Drugs for cancer treatment can be classified into
the categories of chemotherapeutic drugs and targeted therapy
drugs. Examples of chemotherapeutic drugs include alkylating or
alkylating-like agents such as capecitabine, mitotic inhibitors
such as paclitaxel and topoisomerase inhibitors such as irinotecan.
All of these drugs act as cytotoxic or cytostatic agents by killing
rapidly dividing cells.
Targeted therapy agents include endocrine therapy
drugs such as tamoxifen, antigrowth factor drugs such as the
monoclonal antibody trastuzumab against Her2 or the receptor
tyrosine kinase inhibitor (RTKI), gefitinib against the epidermal
growth factor receptor (EGFR) and anti-angiogenesis drugs such as
the monoclonal antibody bevacizumab against vascular endothelial
growth factor (VEGF) or the RTKI sunitinib against VEGF receptors
(VEGFRs) (1). All of these drugs
act by specifically blocking signal transduction pathways involved
in tumor development.
Historically, chemotherapeutic drugs were the first
effective agents used against malignant diseases. In the 1940s it
was discovered that a derivate of a chemical warfare agent,
nitrogen mustard, was effective for treating lymphoma when applied
to patients intravenously (2,3).
Cytotoxic chemotherapies are based on the classical ‘principles of
chemotherapy’ as defined by the observance that tumors exhibit a
sigmoid-shaped Gompertzian growth curve and thus cytoxic drugs are
most effective in killing tumor cells within the logarithmic growth
phase (4). For many cancer
indications, cytotoxic chemotherapeutics are still the recommended
first-line therapy. These agents include the platinum-based drugs
cisplatin or carboplatin for non-small cell lung cancer (NSCLC)
(5) or capecitabine in combination
with oxaliplatin for colorectal cancer (6,7). This
class of drugs does not differentiate between cancerous and normal
cells and thus induces systemic toxicity and adverse reactions
(8,9). The current survival rates of cancer
patients mostly treated with cytotoxic chemotherapeutics and/or
undergoing surgery and/or radiation therapy depends very much on
the site of the primary tumor. Female breast in situ,
uterine corpus and melanoma have 10-year relative survival rates of
up to 100% (female breast in situ). In contrast the 5-year
survival rate of patients with NSCLC and liver cancer is ~15% and
in pancreatic cancer ~5% since cytotoxic chemotherapeutics are
unable to cure metastatic disease even after successful surgical
tumor resection (10,11).
In contrast to cytotoxic chemotherapeutics, the
concept of targeted therapy is aimed to specifically target a
biological pathway that is critical for tumor development or tumor
maintenance and that causes regression or destruction of the
malignant process when it is inhibited (12). Endocrine therapy drugs were
developed for inhibition or modulation of hormone receptors for
hormone-sensitive tumors. The estrogen receptor modulator (SERM)
tamoxifen was the first targeted therapy drug for anticancer
treatment selectively inhibiting estrogen binding to its receptor
(13). When administered as an
adjuvant therapy for primary treatment of estrogen receptor
(ER)-sensitive breast cancer, tamoxifen was shown to reduce relapse
and mortality rates (14), and to
decrease recurrence rates in ER-positive breast cancer patients by
50% 15 years after diagnosis (13).
Another example of targeted therapy drugs are
inhibitors against the EGFR. EGFR is a well characterized example
of a growth factor receptor which plays a central role in tumor
development when becoming overexpressed and/or mutated.
Overexpression and mutation of EGFR leads to proliferation,
invasion of surrounding tissues, angiogenesis and distant
metastasis (15). Activation of
EGFR was also shown to influence resistance to cytotoxic
chemotherapeutic agents. Intracellular signaling leading to all of
these EGFR-mediated processes include the MAP kinase pathway, PI3K
and Akt signaling. Several targeted therapy drugs have been
approved by the FDA and EMEA for blocking the EGFR pathway either
by binding to its ligand EGF (mAb panitumumab) or by inhibiting its
tyrosine kinase activity (mAb cetuximab and RTKIs gefitinib and
erlotinib) (16,17). Gefitinib (Iressa) for example is an
orally available small-molecule RTKI (18) approved for the first-line therapy of
NSCLC patients with activating mutations of the EGFR tyrosine
kinase domain (19). In a subgroup
of the INTEREST trial including patients with activating EGFR
mutations, gefitinib was shown to improve the progression-free
survival (PFS) but not the overall survival (OS) when compared to
docetaxel. The median survival of patients under gefitinib
treatment was 14.2 months compared to 7.6 months in the overall
population (20).
In many diseases of different origins, common
underlying biological mechanisms play central roles. For example,
the dysregulation of EGFR signal transduction is an important
hallmark of certain malignancies as described above (17,34)
and at the same time plays a critical role in poxvirus spreading
(35). Inhibitors against EGFR
developed for anticancer treatment are effective against poxvirus
infections (36). The observation
and the analysis of common mechanisms in different types of
diseases provide the opportunity to draw conclusions from the
treatment of one disease to another. In a holistic model, the
mechanisms of an HIV-1 infection and tumor development are
comparable to a certain extent. These entirely different diseases
have common features that follow the principles of the Darwinian
evolutionary system. HIV-1 comprises a viral genome of ~9,700 bases
of single-stranded RNA (37) and
replicates within an estimated average total of 10.3×109
virions/day (38). Thus, the
disease driver of an HIV infection is primarily the replication
rate (39) in combination with a
high mutation rate introduced by the lack of proof-reading
mechanism of the transcriptase enzyme (40) and secondarily the genetic
variability (41). In tumor cells,
the conditions are exactly opposite. The genome of a malignant
human cell consists of ~3.2×109 base pairs, while its
replication takes ~1–2 days (42,43).
Thus, in tumors, the genetic heterogeneity instead of the
replication rate is the primary evolutionary driver (44–47).
Most importantly, in both diseases, in malignancies and in HIV
infections, the immune system is unable to control the disease due
to the evolutionary drivers that steer the processes of evasion
from immunological responses. In HIV infections and in tumor
development, these mechanisms include the generation of escape
mutants and the suppression of the immune system (43,48–54).
Additionally in tumor development the regulation of rescue pathways
plays a critical role to evade immunological responses (55,56).
Despite the anticipated common underlying biological
mechanisms of HIV infections and tumor development, the development
of an effective therapy concept against HIV was a great success
while there is still no groundbreaking treatment for many types of
cancer. At the beginning of the HIV epidemic, patients were treated
with monotherapy of the nucleoside analogue reverse transcriptase
inhibitor (NRTI) zidovudine developed in 1964. NRTIs selectively
inhibit HIV reverse transcriptase and thereby specifically block
transcription of viral DNA from viral RNA. Even though this
treatment exhibited some effect, the rates of progression-free
survival were still low. In a hemophilic cohort of 111 patients
treated solely with zidovudine and followed up for 11 years, the
progression rates to AIDS, symptoms and death were as high as 85%
(57). In 1996, only 16 years after
the identification of HIV as the cause of AIDS, the breakthrough in
HIV therapy was achieved when targeted therapy drugs were applied
in a combinational protocol introducing the highly active
antiretroviral therapy (HAART) (58). One year later the clinical
superiority of a three-drug regimen over a two-drug regimen by
using the selective protease inhibitor indinavir together with the
NRTIs zidovudine and lamivudine was demonstrated (59). Today, HAART consists of at least
three drugs, including either a protease inhibitor or a non-NRTI
(NNRTI) and two NRTIs. All three different drugs used in HAART are
selective targeted therapy compounds against critical steps in the
HIV-1 replication cycle. The drug class of HIV-1 protease
inhibitors is among the first successful examples of highly
selective targeted therapy drugs (60). A prospective cohort study evaluated
the long-term effectiveness of HAART and showed a reduction in the
progression to AIDS or death by 86% (61).
The successful treatment of HIV by HAART shows that
it is possible to control a disease that follows an evolutionary
concept similar to cancer. In both diseases, primary evolutionary
factors, a high replication rate in HIV and a large genomic
variability in cancer, define the route of evasion from
immunological responses. HAART is successful because it is based on
two key principles of treatment: i) high selectivity of drugs
against disease-specific targets, thus preventing severe
toxicities, adverse reactions and damage of immune system
components; and ii) effective combination of disease-specific
targets, thus preventing evolutionarily driven generation of escape
mutants and drug resistances. Transferring these principles to
cancer treatment would mean to address tumor targets as selective
as possible reducing side-effects and to identify a multi-drug
regimen of targeted therapy drugs for each type of malignancy
preventing tumor cell rescue and drug resistance. However, at
present most of the current anticancer drug therapies consist of
chemotherapeutics that do not follow the principle of drug
selectivity (5–7). These therapies lead to dose-dependent
side-effects and adverse reactions but mostly have a modest to
moderate effect on the malignant disease by prolonging survival
times only by a few months to years at maximum (10). Most of these treatments have toxic
effects on the hematological system and lead to opportunistic
infections that have to be controlled in parallel to the
chemotherapeutic treatment (8,62,63).
Lung cancer is the leading cause of cancer-related
mortality worldwide. Histopathological grading identifies ~85% of
lung cancers as NSCLCs and 15–20% as small-cell lung cancers
(SCLCs). NSCLCs can be subdivided into squamous cell carcinoma and
adenocarcinoma, accounting for 34 and 55% respectively, and into
other subtypes such as large cell carcinoma (64). For NSCLC cases up to grade IIIA, the
standard of care is surgical resection of the primary tumor which
can be supported by adjuvant chemotherapy and/or radiotherapy.
However, most often NSCLC is diagnosed at advanced stages beyond
stage IIIB. These tumors are treated with first-line neoadjuvant
chemotherapies followed by tumor resection or alternatively
combined with radiotherapy. First-line chemotherapies against NSCLC
consist of platinum-based drugs (carboplatin or cisplatin) combined
with third generation cytotoxic drugs such as docetaxel,
paclitaxel, irinotecan, gemcitabin, vinorelbin and pemetrexed
(5). Several targeted therapy drugs
have been approved for the treatment of NSCLC. These include
gefitinib, approved for first line therapy of NSCLC with activating
mutations of the EGFR tyrosine kinase domain (19) and bevacizumab approved for treatment
of non-squamous NSCLC in combination with platinium-based therapy
(65). Another targeted therapy
drug is crizitonib which inhibits the EML4-ALK fusion protein, an
oncogenic driver in a small percentage of NSCLC patients (66). Second-line therapies include
docetaxel, erlotinib, gefitinib, pemetrexed and platinum-based
therapy (5). The 5-year survival
rate for NSCLC patients is only ~15% (10) and thus the medical need for the
development of effective treatment concepts for NSCLC patients is
one of the greatest challenges for health care systems
worldwide.
For the design of a clinical trial for NSCLC
patients without further treatment options based on the concept of
selectivity and adequate target combination as discussed above,
four signaling pathways appear to be suitable targets against which
drugs are already marketed in different indications:
VEGF signaling is a well characterized target
complex with proven importance for tumor angiogenesis and tumor
development, including NSCLC (27).
Among the VEGF receptors VEGFR2 facilitates tumor growth by
inducing angiogenesis in tumor endothelial cells (67). Evidence suggests that VEGFR2
signaling also influences the motility of tumor cells, such as in
pancreatic cancer cells (68).
Signal transduction by VEGFR2 is mediated through the Ras/Raf
kinase pathway connected to MAP kinase signaling and the PI3K/Akt
pathway inducing angiogenesis by mediating cell proliferation and
cell-survival of tumor endothelial cells (69). For blocking the VEGF/VEGFR2
signaling pathway, several targeted therapy drugs are available.
These include the small-molecule RTKI sunitinib and the monoclonal
antibody bevacizumab. Sunitinib is a multi-targeted tyrosine kinase
inhibitor against VEGF receptors that also inhibits the activity of
other tyrosine kinases shown to be involved in tumor growth
(70). Bevacizumab is directed
against VEGF and blocks binding of this ligand to VEGF receptors
(71). Tumors that have been
treated with selective VEGFR2 inhibitors develop hypoxic
microenvironments by a compromised blood supply. It was shown that
this effect is countered by upregulation of growth factors which
have the capacity to replace VEGF and stimulate new blood vessel
growth such as EGF and SDF1α as well as their receptors (30).
The observation that treatment resistance to the
blockage of VEGFR2 is mediated by EGF and SDF1α and their receptors
qualifies these pathways as further potential targets for a
multi-drug-regimen targeted therapy for NSCLC treatment. SDF1α and
its receptors CXCR4 and CXCR7 were previously shown to be involved
in tumor development and tumor metastasis (72–74).
SDF1α signal transduction through CXCR4 is a well described pathway
that leads to activation of JAK/Stat, MAPK/ERK and PI3K with
phosphorylation of Akt (75,76).
CXCR4 activation plays a role in tumor metastasis, induction of
tumor growth and rescue of tumor cells from apoptosis (77,78).
CXCR7 was recently identified as a receptor that affects tumor cell
survival (79). SDF1α was shown to
mediate the homing of hematologic stem cells to the bone marrow via
CXCR4 signaling (80). High
expression of CXCR4 correlates with insensitivity against treatment
with sunitinib in mRCC and thus represents a possible mediator of
therapy resistance in tumors (81).
In addition, evidence indicates that SDF1α/CXCR4 signaling induces
EGFR activation in human trophoblast cells (82). Interestingly, a selective targeted
therapy drug, plerixafor, against CXCR4 has been approved by the
FDA for mobilization of hematopoietic stem cells from the bone
marrow for collection from peripheral blood for autologous
stem-cell transplantation in patients with non-Hodgkin’s lymphoma
(NHL) or multiple myeloma (MM) (83). Plerixafor shows an excellent safety
profile even when administered in combination with cytotoxic
chemotherapy in cancer patients (84). Most importantly, in an experimental
setting, plerixafor was shown to be effective for the treatment of
metastatic lung cancer, including NSCLC (72) and in inhibition of invasiveness of
human CRC cells (85).
A further candidate of a targeted therapy multi-drug
regimen is the EGFR oncogene pathway. Evidence for the increased
expression of EGF in tumors treated with VEGFR inhibitors has been
provided (30). As described above,
inhibitors against EGFR signaling have already been approved for
the treatment of EGFR-activated NSCLCs (19). Furthermore EGFR signal transduction
steers similar intracellular signaling cascades such as the
SDF1α/CXCR4 axis, PI3K/Akt and MAPK (ERK1/2) (17) and thus may also be involved in drug
resistance. In addition, EGFR was found to increase the expression
of angiogenic factors such as the enzyme cyclooxygenease 2 (COX2)
(86).
The COX2 metabolic pathway results in the production
of prostaglandin E2 (PGE2) which activates G-protein coupled EP.
COX2 is increased in cancer and other pathological conditions and
is suspected to participate in carcinogenesis and in tumor
angiogenesis. It was shown that the COX2 pathway indirectly induces
the upregulation of VEGF via the PKC pathway in NSCLC and in human
lung fibroblasts (87,88). COX2 was reported to increase the
expression of SDF1α and CXCR4 via PGE2 in myeloid-derived
suppressor cells in ascites from ovarian cancer patients (89) and that it contributes to cell
survival of human bladder cancer cells (90). In an experimental setting, the
selective inhibition of COX2 reduced the growth of colon carcinoma
cells in vivo(91). In
malignant gliomas, COX2 inhibitors are currently been discussed for
use in cancer treatment (92).
Several selective targeted therapy drugs against COX2 [such as
celecoxib (Celebrex) and etoricoxib (Arcoxia)] have been approved
for the treatment of arthritis, osteoarthritis, dysmenorrhea and
acute pain. The possible involvement in tumor development and the
connection between COX2 signaling and the induction of VEGF, SDF1α
and CXCR4 qualifies this enzyme as a potential candidate for a
multi-drug regimen therapy for cancer treatment.
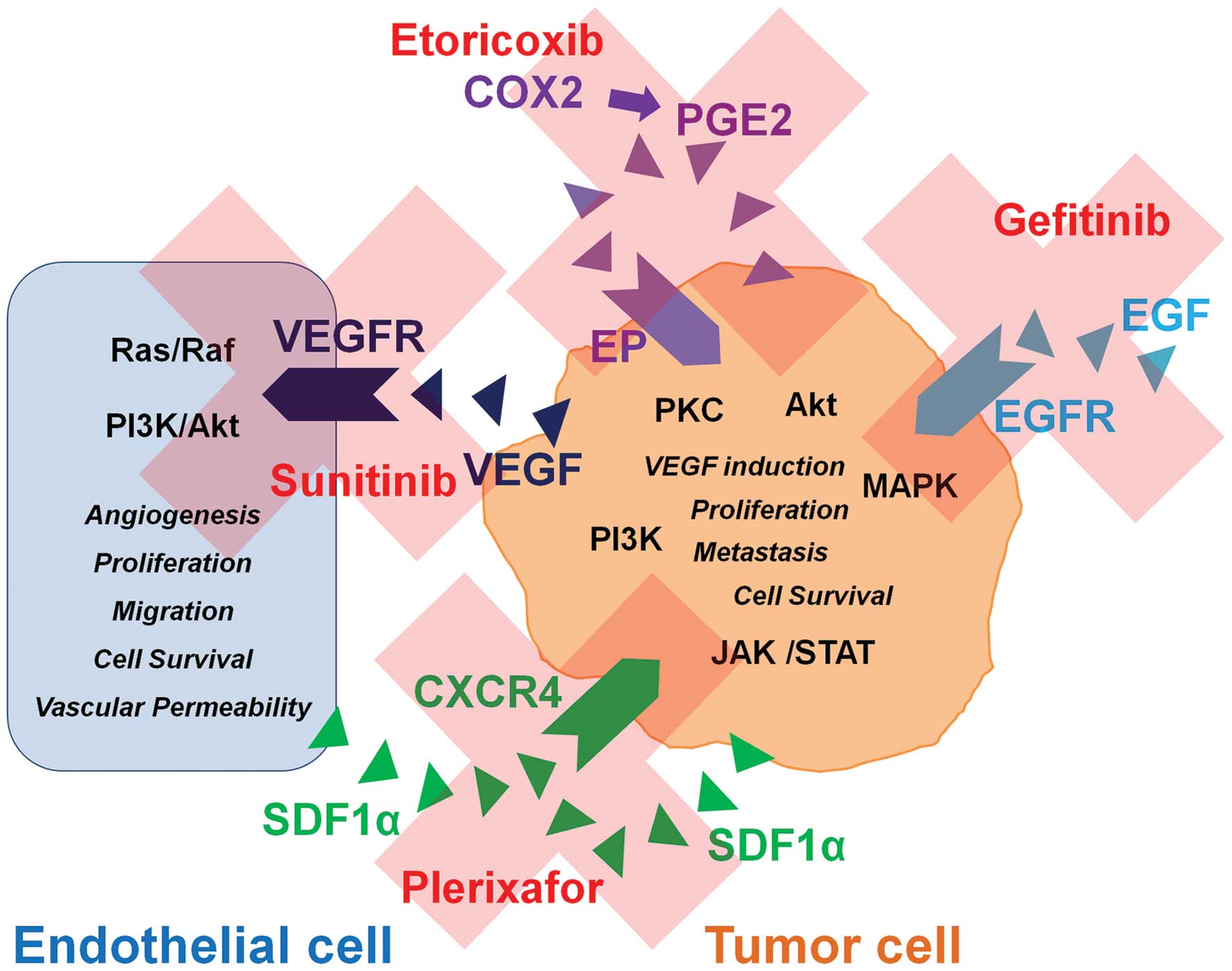
The combined use of inhibitors against VEGFR
signaling, such as sunitinib or bevacizumab combined with the CXCR4
inhibitor plerixafor and an inhibitor of EGFR signaling such as
gefitinib and the COX2 inhibitor etoricoxib would block multiple
signaling pathways in NSCLC. These pathways would be blocked at the
receptor level while inhibiting multiple intracellular connected
downstream pathways involved in tumor development and treatment
resistance (Ras/Raf, PI3K/Akt, Jak/Stat, MAPK, PKC) (Fig. 1). Thus, the simultaneous inhibition
of intracellular connected pathways in NSCLC tumors may overcome
resistance mechanisms to targeted therapy drugs commonly noted in
monotherapies or in a combination of targeted therapies with
chemotherapeutics. All of the mentioned drugs are approved for
different indications, and therefore their clinical profiles such
as pharmacokinetics, pharmacodynamics and toxicities are well
known. They are readily available as study drugs for usage in a
clinical trial.
In order to identify NSCLC patient subgroups that
may respond to this therapeutic concept, inclusion criteria should
require the proof of expression of the respective targets.
Bronchoscopy is a standard diagnostic procedure in NSCLC that
allows biopsy of tumor tissue. The collected tumor specimens can
easily be used for mRNA expression analysis of the respective
targets by specific RT-real time PCR or by a customized gene
expression array.
Similar to treatment of HIV by HAART, the herein
proposed combination of targeted therapy drugs for antitumor
treatment would fulfill the requirement of i) being highly
selective and ii) inhibiting multiple targets involved in disease
mechanism simultaneously. This may offer the chance for NSCLC
patients beyond treatment to achieve an antitumor effect.
|
1
|
Wang B, Rosano JM, Cheheltani R, Achary MP
and Kiani MF: Towards a targeted multi-drug delivery approach to
improve therapeutic efficacy in breast cancer. Expert Opin Drug
Deliv. 7:1159–1173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gilman A: The initial clinical trial of
nitrogen mustard. Am J Surg. 105:574–578. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Papac RJ: Origins of cancer therapy. Yale
J Biol Med. 74:391–398. 2001.
|
|
4
|
Dorr RT and Von Hoff DD: The Cancer
Chemotherapy Handbook. 2nd edition. Appleton & Lange; East
Norwalk, CT: pp. 51994
|
|
5
|
Azzoli CG, Temin S and Giaccone G: 2011
focused update of 2009 American Society of Clinical Oncology
clinical practice guideline update on chemotherapy for stage IV
non-small-cell lung cancer. J Oncol Pract. 8:63–66. 2012.
View Article : Google Scholar
|
|
6
|
Porschen R, Arkenau HT, Kubicka S, Greil
R, Seufferlein T, Freier W, Kretzschmar A, Graeven U, Grothey A,
Hinke A, Schmiegel W and Schmoll HJ: Phase III study of
capecitabine plus oxaliplatin compared with fluorouracil and
leucovorin plus oxaliplatin in metastatic colorectal cancer: a
final report of the AIO Colorectal Study Group. J Clin Oncol.
25:4217–4223. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Madi A, Fisher D, Wilson RH, Adams RA,
Meade AM, Kenny SL, Nichols LL, Seymour MT, Wasan H, Kaplan R and
Maughan TS: Oxaliplatin/capecitabine vs oxaliplatin/infusional 5-FU
in advanced colorectal cancer: the MRC COIN trial. Br J Cancer.
107:1037–1043. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hassett MJ, O’Malley AJ, Pakes JR,
Newhouse JP and Earle CC: Frequency and cost of
chemotherapy-related serious adverse effects in a population sample
of women with breast cancer. J Natl Cancer Inst. 98:1108–1117.
2006. View Article : Google Scholar
|
|
9
|
Altena R, Perik PJ, van Veldhuisen DJ, de
Vries EG and Gietema JA: Cardiovascular toxicity caused by cancer
treatment: strategies for early detection. Lancet Oncol.
10:391–399. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Howlader N, Noone A, Krapcho M, Neyman N,
Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z,
Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ and
Cronin KA: SEER Cancer Statistics Review, 1975–2009. Vintage 2009
Populations. National Cancer Institute; Bethesda, MD: 2012
|
|
11
|
Ardizzoni A, Boni L, Tiseo M, Fossella FV,
Schiller JH, Paesmans M, Radosavljevic D, Paccagnella A, Zatloukal
P, Mazzanti P, Bisset D and Rosell R; CISCA (CISplatin versus
CArboplatin) Meta-analysis Group. Cisplatin- versus
carboplatin-based chemotherapy in first-line treatment of advanced
non-small-cell lung cancer: an individual patient data
meta-analysis. J Natl Cancer Inst. 99:847–857. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ross JS, Schenkein DP, Pietrusko R, Rolfe
M, Linette GP, Stec J, Stagliano NE, Ginsburg GS, Symmans WF,
Pusztai L and Hortobagyi GN: Targeted therapies for cancer 2004. Am
J Clin Pathol. 122:598–609. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Swaby RF, Sharma CG and Jordan VC: SERMs
for the treatment and prevention of breast cancer. Rev Endocr Metab
Disord. 8:229–239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Early Breast Cancer Trialists’
Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal
therapy for early breast cancer on recurrence and 15-year survival:
an overview of the randomised trials. Lancet. 365:1687–1717.
2005.
|
|
15
|
Grandis JR and Sok JC: Signaling through
the epidermal growth factor receptor during the development of
malignancy. Pharmacol Ther. 102:37–46. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dutta PR and Maity A: Cellular responses
to EGFR inhibitors and their relevance to cancer therapy. Cancer
Lett. 254:165–177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okamoto I: Epidermal growth factor
receptor in relation to tumor development: EGFR-targeted anticancer
therapy. FEBS J. 277:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harandi A, Zaidi AS, Stocker AM and Laber
DA: Clinical efficacy and toxicity of anti-EGFR therapy in common
cancers. J Oncol. 2009:5674862009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sequist LV: First-generation epidermal
growth factor receptor tyrosine kinase inhibitors in EGFR mutation:
positive non-small cell lung cancer patients. J Thorac Oncol.
3:143–145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Douillard JY, Shepherd FA, Hirsh V, et al:
Molecular predictors of outcome with gefitinib and docetaxel in
previously treated non-small-cell lung cancer: data from the
randomized phase III INTEREST trial. J Clin Oncol. 28:744–752.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Folkman J: Tumor angiogenesis: therapeutic
implications. New Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Folkman J: Angiogenesis: an organizing
principle for drug discovery? Nat Rev Drug Discov. 6:273–286. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
National Cancer Institute. Angiogenesis
inhibitors. FactSheet. http://www.cancer.gov/cancertopics/factsheet/Therapy/angiogenesis-inhibitors.
Revised October 7, 2011.
|
|
24
|
Heath VL and Bicknell R: Anticancer
strategies involving the vasculature. Nat Rev Clin Oncol.
6:395–404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ivy SP, Wick JY and Kaufman BM: An
overview of small-molecule inhibitors of VEGFR signaling. Nat Rev
Clin Oncol. 6:569–579. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jayson GC, Hicklin DJ and Ellis LM:
Antiangiogenic therapy - evolving view based on clinical trial
results. Nat Rev Clin Oncol. 9:297–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ebos JM and Kerbel RS: Antiangiogenic
therapy: impact on invasion, disease progression, and metastasis.
Nat Rev Clin Oncol. 8:210–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oudard S, Beuselinck B, Decoene J and
Albers P: Sunitinib for the treatment of metastatic renal cell
carcinoma. Cancer Treat Rev. 37:178–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hurwitz H, Fehrenbacher L, Novotny W,
Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, et al:
Bevacizumab plus irinotecan, fluorouracil, and leucovorin for
metastatic colorectal cancer. N Engl J Med. 350:2335–2342. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shojaei F: Anti-angiogenesis therapy in
cancer: current challenges and future perspectives. Cancer Lett.
320:130–137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tanne JH: FDA cancels approval for
bevacizumab in advanced breast cancer. BMJ. 343:d76842011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Goulart BH, Clark JW, Pien HH, Roberts TG,
Finkelstein SN and Chabner BA: Trends in the use and role of
biomarkers in phase I oncology trials. Clin Cancer Res.
13:6719–6726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yarden Y: The EGFR family and its ligands
in human cancer. Signaling mechanisms and therapeutic
opportunities. Eur J Cancer. 37:S3–S8. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Buller RM, Chakrabarti S, Moss B and
Fredrickson T: Cell proliferative response to vaccinia virus is
mediated by VGF. Virology. 164:182–192. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Langhammer S, Koban R, Yue C and Ellerbrok
H: Inhibition of poxvirus spreading by the anti-tumor drug
Gefitinib (Iressa). Antiviral Res. 89:64–70. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ratner L, Fisher A, Jagodzinski LL,
Mitsuya H, Liou RS, Gallo RC and Wong-Staal F: Complete nucleotide
sequences of functional clones of the AIDS virus. AIDS Res Hum
Retroviruses. 3:57–69. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Perelson AS, Neumann AU, Markowitz M,
Leonard JM and Ho DD: HIV-1 dynamics in vivo: virion clearance
rate, infected cell life-span, and viral generation time. Science.
271:1582–1586. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Campbell TB, Schneider K, Wrin T,
Petropoulos CJ and Connick E: Relationship between in vitro human
immunodeficiency virus type 1 replication rate and virus load in
plasma. J Virol. 77:12105–12112. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ho DD, Neumann AU, Perelson AS, Chen W,
Leonard JM and Markowitz M: Rapid turnover of plasma virions and
CD4 lymphocytes in HIV-1 infection. Nature. 373:123–126. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hemelaar J: The origin and diversity of
the HIV-1 pandemic. Trends Mol Med. 18:182–192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Klein CA: Parallel progression of primary
tumors and metastases. Nat Rev Cancer. 9:302–312. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Greaves M and Maley CC: Clonal evolution
in cancer. Nature. 481:306–313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Merlo LM, Pepper JW, Reid BJ and Maley CC:
Cancer as an evolutionary and ecological process. Nat Rev Cancer.
6:924–935. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Maley CC, Galipeau PC, Finley JC,
Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques
RA, Rabinovitch PS and Reid BJ: Genetic clonal diversity predicts
progression to esophageal adenocarcinoma. Nat Genet. 38:468–473.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pepper J, Findlay CS, Kassen R, Spencer S
and Maley CC: Cancer research meets evolutionary biology. Evol
Appl. 2:62–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
González-García I, Solé RV and Costa J:
Metapopulation dynamics and spatial heterogeneity in cancer. Proc
Natl Acad Sci USA. 99:13085–13089. 2002.
|
|
48
|
Staveley-O’Corrol K, Sotomayor E,
Mongomery J, Borrello I, Hwang L, Fein S, Pardoll D and Levitsky H:
Induction of antigen-specific T cell anergy: an early event in the
course of tumor progression. Proc Natl Acad Sci USA. 95:1178–1183.
1998.PubMed/NCBI
|
|
49
|
Finke J, Ferrone S, Frey A, Mufson A and
Ochoa A: Where have all the T cells gone? Mechanisms of immune
evasion by tumors. Immunol Today. 20:158–160. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Levy LS: Retrovirus-induced
immunodeficiency and cancer. Retroviruses and Insights into Cancer.
Dudley J: Springer; New York: pp. 259–283. 2011
|
|
51
|
Dunn GP, Bruce AT, Ikeda H, Old LJ and
Schreiber RD: Cancer immunoediting: from immunosurveillance to
tumor escape. Nat Immunol. 3:991–998. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zitvogel L, Tesniere A and Kroemer G:
Cancer despite immunosurveillance: immunoselection and
immunosubversion. Nat Rev Immunol. 6:715–727. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Leslie AJ, Pfafferott KJ, Chetty P,
Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado
JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA,
St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall
H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P,
Walker BD and Goulder PJ: HIV evolution: CTL escape mutation and
reversion after transmission. Nat Med. 10:282–289. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Klenerman P, Wu Y and Phillips R: HIV:
current opinion in escapology. Curr Opin Microbiol. 5:408–413.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang T, Niu G, Kortylewski M, Burdelya L,
Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola
D, Dalton W, Jove R, Pardoll D and Yu H: Regulation of the innate
and adaptive immune responses by Stat-3 signaling in tumor cells.
Nat Med. 10:48–54. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
56
|
Oosterhoff D, Lougheed S, van de Ven R,
Lindenberg J, van Cruijsen H, Hiddingh L, Kroon J, van den Eertwegh
AJ, Hangalapura B, Scheper RJ and de Gruijl TD: Tumor-mediated
inhibition of human dendritic cell differentiation and function is
consistently counteracted by combined p38 MAPK and STAT3
inhibition. Oncoimmunology. 1:649–658. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lee CA, Phillips AN, Elford J, Janossy G,
Griffiths P and Kernoff P: Progression of HIV disease in a
haemophilic cohort followed for 11 years and the effect of
treatment. BMJ. 303:1093–1096. 1991.Erratum in: BMJ 303: 1446,
1991.
|
|
58
|
Hammer SM, Katzenstein DA, Hughes MD,
Gundacker H, Schooley RT, Haubrich RH, Henry WK, Lederman MM, Phair
JP, Niu M, Hirsch MS and Merigan TC: A trial comparing nucleoside
monotherapy with combination therapy in HIV-infected adults with
CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical
Trials Group Study 175 Study Team. N Engl J Med. 335:1081–1090.
1996. View Article : Google Scholar
|
|
59
|
Hammer SM, Squires KE, Hughes MD, Grimes
JM, Demeter LM, Currier JS, Eron JJ Jr, Feinberg JE, Balfour HH Jr,
Deyton LR, Chodakewitz JA and Fischl MA: A controlled trial of two
nucleoside analogues plus indinavir in persons with human
immunodeficiency virus infection and CD4 cell counts of 200 per
cubic millimeter or less. AIDS Clinical Trials Group 320 Study
Team. N Engl J Med. 337:725–733. 1997. View Article : Google Scholar
|
|
60
|
Leung D, Abbenante G and Fairlie DP:
Protease inhibitors: current status and future prospects. J Med
Chem. 43:305–341. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sterne JA, Hernán MA, Ledergerber B,
Tilling K, Weber R, Sendi P, Rickenbach M, Robins JM and Egger M;
Swiss HIV Cohort Study. Long-term effectiveness of potent
antiretroviral therapy in preventing AIDS and death: a prospective
cohort study. Lancet. 366:378–384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Saif MW, Katirtzoglou NA and Syrigos KN:
Capecitabine: an overview of the side effects and their management.
Anticancer Drugs. 19:447–464. 2008.PubMed/NCBI
|
|
63
|
Hatziveis K, Tourlakis D, Hountis P,
Kyriazanos I, Sougleri M, Ginopoulos P and Camoutsis C: Effects on
the immune system and toxicity of carboplatin/paclitaxel
combination chemotherapy in patients with stage III–IV ovarian and
non-small cell lung cancer and its role in survival and toxicity. J
BUON. 17:143–148. 2012.PubMed/NCBI
|
|
64
|
Li T, Kung HJ, Mack PC and Gandara DR:
Genotyping and genomic profiling of non-small-cell lung cancer:
implications for current and future therapies. J Clin Oncol.
31:1039–1049. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cohen MH, Gootenberg J, Keegan P and
Pazdur R: FDA drug approval summary: bevacizumab (Avastin) plus
Carboplatin and Paclitaxel as first-line treatment of
advanced/metastatic recurrent nonsquamous non-small cell lung
cancer. Oncologist. 12:713–718. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Shaw AT, Solomon B and Kenudson MM:
Crizotinib and testing for ALK. J Natl Compr Canc Netw.
9:1335–1341. 2011.PubMed/NCBI
|
|
67
|
Takahashi S: Vascular endothelial growth
factor (VEGF), VEGF receptors and their inhibitors for
antiangiogenic tumor therapy. Biol Pharm Bull. 34:1785–1788. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Doi Y, Yashiro M, Yamada N, Amano R, Noda
S and Hirakawa K: VEGF-A/VEGFR-2 signaling plays an important role
for the motility of pancreas cancer cells. Ann Surg Oncol.
19:2733–2743. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Abrams TJ, Murray LJ, Pesenti E, Holway
VW, Colombo T, Lee LB, Cherrington JM and Pryer NK: Preclinical
evaluation of the tyrosine kinase inhibitor SU11248 as a single
agent and in combination with ‘standard of care’ therapeutic agents
for the treatment of breast cancer. Mol Cancer Ther. 2:1011–1021.
2003.
|
|
71
|
Ferrara N, Hillan KJ, Gerber HP and
Novotny W: Discovery and development of bevacizumab, an anti-VEGF
antibody for treating cancer. Nat Rev Drug Discov. 3:391–400. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Burger JA, Stewart DJ, Wald O and Peled A:
Potential of CXCR4 antagonists for the treatment of metastatic lung
cancer. Expert Rev Anticancer Ther. 11:621–630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhao BC, Wang ZJ, Mao WZ, Ma HC, Han JG,
Zhao B and Xu HM: CXCR4/SDF-1 axis is involved in lymph node
metastasis of gastric carcinoma. World J Gastroenterol.
17:2389–23896. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hou KL, Hao MG, Bo JJ and Wang JH: CXCR7
in tumorigenesis and progression. Chin J Cancer. 29:456–459. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Burger M, Glodek A, Hartmann T,
Schmitt-Gräff A, Silberstein LE, Fujii N, Kipps TJ and Burger JA:
Functional expression of CXCR4 (CD184) on small-cell lung cancer
cells mediates migration, integrin activation, and adhesion to
stromal cells. Oncogene. 22:8093–80101. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Soriano SF, Serrano A, Hernanz-Falcón P,
Martín de Ana A, Monterrubio M, Martinez C, Rodríguez-Frade JM and
Mellado M: Chemokines integrate JAK/STAT and G-protein pathways
during chemotaxis and calcium flux responses. Eur J Immunol.
33:1328–1333. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kijima T, Maulik G, Ma PC, Tibaldi EV,
Turner RE, Rollins B, Sattler M, Johnson BE and Salgia R:
Regulation of cellular proliferation, cytoskeletal function, and
signal transduction through CXCR4 and c-Kit in small cell lung
cancer cells. Cancer Res. 62:6304–6311. 2002.PubMed/NCBI
|
|
78
|
D’Alterio C, Barbieri A, Portella L, Palma
G, Polimeno M, Riccio A, Ieranò C, Franco R, Scognamiglio G, Bryce
J, Luciano A, Rea D, Arra C and Scala S: Inhibition of stromal
CXCR4 impairs development of lung metastases. Cancer Immunol
Immunother. 61:1713–1720. 2012.PubMed/NCBI
|
|
79
|
Burns JM, Summers BC, Wang Y, Melikian A,
Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ,
Wei K, McMaster BE, Wright K, Howard MC and Schall TJ: A novel
chemokine receptor for SDF-1 and I-TAC involved in cell survival,
cell adhesion, and tumor development. J Exp Med. 203:2201–2213.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sharma M, Afrin F, Satija N, Tripathi RP
and Gangenahalli GU: Stromal-derived factor-1/CXCR4 signaling:
indispensable role in homing and engraftment of hematopoietic stem
cells in bone marrow. Stem Cells Dev. 20:933–946. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
D’Alterio C, Portella L, Ottaiano A, Rizzo
M, Carteni G, Pignata S, Facchini G, Perdona S, Di Lorenzo G,
Autorino R, Franco R, La Mura A, Nappi O, Castello G and Scala S:
High CXCR4 expression correlates with sunitinib poor response in
metastatic renal cancer. Curr Cancer Drug Targets. 12:693–702.
2012.PubMed/NCBI
|
|
82
|
Zhao HB, Tang CL, Hou YL, Xue LR, Li MQ,
Du MR and Li DJ: CXCL12/CXCR4 axis triggers the activation of EGF
receptor and ERK signaling pathway in CsA-induced proliferation of
human trophoblast cells. PLoS One. 7:e383752012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Keating GM: Plerixafor: a review of its
use in stem-cell mobilization in patients with lymphoma or multiple
myeloma. Drugs. 71:1623–1647. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Attolico I, Pavone V, Ostuni A, Rossini B,
Musso M, Crescimanno A, Martino M, Iacopino P, Milone G, Tedeschi
P, Coluzzi S, Nuccorini R, Pascale S, Di Nardo E and Olivieri A:
Plerixafor added to chemotherapy plus G-CSF is safe and allows
adequate PBSC collection in predicted poor mobilizer patients with
multiple myeloma or lymphoma. Biol Blood Marrow Transplant.
18:241–249. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Li JK, Yu L, Shen Y, Zhou LS, Wang YC and
Zhang JH: Inhibition of CXCR4 activity with AMD3100 decreases
invasion of human colorectal cancer cells in vitro. World J
Gastroenterol. 14:2308–2313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Garnier D, Magnus N, D’Asti E, Hashemi M,
Meehan B, Milsom C and Rak J: Genetic pathways linking hemostasis
and cancer. Thromb Res. 129(Suppl 1): S22–S29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Nakanishi M, Sato T, Li Y, Nelson AJ,
Farid M, Michalski J, Kanaji N, Wang X, Basma H, Patil A, Goraya J,
Liu X, Togo S, Toews LM, Holz O, Muller KC, Magnussen H and Rennard
SI: Prostaglandin E2 stimulates the production of vascular
endothelial growth factor through the E-prostanoid-2 receptor in
cultured human lung fibroblasts. Am J Respir Cell Mol Biol.
46:217–223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Luo H, Chen Z, Jin H, Zhuang M, Wang T, Su
C, Lei Y, Zou J and Zhong B: Cyclooxygenase-2 up-regulates vascular
endothelial growth factor via a protein kinase C pathway in
non-small cell lung cancer. J Exp Clin Cancer Res. 30:62011.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Obermajer N, Muthuswamy R, Odunsi K,
Edwards RP and Kalinski P: PGE(2)-induced CXCL12 production and
CXCR4 expression controls the accumulation of human MDSCs in
ovarian cancer environment. Cancer Res. 71:7463–7470. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Shimada K, Anai S, Marco DA, Fujimoto K
and Konishi N: Cyclooxygenase 2-dependent and independent
activation of Akt through casein kinase 2α contributes to human
bladder cancer cell survival. BMC Urol. 11:82011.PubMed/NCBI
|
|
91
|
Pozzi A, Yan X, Macias-Perez I, Wei S,
Hata AN, Breyer RM, Morrow JD and Capdevila JH: Colon carcinoma
cell growth is associated with prostaglandin E2/EP4 receptor-evoked
ERK activation. J Biol Chem. 279:29797–29804. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Schönthal AH: Exploiting
cyclooxygenase-(in)dependent properties of COX-2 inhibitors for
malignant glioma therapy. Anticancer Agents Med Chem. 10:450–461.
2010.PubMed/NCBI
|