMV-6mt were gifts from Dr R. Kopan
(http://devbio.wustl.edu/kopannulab/plasmids.htm)
(16). Anti-Notch1 (C-20)-R and
(C-10), -HES1, -HERP, -c-Myc were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA), and antibodies, unless
otherwise stated, were obtained from Cell Signaling Technology
(Danvers, MA, USA). For cell cycle analysis, cells were serum
starved for 36 h and then released from cell cycle arrest by
replacing the RPMI media supplemented with 10% FBS. Cells were
stained with propidium iodide and analyzed using a FACSCanto II
flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
Animal model and immunohistochemistry
(IHC)
Lox-Stop-Lox (LSL) K-ras G12D mice were obtained
from the NCI mouse repository (http://mouse.ncifcrf.gov/), bred, and genotyped
according to the supplier's guidelines. This animal study was
approved by our Institutional Animal Care and Use Committee,
following the guidelines of the American Association for the
Assessment and Accreditation of Laboratory Animal Care. AdCre virus
was obtained from the Gene Transfer Vector Core of the University
of Iowa (Iowa City, IA, USA). Eight-week-old heterozygotes were
intranasally infected with 5×108 PFU of the AdCre virus
according to a protocol described elsewhere (17). Six, 12 and 16 weeks after infection,
mice were sacrificed, and expression of E-cadherin and Notch1 were
analyzed using IHC. IHC was performed using the LABS®2
System (Dako, Carpinteria, CA, USA) according to the manufacturer's
instructions. Briefly, sections were deparaffinization, rehydrated,
immersed in H2O2-methanol solution, and then
incubated overnight with primary anti-E-cadherin or -Notch1
antibodies in antibody diluent (Dako) at a 1:100 dilution. Sections
were incubated for 10 min with biotinylated linker and processed
using avidin-biotin IHC techniques. 3,3′-Diaminobenzidine (DAB) was
used as a chromogen in conjunction with the Liquid DAB Substrate
kit (Novacastra, UK).
Quantitative real time (RT)-PCR
Total RNA was extracted using TRI
reagent® (Ambion, Austin, TX, USA). Quantitative RT-PCR
analysis was conducted using TaqMan® Gene Expression
assay reagents and the StepOnePlus™ Real-Time PCR system (Applied
Biosystems, Carlsbad, CA, USA) using an inventoried primer-probe
set described in the company's websites (http://bioinfo.appliedbiosystems.com/genome-database/gene-expression.html).
EDTA treatment and siRNA experiment
Induction of N1ICD and activation of Notch signaling
using calcium treatment was described elsewhere (10,18).
Briefly, cells were washed two times with Ca2+- and
Mg2+-free DPBS and then incubated with DPBS containing
2.5 mM EDTA for 5 min. The media were then replaced with full
growth media, and cells were incubated for the indicated times.
MISSON® Predesigned siRNA against E-cadherin was
obtained from Sigma-Aldrich (St. Louis, MO, USA) and transfected
using Lipofectamine® RNAiMAX (Invitrogen, Carlsbad, CA,
USA) reagents as per the manufacturer's recommendations.
Western blotting
Cells were harvested using 2X LSB lysis buffer
containing protease inhibitor cocktail and phosphatase inhibitor
cocktail (Sigma-Aldrich) on ice. After sonication, the BCA protein
assay reagent (Thermo Scientific, Rockford, IL, USA) was used for
protein quantification. Protein lysates (30–50 mg) were separated
by gel electrophoresis on 7.5 to 12% polyacrylamide gels and
analyzed by western blot analysis using nitrocellulose membranes
(Bio-Rad Laboratories, Inc., Richmond, CA, USA). The expression
level of each protein was measured using ImageJ (http://rsbweb.nih.gov/ij/) and quantified relative to
that of β-actin.
Immunocytochemistry
Cells (5×105) were plated in 6-well
plates containing a sterilized coverslip. On the following day,
cells were fixed with 4% formaldehyde in PBS, incubated in blocking
solution containing 5% BSA in PBS, and then anti-c-Myc mouse
(1:100) and α-E-catenin, β-catenin, δ1-catenin rabbit antibodies
(1:200) were added for 16 h. On the following day, the cells were
washed, and anti-mouse IgG (Alexa Fluor 555 conjugate) and
anti-rabbit IgG (Alexa Flour 488 conjugate) secondary antibodies
were added. The nuclei were counterstained with DAPI (1:1,000) in
PBS and imaged using an LMS 510 confocal microscope (Carl Zeiss,
Oberkochen, Germany).
Statistical analysis
The independent sample t-test was used for
univariate analysis of continuous variables. All statistical
analyses were two-tailed.
Results
Inverse relationship between E-cadherin
and Notch1 expression in a lung cancer mouse model
E-cadherin is an active suppressor of tumor growth
and loss of its expression is related to the invasion, metastasis
and poor prognosis of cancer whereas the activity of Notch1
signaling correlates with increased intercellular interaction and a
hypoxic condition which are related to tumor progression (4,19–21).
Therefore, we postulated that there is an inverse relationship
between E-cadherin and Notch1 expression. To investigate this,
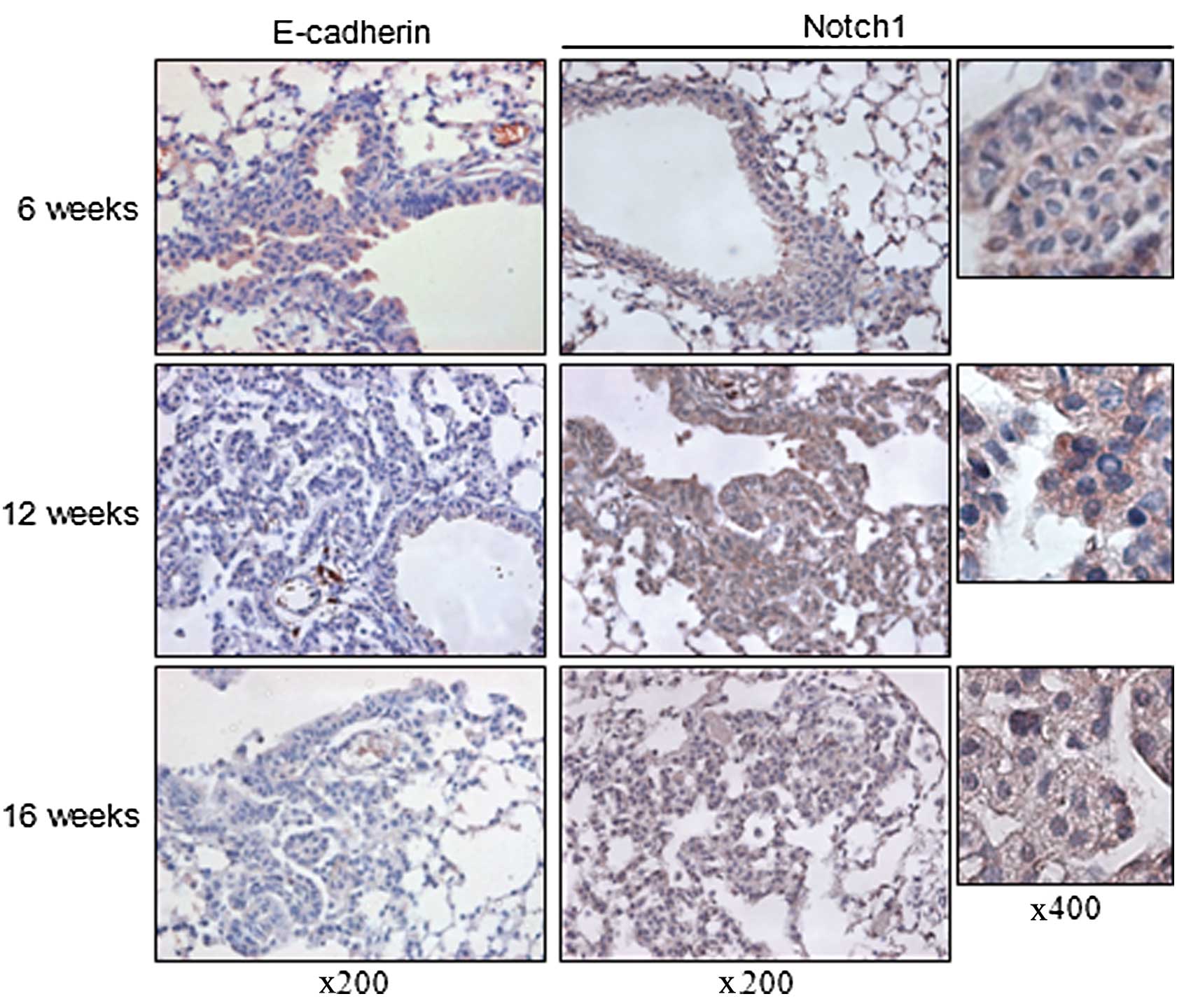
8-week-old LSL K-ras G12D mice were administered the AdCre virus
intranasally and sacrificed at 6, 12 and 16 weeks after viral
inhalation. E-cadherin expression decreased with the progression of
tumors over time. Compared to the hyperplastic lesions of the mice
sacrificed at 6 weeks after AdCre virus inhalation, lesions of the
mice sacrificed at 12 and 16 weeks after inhalation showed loss of
E-cadherin expression. Notch1 was ubiquitously expressed in the
membranous lesions of the normal-appearing bronchial epithelial
cells and hyperplastic lesions of the lungs of mice sacrificed at 6
weeks after AdCre virus inhalation. In the neoplastic lesions from
the mice which were sacrificed at 12 weeks after virus inhalation,
there was increased cytoplasmic expression of Notch1. More frequent
nuclear expression of Notch1, which is considered as biologically
active, was detected in the neoplastic lesions of the lungs from
the mice sacrificed at 16 weeks after inhalation. These findings
suggest an inverse relationship between E-cadherin and Notch1
expression with the progression of tumors of LSL K-ras G12D mice
(Fig. 1).
Transduction of N1ICD induces changes in
the components of adherens junctions
To evaluate the role of Notch1 signaling on adherens
junctions, we first transduced either the human N1ICD-myc or the
control vector into NSCLC cells and assessed the expression of
components of adherens junctions by immunoblotting. The
characteristics of the MSCV-N1ICD-myc and MSCV-control vectors are
described in Lim et al(5).
Transduction of N1ICD was confirmed by expression of the myc tag at
the N-terminal of the insert and the overexpression of HES1 and
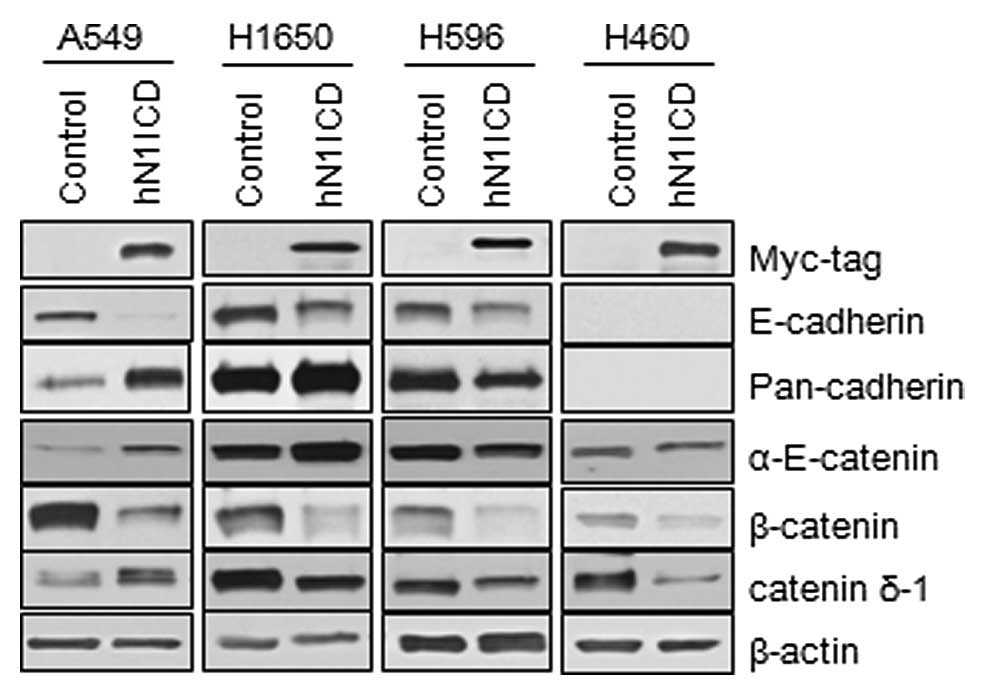
Herp (data not shown). A549, H1650 and H596 cells, which express
E-cadherin, and H460 cells, which do not express E-cadherin, showed
inhibition of E-cadherin and β-catenin expression. Other components
of the adherens junction, α-E-catenin and δ1-catenin, showed
different changes depending on the cell lines (Fig. 2). To further evaluate the effect of
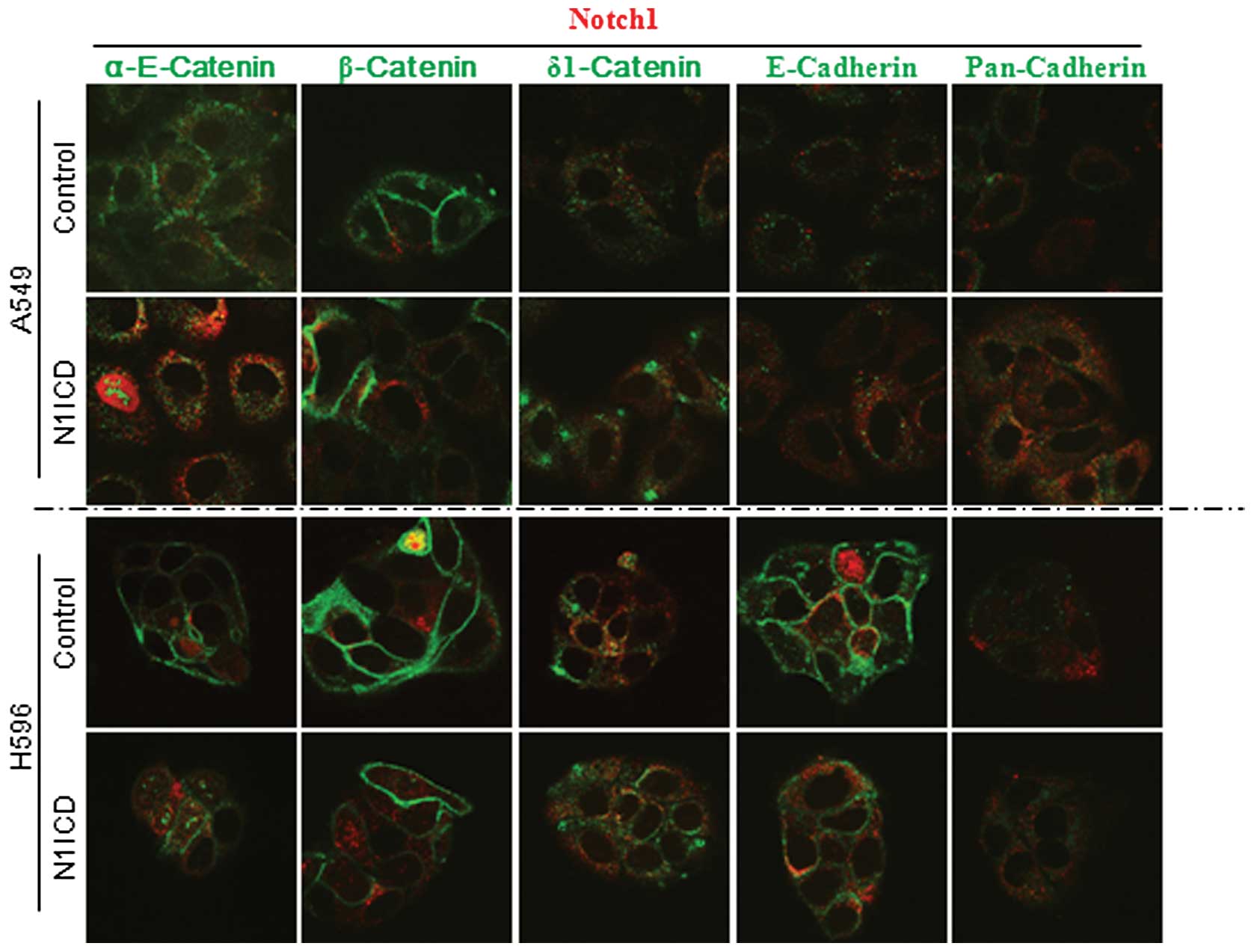
Notch signaling on adherens junctions, we performed an
immunocytochemical study using N1ICD- and control vector-transduced
cells. Transduction of N1ICD showed decreased membranous
localization of α-E-catenin, β-catenin and E-cadherin in A549, H596
(Fig. 3) and H1650 NSCLC cells
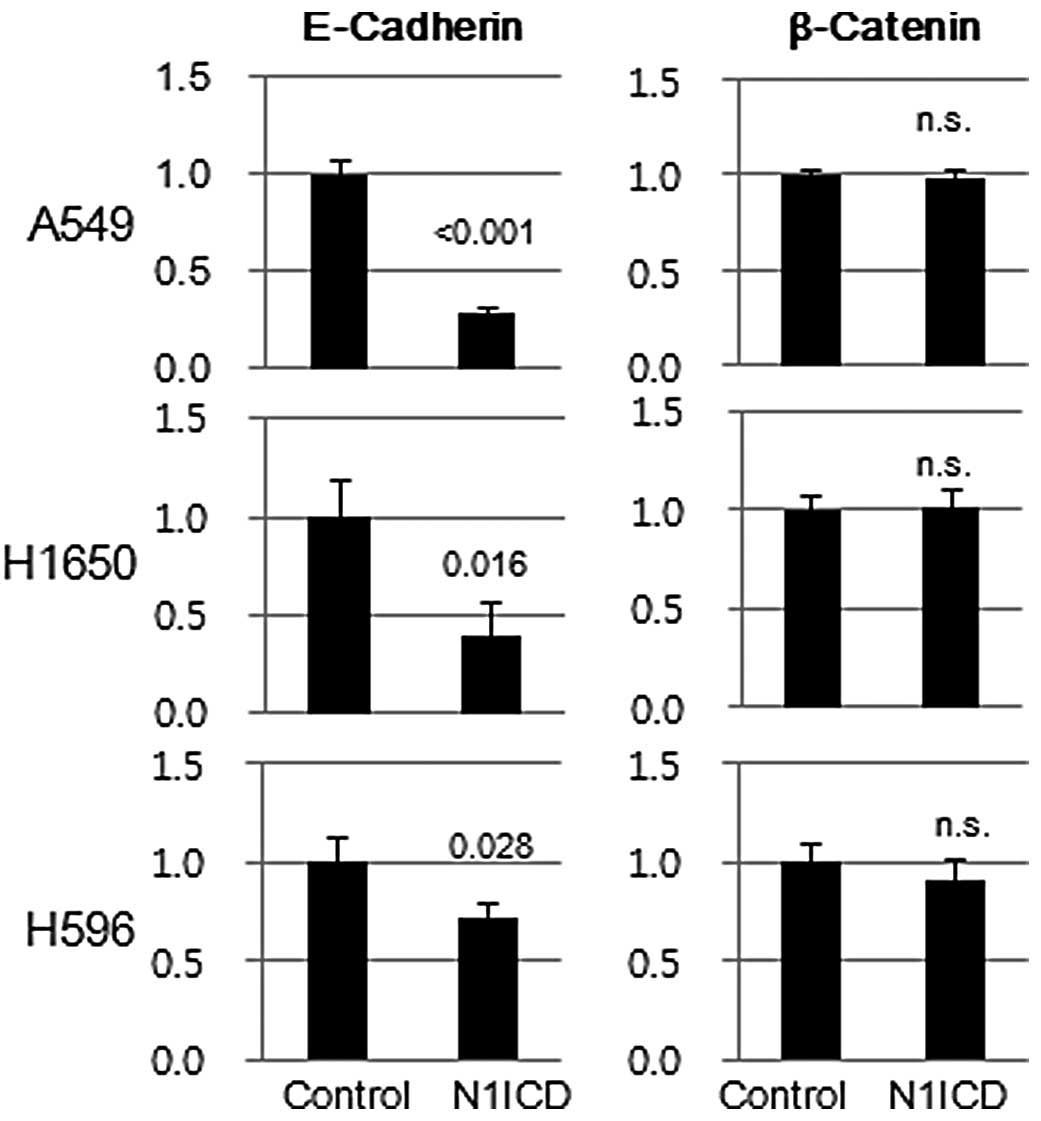
(data not shown). Transduction of N1ICD inhibited both protein and
mRNA expression of E-cadherin in A5459, H1650 and H596 cells.
However, there was no difference in β-catenin mRNA expression
between N1ICD- and control vector-transduced cells, which suggests
post-translational regulation of β-catenin expression (Fig. 4).
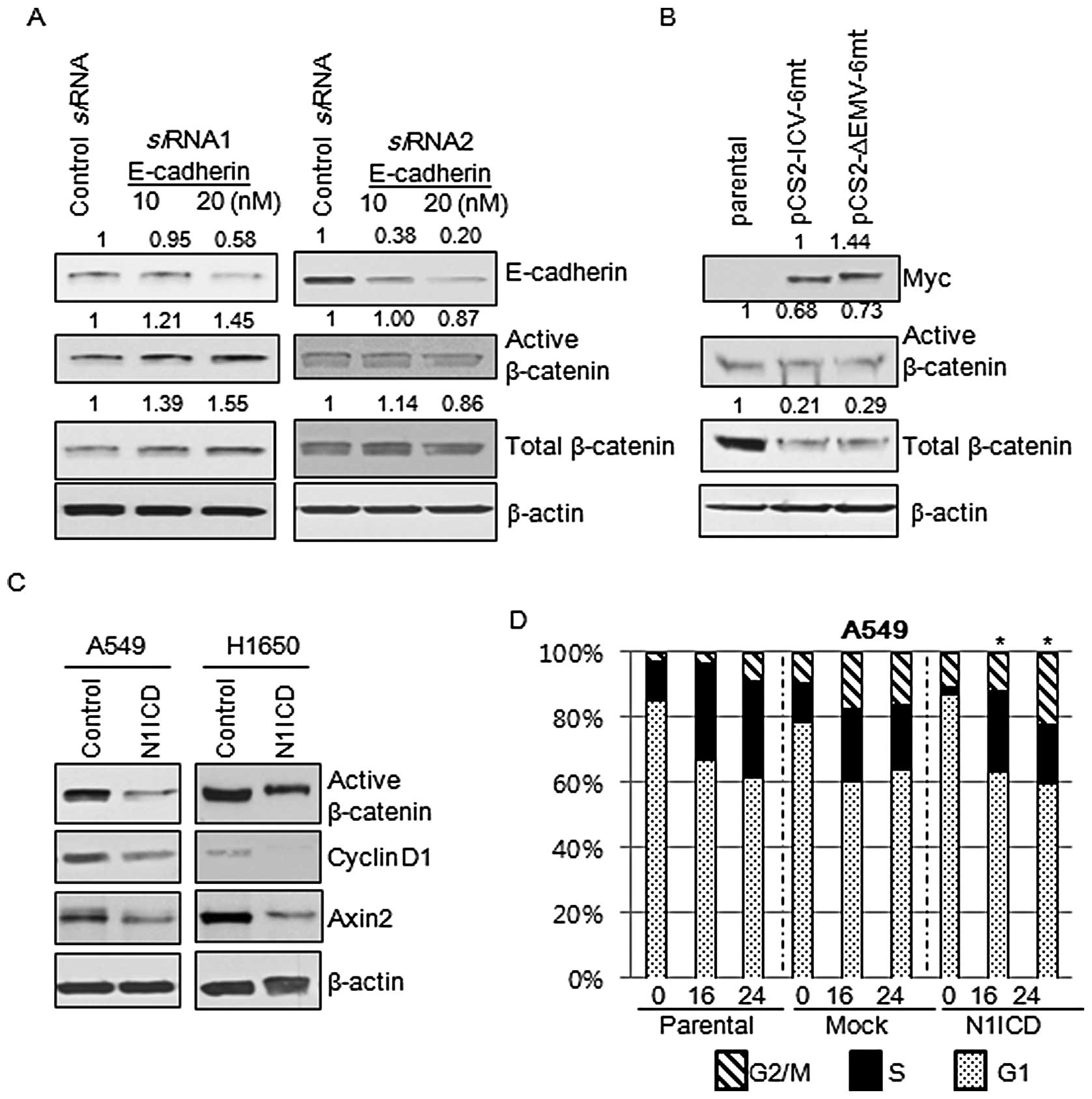
Notch1 inhibits E-cadherin expression
through Snail family of transcriptional complex
Calcium chelating agents interfere with the
non-covalent binding of the Notch1 ectodermal domain to the Notch1
transmembrane intracellular (NTMIC) domain and facilitate S2 and S3
cleavage of Notch1 to activate Notch signaling (10). Brief exposure to the millimolar
concentrations of EDTA resulted in the robust induction of N1ICD in
a short period of time, and the signal was propagated to Notch
target molecules, namely HES1 and Herp. Pretreatment of cells with
compound E, a γ-secretase inhibitor, inhibited the induction of
N1ICD and its target molecules (Fig.
5A). HES1 expression was upregulated at the transcriptional
level (Fig. 5B). Since induction of
E-cadherin repressor complexes is one of the important mechanisms
that induces E-cadherin loss, we evaluated the mRNA expression of
molecules that comprise the E-cadherin repressor complex by RT-PCR.
Among the E-cadherin repressor complex molecules, both Snail and
Slug were the E-cadherin repressor complex that showed consistent
increase in response to N1ICD induction (Fig. 5C).
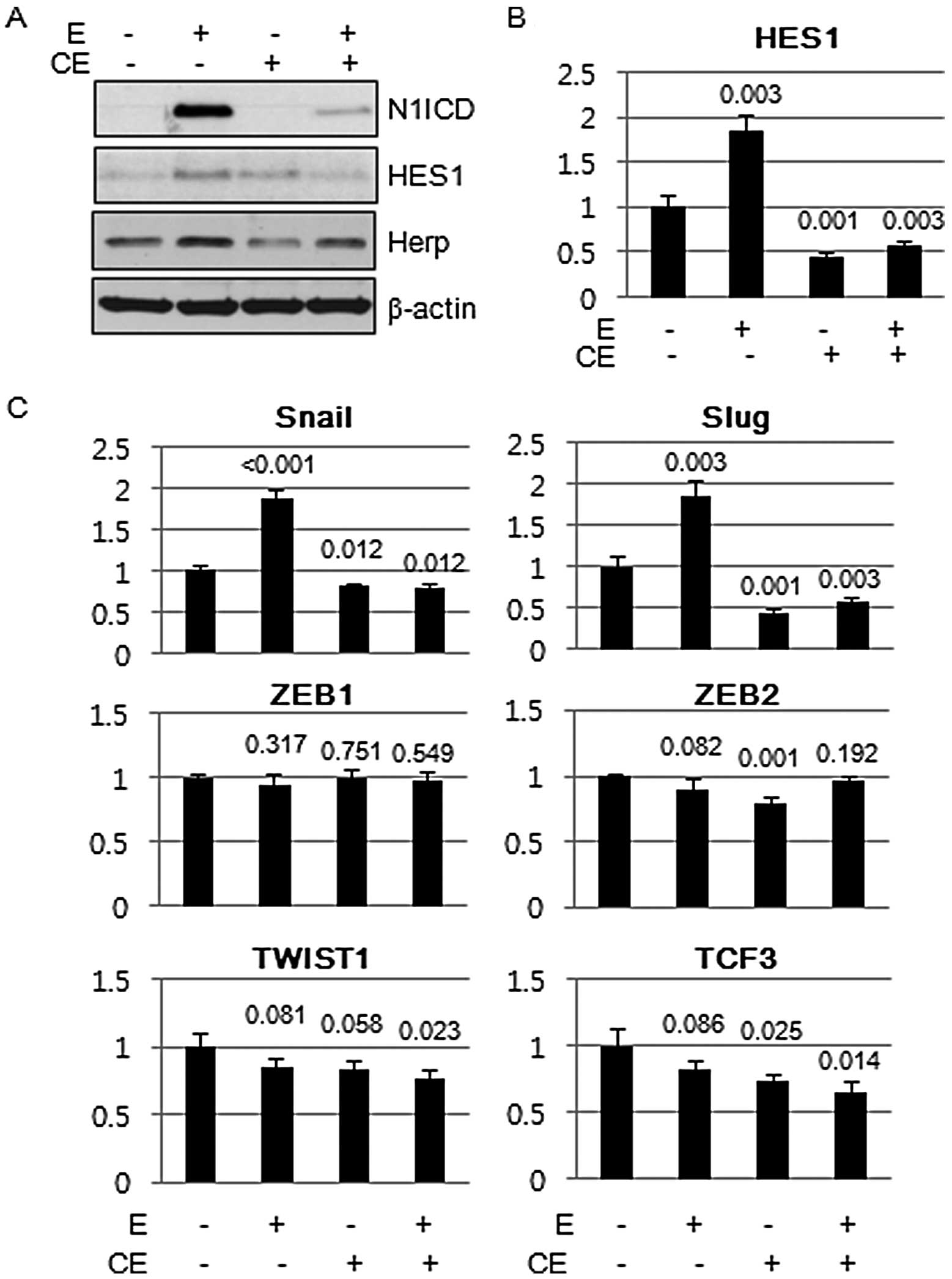 | Figure 5Snail family of E-cadherin repressor
complex, Snail and Slug, are induced by Notch1 intracellular domain
(N1ICD). (A) A549 cells were exposed to EDTA (2.5 mM) for 5 min and
expression of N1ICD and its downstream readouts of Notch1
signaling, HES1 and Herp, were evaluated by immunoblotting. Brief
exposure to the calcium chelating agent, EDTA, induced the
expression of N1ICD and its downstream targets, HES1 and Herp,
whereas pretreatment of a γ-secretase inhibitor, compound E,
inhibited the expression of N1ICD and its downstream targets. (B)
Real-time PCR showed that induction of HES1 by EDTA and inhibition
by compound E were regulated at the transcriptional level. (C)
Real-time PCR of members of the E-cadherin repression complexes.
Transcript expression of Snail and Slug was consistently increased
by EDTA treatment and was inhibited by compound E. Comparisons were
made to that of cells treated with vehicle. P-value was obtained by
Student's t-test. CE, compound E; E, EDTA. |
N1ICD-induced inhibition of β-catenin is
E-cadherin independent
Since the transduction of N1ICD into H460 cells,
which do not express E-cadherin, showed a decrease in β-catenin
expression, we aimed to ascertain whether the Notch1-mediated
inhibition of β-catenin expression was E-cadherin dependent. We
treated A549 cells with siRNA against E-cadherin and then evaluated
the expression of total and active β-catenin. Active β-catenin,
which is not phosphorylated at Ser33/37 and Thr41, is functionally
active in cell-to-cell adhesion and the canonical Wnt signaling
pathway. A 48-h siRNA treatment revealed that a decrease in
E-cadherin expression did not result in decreases in expression of
total and active β-catenin (Fig.
6A). These findings suggest that Notch1 inhibits the expression
of active and total β-catenin in an E-cadherin-independent manner.
Then we questioned whether inhibition of expression of total and
active β-catenin was a consequence of the activation of Notch
signaling. The membrane-tethered Notch is transcriptionally
inactive and considered biologically inert. To be biologically
active, Notch1 requires intramembranous cleavage between amino acid
Gly1743 and Val1744 mediated by the γ-secretase complex. We
transfected A549 cells with biologically active Notch
(pCS2-IVC-6mt) and membrane tethered Notch (pCS2-ΔEMV-6mt) and then
evaluated the expression of total and active β-catenin. Expression
of total and active β-catenin was inhibited by both Notch
constructs (Fig. 6B).
Wnt/β-catenin signaling is deregulated by
the Notch1 pathway
Since Notch1 is a transmembrane protein that relays
signals from cellular interactions and interacts with β-catenin,
one of the important components of adherens junctions, we reasoned
that Notch1 is involved in the Wnt/β-catenin signaling pathway.
Active β-catenin, cyclin D1 and Axin2 are the representative
markers of activation of Wnt/β-catenin signaling. Markers for
activation of Wnt/β-catenin signaling were decreased by
transduction with N1ICD (Fig. 6C).
Since the Wnt/β-catenin pathway is one of the important regulators
of cell cycle progression, we evaluated whether N1ICD influences
cell cycle propagation through regulation of the Wnt/β-catenin
pathway. Cell cycle progression of parental, control, and
N1ICD-transduced cells was measured by flow cytometry after PI
staining and comparisons were made to the proportion of cells in
the G2/M and S phases between the control and N1ICD-transduced
cells at the same time points. There were no statistical
differences in the cell cycle progression between the control and
N1ICD-transduced A549 (Fig. 6D) and
H1650 cells (data not shown).
Discussion
The adherens junction complex, in addition to its
adhesive function of maintaining the integrity of epithelial
cell-to-cell contacts, plays a crucial role in regulating
Wnt/β-catenin signaling. Loss of cadherin-mediated cell adhesion
promotes Wnt/β-catenin signaling, and deregulated Wnt/β-catenin
signaling due to unstable E-cadherin/β-catenin interaction induces
expression of oncogenes such as c-myc and cyclin
D1(22,23). Kwon et al(15) showed that NTMIC interacts with
β-catenin through an RAM domain and negatively regulates
Wnt/β-catenin signaling in a Numb-dependent manner, further
complicating the signaling cascades mediated by adherens
junctions.
The addition of any soluble ligands into culture
media does not activate Notch1 signaling. To active Notch1
signaling, Notch1 ligands must be immobilized on a culture plate,
aggregated with macromolecules and overexpressed on co-cultured
ligand cells (24). Otherwise
active Notch1 molecules have to be either transfected or transduced
or culture medium has to be briefly depleted of calcium to
dissociate Notch1 heterodimers (10). In the present study, we used the
calcium chelating agent EDTA to induce N1ICD. Although multiple
targets may be affected by calcium chelation, it is clear that
calcium chelation induces the biologically active Notch1 molecule
N1ICD, and propagates Notch1 signaling through transcriptional
activation of downstream targets. During the period when N1ICD is
detected by immunoblotting, we did not observe changes in the mRNA
levels of E-cadherin, indicating that N1ICD may not directly
repress E-cadherin expression (data not shown).
E-cadherin expression can be caused by promoter
hypermethylation (25), CDH1 gene
mutations (26), or induction of
E-cadherin transcriptional repressors, including the bHLH family
members Twist1 and Twist2, the E2A gene product E12/47, the Snail
gene family of transcription factors, and the ZFH family repressors
ZEB1 and ZEB2 (27). There are a
few reports of the involvement of Notch1 signaling in the loss of
E-cadherin and the EMT through upregulation of either Snail (SNAI1)
(28,29) or Slug (SNAI2) (30). It has also been reported that Notch1
induces an EMT phenotype through activation of TGF-β/Smad3 pathways
(31) or directly induces the
expression of smooth muscle α-actin (SMA) (32). Although there are two CSL
consensus-binding sites in the SMA gene promoters (32,33),
we did not observe increased mRNA expression of SMA in
N1ICD-transduced and EDTA-exposed NSCLC cells (data not shown). We
were not able to detect morphological differences among N1ICD-,
control vector-transduced, and parental cells under a phase
contrast microscope, suggesting that Notch1-mediated E-cadherin
repression may be an early event in Notch1-mediated EMT.
There is mutual compensation between Notch1 and
Wnt/β-catenin signaling. c-Myc is a common target molecule of the
Notch1 and Wnt/β-catenin signaling pathways. Among the cyclin D
family members, cyclin D1 is a target of the Wnt/β-catenin pathway
and cyclin D3 is a target of the Notch1 pathway. Notch1 has
conflicting roles in Wnt/β-catenin signaling. The physical
interaction between Notch1 and β-catenin negatively regulates
Wnt/β-catenin signaling. In contrast, Notch1 participates in the
deregulation of Wnt/β-catenin signaling by inhibiting the
expression of E-cadherin. Furthermore, N1ICD transduction was found
to result in cell cycle progression and aneuploidy, which suggests
another role for Notch1 in cancer progression. Sarmento et
al(34) reported that forced
induction of the N1ICD upregulated the expression of the S phase
kinase-associated protein 2 (SKP2) and caused premature entry into
S phase. In the present study, transduction of cells with N1ICD
decreased active β-catenin, cyclin D1 and Axin2 expression,
indicating that the active form of Notch1 acts as an inhibitor of
the Wnt/β-catenin pathway. However, negative regulation of
Wnt/β-catenin signaling through Notch1 did not result in a
difference in cell cycle progression between the control vector and
N1ICD-transduced cells. Taken together, activation of Wnt/β-catenin
signaling mediated by E-cadherin loss is compensated for by the
inhibitory effect of Notch1 on β-catenin.
In conclusion, activation of Notch1 signaling
destabilizes adherens junction by inhibition of E-cadherin and
β-catenin expression. Transduction of the transcriptionally active
domain of Notch1, namely N1ICD, into NSCLC cells decreased
expression of E-cadherin through induction of a Snail family of
E-cadherin repression complex. N1ICD also decreased the expression
of β-catenin in an E-cadherin-independent manner, which resulted in
a decrease in the expression of markers of activation of
Wnt/β-catenin. Despite the inhibition of Wnt/β-catenin signaling in
N1ICD-transduced cells, cells transduced with N1ICD showed no
difference in cell cycle progression when compared with the control
vector-transduced cells, suggesting that Notch1 signaling may
compensate for the inhibition of the Wnt/β-catenin pathway.
Acknowledgements
We thank Dr R. Kopan (Washington University) and Dr
G. Jung (Seoul National University) for providing the Notch
deletion/tethered Notch construct and the MSCV-N1ICD construct,
respectively. The present study was supported by an institutional
grant from Yonsei University College of Medicine (6-2012-0104)
awarded to Y.S.C.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Kopan R and Ilagan MX: The canonical Notch
signaling pathway: unfolding the activation mechanism. Cell.
137:216–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gustafsson MV, Zheng X, Pereira T, et al:
Hypoxia requires notch signaling to maintain the undifferentiated
cell state. Dev Cell. 9:617–628. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y, De Marco MA, Graziani I, et al:
Oxygen concentration determines the biological effects of NOTCH-1
signaling in adenocarcinoma of the lung. Cancer Res. 67:7954–7959.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lim SO, Kim HS, Quan X, et al: Notch1
binds and induces degradation of Snail in hepatocellular carcinoma.
BMC Biol. 9:832011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joshi I, Minter LM, Telfer J, et al: Notch
signaling mediates G1/S cell-cycle progression in T cells via
cyclin D3 and its dependent kinases. Blood. 113:1689–1698. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morimura T, Goitsuka R, Zhang Y, Saito I,
Reth M and Kitamura D: Cell cycle arrest and apoptosis induced by
Notch1 in B cells. J Biol Chem. 275:36523–36531. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sriuranpong V, Borges MW, Ravi RK, et al:
Notch signaling induces cell cycle arrest in small cell lung cancer
cells. Cancer Res. 61:3200–3205. 2001.PubMed/NCBI
|
|
9
|
Qi R, An H, Yu Y, et al: Notch1 signaling
inhibits growth of human hepatocellular carcinoma through induction
of cell cycle arrest and apoptosis. Cancer Res. 63:8323–8329.
2003.PubMed/NCBI
|
|
10
|
Rand MD, Grimm LM, Artavanis-Tsakonas S,
et al: Calcium depletion dissociates and activates heterodimeric
notch receptors. Mol Cell Biol. 20:1825–1835. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
King AM, Van der Put E, Blomberg B and
Riley RL: Accelerated Notch-dependent degradation of E47 proteins
in aged B cell precursors is associated with increased ERK MAPK
activation. J Immunol. 178:3521–3529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bryant DM and Mostov KE: From cells to
organs: building polarized tissue. Nat Rev Mol Cell Biol.
9:887–901. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Behrens J, Vakaet L, Friis R, et al: Loss
of epithelial differentiation and gain of invasiveness correlates
with tyrosine phosphorylation of the E-cadherin/beta-catenin
complex in cells transformed with a temperature-sensitive v-SRC
gene. J Cell Biol. 120:757–766. 1993. View Article : Google Scholar
|
|
14
|
Taddei ML, Chiarugi P, Cirri P, et al:
Beta-catenin interacts with low-molecular-weight protein tyrosine
phosphatase leading to cadherin-mediated cell-cell adhesion
increase. Cancer Res. 62:6489–6499. 2002.PubMed/NCBI
|
|
15
|
Kwon C, Cheng P, King IN, et al: Notch
post-translationally regulates β-catenin protein in stem and
progenitor cells. Nat Cell Biol. 13:1244–1251. 2011.
|
|
16
|
Schroeter EH, Kisslinger JA and Kopan R:
Notch-1 signalling requires ligand-induced proteolytic release of
intracellular domain. Nature. 393:382–386. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
DuPage M, Dooley AL and Jacks T:
Conditional mouse lung cancer models using adenoviral or lentiviral
delivery of Cre recombinase. Nat Protoc. 4:1064–1072. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Tetering G, van Diest P, Verlaan I,
van der Wall E, Kopan R and Vooijs M: Metalloprotease ADAM10 is
required for Notch1 site 2 cleavage. J Biol Chem. 284:31018–31027.
2009.PubMed/NCBI
|
|
19
|
Hazan RB, Qiao R, Keren R, Badano I and
Suyama K: Cadherin switch in tumor progression. Ann NY Acad Sci.
1014:155–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bremnes RM, Veve R, Gabrielson E, et al:
High-throughput tissue microarray analysis used to evaluate biology
and prognostic significance of the E-cadherin pathway in
non-small-cell lung cancer. J Clin Oncol. 20:2417–2428. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sahlgren C, Gustafsson MV, Jin S,
Poellinger L and Lendahl U: Notch signaling mediates
hypoxia-induced tumor cell migration and invasion. Proc Natl Acad
Sci USA. 105:6392–6397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He TC, Sparks AB, Rago C, et al:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moloney DJ, Panin VM, Johnston SH, et al:
Fringe is a glycosyltransferase that modifies Notch. Nature.
406:369–375. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Graff JR, Herman JG, Lapidus RG, et al:
E-cadherin expression is silenced by DNA hypermethylation in human
breast and prostate carcinomas. Cancer Res. 55:5195–5199.
1995.PubMed/NCBI
|
|
26
|
Berx G, Becker KF, Höfler H and van Roy F:
Mutations of the human E-cadherin (CDH1) gene. Hum Mutat.
12:226–237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schmalhofer O, Brabletz S and Brabletz T:
E-cadherin, beta-catenin, and ZEB1 in malignant progression of
cancer. Cancer Metastasis Rev. 28:151–166. 2009.PubMed/NCBI
|
|
28
|
Saad S, Stanners SR, Yong R, Tang O and
Pollock CA: Notch mediated epithelial to mesenchymal transformation
is associated with increased expression of the Snail transcription
factor. Int J Biochem Cell Biol. 42:1115–1122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsuno Y, Coelho AL, Jarai G, Westwick J
and Hogaboam CM: Notch signaling mediates TGF-β1-induced
epithelial-mesenchymal transition through the induction of Snai1.
Int J Biochem Cell Biol. 44:776–789. 2012.
|
|
30
|
Niessen K, Fu Y, Chang L, Hoodless PA,
McFadden D and Karsan A: Slug is a direct Notch target required for
initiation of cardiac cushion cellularization. J Cell Biol.
182:315–325. 2008.PubMed/NCBI
|
|
31
|
Aoyagi-Ikeda K, Maeno T, Matsui H, et al:
Notch induces myofibroblast differentiation of alveolar epithelial
cells via transforming growth factor-{beta}-Smad3 pathway. Am J
Respir Cell Mol Biol. 45:136–144. 2011.PubMed/NCBI
|
|
32
|
Noseda M, Fu Y, Niessen K, et al: Smooth
muscle alpha-actin is a direct target of Notch/CSL. Circ Res.
98:1468–1470. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang Y, Urs S, Boucher J, et al: Notch and
transforming growth factor-beta (TGFbeta) signaling pathways
cooperatively regulate vascular smooth muscle cell differentiation.
J Biol Chem. 285:17556–17563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sarmento LM, Huang H, Limon A, et al:
Notch1 modulates timing of G1-S progression by inducing SKP2
transcription and p27 Kip1 degradation. J Exp Med. 202:157–168.
2005. View Article : Google Scholar : PubMed/NCBI
|