Introduction
The cancer chemopreventive properties of selenium
have been studied for over 20 years, and most studies have used
rodent models of mammary carcinogenesis (1). The anticancer activity of both the
inorganic and organic forms of selenium compounds has been studied.
The prototype form, sodium selenite, exhibited a major drawback
when its chemopreventive activity was tested; it is rapidly
converted to hydrogen selenide, which generates DNA strand breaks
and subsequent cytotoxicity (2–4). Among
the organic forms of selenium, selenomethionine (SeMet) has been
reported to have anticancer effects (2–6) and
oral administration to humans as an anticancer drug has been
approved. The efficacy of selenium, vitamin E, and β-carotene alone
and in combination in the prevention of prostate cancer is
currently being assessed in population-based clinical trials
(7,8). These studies have been based on
secondary analyses of large-scale chemoprevention trials for other
cancers (9,10).
Recently, the chemopreventive mechanism of SeMet has
been investigated via in vitro studies. Organic selenium
SeMet compounds have been reported to activate tumor suppressor
p53, and this activation may be one of the alternative
chemopreventive mechanisms mediated by organic selenium (11). The protein p53 has been frequently
referred to as the gatekeeper of the genome; it plays a
well-established role in maintaining the stability of the genome by
inducing either cell cycle arrest or apoptosis (12–14).
The expression of p53 is induced by cellular stresses that cause
DNA damage, and this increase in p53 expression promotes either
cell cycle arrest or DNA repair (15). If the damage is too severe,
apoptosis is induced (16). It has
been established that p53 increases global DNA repair but not
transcription-coupled nucleotide excision repair (NER) (17). Our previous study suggested that
activation of p53 by SeMet plays a role in protecting cells against
DNA damage induced by ultraviolet (UV) irradiation (11). Furthermore, our more recent study
suggested that p53 and its downstream gene gadd45a are
activated in cells following treatment with organic selenium, and
this response may participate in restoring apurinic/apyrimidinic
endonuclease (AP) sites during methyl methanesulfonate
(MMS)-induced base excision repair (BER) (18).
Gadd45 (or Gadd45a, growth arrest and DNA
damage-inducible gene) and p21 (Waf1/Cip1, a cyclin-dependent
kinase inhibitor) are two well-established p53-regulated genes.
Gadd45a binds to UV-damaged chromatin and affects the accessibility
of sites of DNA damage to DNA repair machinery (19). An early study by Smith et
al(20) showed that Gadd45a
interacts with proliferating cell nuclear antigen (PCNA) and
participates in NER; however, the precise mechanism and function of
this interaction requires further characterization. A recent study
suggested that PCNA interacts with APE1/Ref1, a key component of
the BER pathway, in the nucleus (21). Taken together, these data suggest
that Gadd45a affects BER activity via its interaction with PCNA and
APE1/Ref-1. The BER pathway corrects DNA damage generated by
ionizing radiation, simple alkylating agents, and endogenous
hydrolytic and oxidative processes. BER is initiated by a
monofunctional glycosylase, followed by AP endonuclease
(APE)-mediated strand cleavage 5′ to the apurinic/apyrimidinic (AP)
site. p53 has been reported to enhance methyl methanesulfonate
(MMS)-induced BER activity. Our previous study suggested that
gadd45a, a gene downstream of p53, participates in the BER
pathway by interacting with BER-related proteins, such as PCNA and
APE1/Ref-1 (22).
In the present study, we provide initial evidence
that the p53-dependent interaction of Gadd45a with repair proteins
is involved in the activation of BER in response to the organic
selenium compound SeMet. Our study identified a novel
chemopreventive property of the antioxidant selenium.
Materials and methods
Cell culture and treatment
We used isogenic human colon cancer cell lines
carrying wild-type p53 and mutant p53 derivatives, in which p53
function was abrogated by the introduction of a dominant-negative
p53 mutant allele (codon 143; valine to alanine) (23). Cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS) (both from
Gibco-BRL, Carlsbad, CA, USA) and antibiotics. RKO cells were
treated with organic selenium (20 and 40 μM) for 16 h at 37°C and
incubated in 5% CO2:95% air.
Preparation of DNA substrates
Oligonucleotides were 17-mers containing either
tetrahydrofuran (THF) or a normal nucleotide (dA) at position 9.
Complementary oligos with a T opposite the THF were also used. The
sequences were as follows: 5′-AGCATTCGXGACTGGGT-3′, in which the
X indicates THF,
and dA; 5′-ACCCAGTCTCGAATGCT-3′ was the complementary strand.
Spacer CE phosphoramidite (a tetrahydrofuran derivative) was
purchased from Integrated DNA Technologies (IDT) Inc. (USA). The
oligonucleotides were radiolabeled at their 5′ end using
[γ-32P]-ATP (Amersham Life Sciences, USA) and T4
polynucleotide kinase (Promega Corporation, Madison, WI, USA). The
oligonucleotides were annealed by heating them at 90°C, followed by
cooling to room temperature for several hours.
Assay measuring AP endonuclease activity
in cell lysates
Crude cell extracts were assayed for AP endonuclease
activity using the double-stranded oligonucleotide as a substrate.
Reactions were performed in 10 mM Tris-HCl at pH 7.5, 50 mM NaCl
and 1 mM EDTA. The reaction mixtures containing 25 μg total protein
and 5′ radiolabeled substrate (104 cpm, ≈0.75 pmol) were
incubated at 37°C for 30 min. The stop solution (98% formamide, 10
mM EDTA, 0.025% bromophenol blue and 0.025% xylene cyanol) was
added to the reactions, followed by incubation on ice. The products
were separated on 20% denaturing polyacrylamide gels, and
autoradiography was carried out for 15 h at −70°C using Kodak
X-Omat AR film (Eastman Kodak, Rochester, NY, USA).
Transfection of p53 siRNA
siRNA duplexes targeting p53 were designed and
synthesized by Dharmacon Inc. (Chicago, IL, USA). RKO cells were
transfected using Oligofectamine reagent (Invitrogen GmbH,
Karlsruhe, Germany), according to the manufacturer’s instructions,
with single-strand sense and antisense RNA oligonucleotides (human
p53 sense RNA, 5′-GAGGUUGGCUCUGACUGUAUU-3′ and antisense RNA,
5′-UACAGUCAGAGCCAACCUCUU-3′). For each well, 5 μl of 20 nmol of the
oligonucleotides was diluted with 175 μl of serum-free RPMI-1640
medium. Then 4 μl of Oligofectamine (Invitrogen GmbH) was diluted
and incubated for 10 min in 15 μl of serum-free RPMI-1640 medium.
The 2 solutions were combined and incubated at room temperature for
15 min. This solution was incubated with RKO cells for 4 h, and
then the medium was replaced with RPMI-1640 containing 10% FBS.
APE1/Ref1 immunostaining
For APE1/Ref1 staining, cells were grown on
coverslips and fixed with ice-cold 100% methanol for 30 min at
−20°C. The cells were then dehydrated with ice-cold 100% acetone
for 1 sec and washed 3 times with PBS. The fixed cells were
incubated with an APE1/Ref-1 mouse antibody (clone Ab82)
(NeoMarkers, Fremont, CA, USA) diluted 1:3,000 in BSA solution
(0.5% bovine serum albumin in PBS) for 4 h at room temperature.
After incubation with the primary antibody, the coverslips were
washed 4 times for 5 min each with PBS containing 0.1% Tween-20.
Then the cells were incubated with fluorescein isothiocyanate
(FITC)-conjugated anti-mouse secondary antibody (Vector
Laboratories, Burlingame, CA, USA) for 1 h at room temperature,
followed by 6 5-min washes with shaking. Coverslips were mounted on
standard microscope slides with mounting media containing DAPI
(Vector Laboratories). Images were viewed and captured using a
fluorescence microscope (Nikon Eclipse 50i and NIS-Elements F 2.20,
Nikon, Japan).
Immunoblotting and
immunoprecipitation
All western blotting was conducted as previously
described by Smith et al(24). The following antibodies were used:
APE1/Ref1 (ab82) (NeoMarkers); PCNA (PC10); Gadd45a (Ab H165) (both
from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (LabFrontier,
Seoul, Korea). The proteins were detected using horseradish
peroxidase-conjugated secondary antibodies (Sigma-Aldrich, St.
Louis, MO, USA), followed by enhanced chemiluminescence (Pierce
Biotechnology, Inc., Rockford, IL, USA).
Cells were lysed in immunoprecipitation (IP) buffer
[1% Nonidet P40, 0.1% SDS, 50 mM Tris-HCl (pH 7.8), 150 mM NaCl, 1
mM DTT and 0.5 mM EDTA containing protease inhibitors (Complete™;
Roche Molecular Biochemicals, Mannheim, Germany)] for 30 min at
4°C. The samples were sonicated for 5 sec. Equal amounts of protein
extracts were precleared by incubating them with 30 μl of protein
A/G agarose beads (Santa Cruz Biotechnology, Inc.) for 1 h at 4°C
with shaking. The precleared supernatants were aliquoted for IP
with 1 μg of either rabbit anti-Gadd45a antibody, mouse anti-PCNA
antibody, or rabbit anti-APE1/Ref-1 antibody for 4 h at 4°C. These
antibody-containing lysates were incubated with 40 μl of protein
A/G agarose beads (Santa Cruz Biotechnology, Inc.) overnight at
4°C. The beads were washed 4 times with IP buffer. The
immunoprecipitated proteins were resuspended in 4X sample buffer,
resolved by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), and electrotransferred to membranes
before western blotting with the indicated antibodies.
Results
Increased interaction of PCNA and
APE1/Ref-1 with Gadd45a in response to SeMet
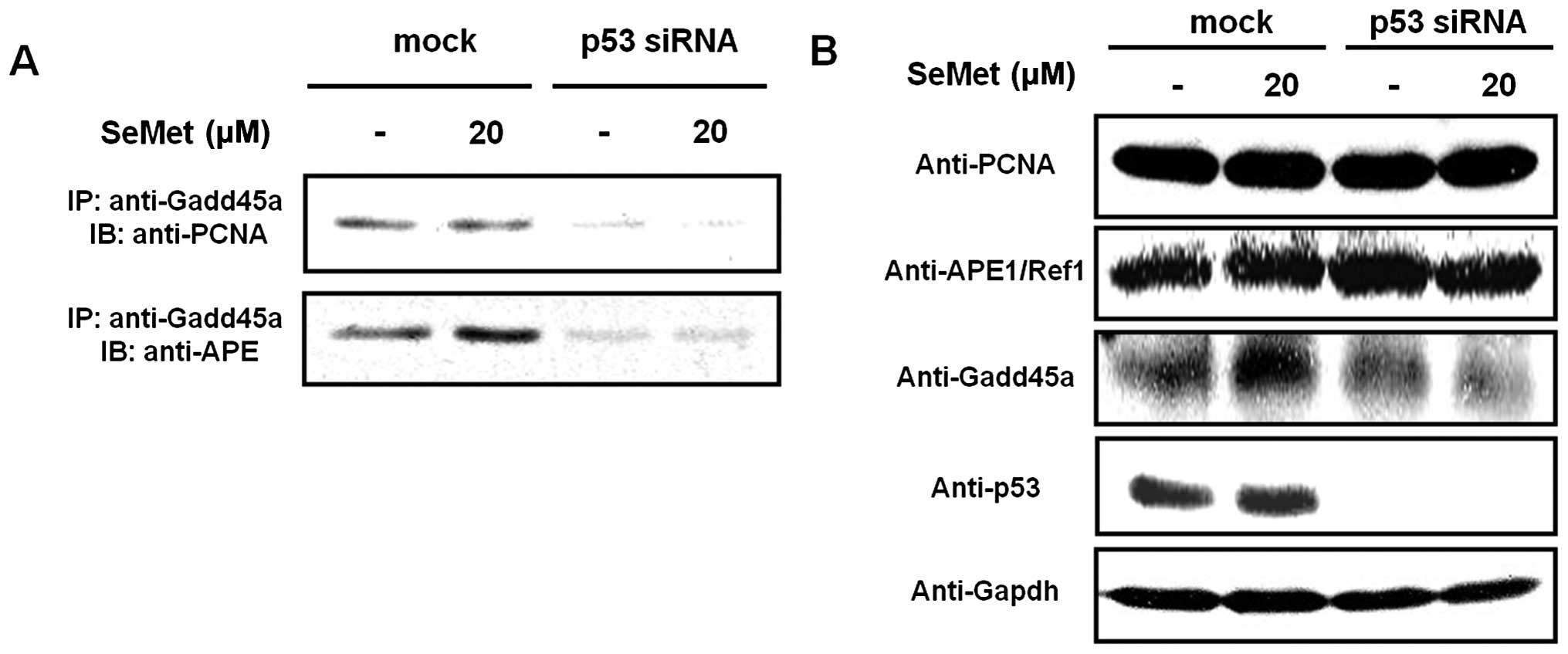
We first examined whether an interaction exists
between Gadd45a, APE1/Ref1 and PCNA following the treatment of
SeMet (20 μM as a non-genotoxic dose in which apoptosis and cell
cycle arrest are not induced, data not shown). We used the
technique of immunoprecipitation from human colon cancer RKO cell
lysates to evaluate this potential interaction. An interaction
between Gadd45a and PCNA was previously demonstrated; therefore, we
extended this study to determine whether Gadd45a also interacts
with PCNA and APE1/Ref1 in the context of BER activation. Gadd45a
strongly interacted with PCNA in mock-treated RKO cells in response
to organic selenium (Fig. 1),
whereas almost no interaction took place between Gadd45a and PCNA
in p53 siRNA-treated RKO cells treated with organic selenium. In
addition, the interaction between Gadd45a and APE1/Ref-1 was
dramatically reduced in p53 siRNA-treated cells (Fig. 1A). The expression of Gadd45a,
APE1/Ref-1 and PCNA in the whole cell lysates and siRNA-mediated
knockdown of p53 were confirmed by western blotting (Fig. 1B). Our data suggest that the
interaction between Gadd45a, PCNA and APE1/Ref-1 is promoted in a
p53-dependent manner in response to organic selenium.
Increased p53-dependent AP endonuclease
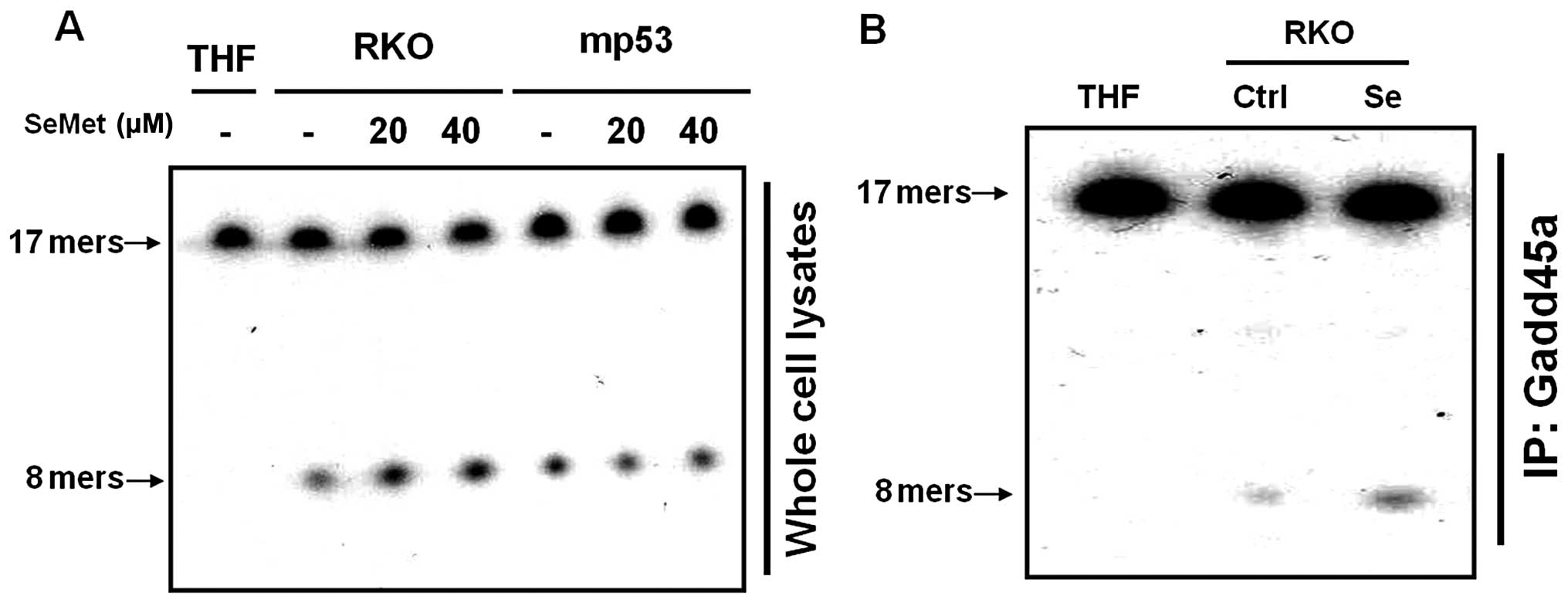
activity in response to SeMet
We examined cell extracts for the presence of
enzymes that could act on damaged bases or their analogues.
Tetrahydrofuran (THF)-containing oligonucleotide duplexes were used
as substrates to assess AP endonuclease activity. In mammalian
cells, APE1, the major AP endonuclease, is able to cleave DNA
duplexes containing THF. The cleavage of 17-mer THF-containing
duplex oligonucleotides into 8-mers by APE1 was dramatically
increased in the SeMet-treated cells, as compared to untreated
cells. These data suggest that the organic selenium-treated cells
had greater APE1 activity than untreated cells (Fig. 2A). Furthermore, the cells that
expressed mutant p53 did not exhibit any alteration in the level of
APE1 activity in the presence of SeMet (Fig. 2A). In addition, we demonstrated that
the interaction of Gadd45a with APE1, as assessed by
immunoprecipitation, increased the activity of APE1 in response to
SeMet (Fig. 2B). Taken together,
these data suggest that the AP endonuclease activity required to
remove the abasic site during BER increased as a result of its
interaction with Gadd45a in response to the antioxidant organic
selenium.
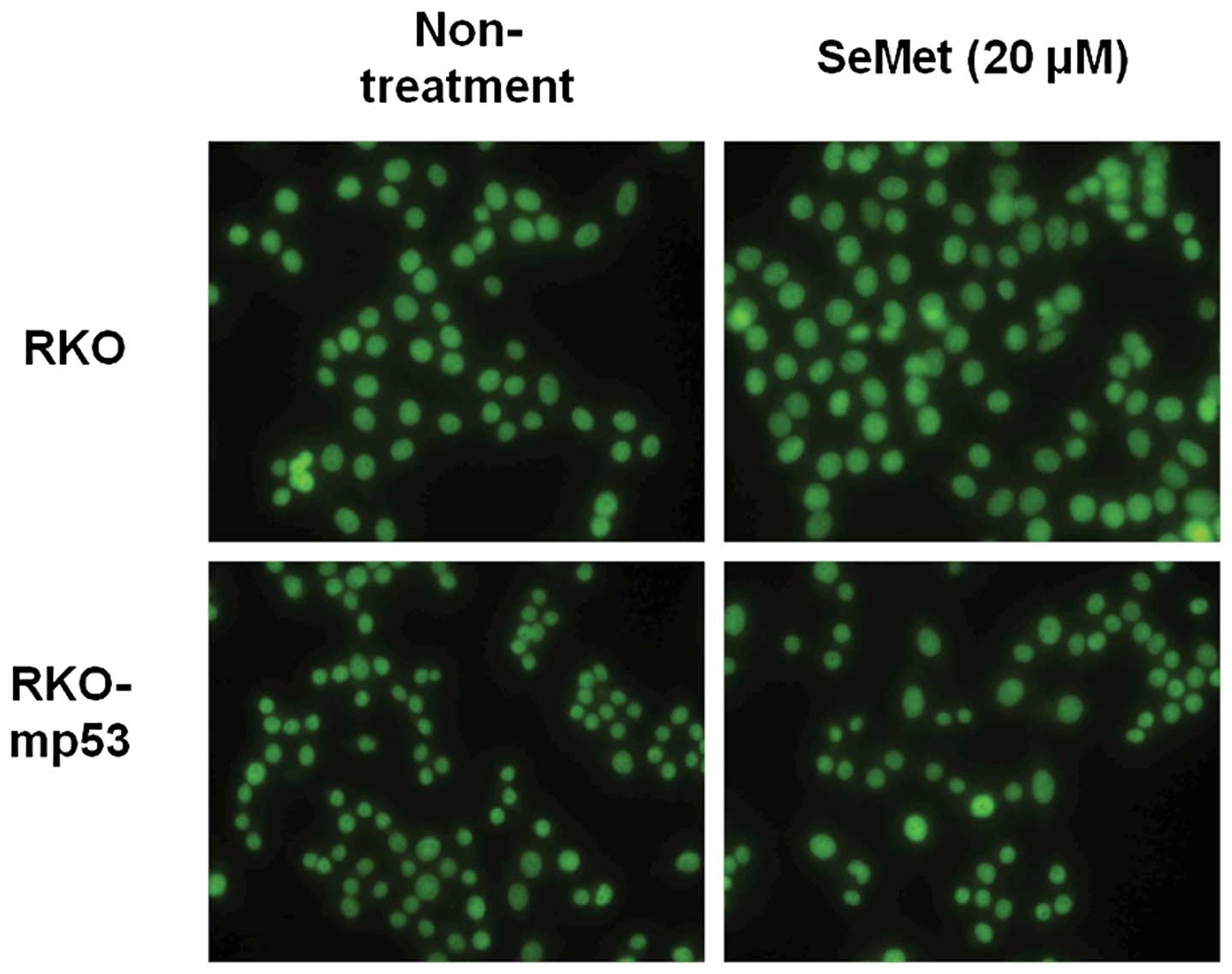
APE1 localization is not altered in the
presence of SeMet
APE1/Ref-1 is recruited to sites of DNA damage and
becomes associated with nuclear substructures. We examined whether
the localization of APE1/Ref-1 is regulated by p53 in response to
SeMet using an isogenic pair of RKO cell lines with wild-type and
mutant p53. APE1/Ref-1 immunostaining showed that APE1/Ref-1 was
localized to the nucleus in both the wild-type and p53 mutant RKO
cell lines irrespective of organic selenium treatment (Fig. 3). These data suggest that altered
localization of APE1 is not involved in the p53-mediated promotion
of BER activity in response to organic selenium.
Discussion
BER activity has been reported to increase when DNA
damage is induced by ionizing radiation and simple alkylating
agents, as well as free radicals generated from endogenous
hydrolytic and oxidative processes (25). BER converts diverse base lesions
into common intermediate apurinic/apyrimidinic (AP) sites. These AP
sites have been reported to spontaneously arise at a substantial
rate and are thought to be one of the most frequent types of DNA
lesions (26). It is estimated that
the spontaneous hydrolytic loss of purines generates ~10,000 AP
sites per day in mammalian cells (27,28).
Therefore, the BER pathway is critical for handling the diverse
lesions produced as a result of the intrinsic instability of DNA or
by the insults of various endogenous and exogenous reactive oxygen
species. Defects in the BER process are associated with an
increased susceptibility to cancer and neurodegenerative
disorders.
Although the SELECT study, one of the largest human
cancer prevention clinical trials, did not find evidence that SeMet
prevents prostate cancer (29),
increasing evidence indicates that other micronutrient combinations
may have beneficial effects. Recently, Qiao et al(7) suggested that the beneficial effects of
Se, vitamin E, and β-carotene on total and gastric cancer mortality
were still evident up to 10 years after supplementation and were
consistently greater in participants younger than 55. This general
population nutrition intervention trial, conducted in Linxian,
China, where nutritional deficiencies and high cancer rates are
common, suggest that the basal level of minerals including Se is
critical to cancer incidence.
The interaction of BER repair proteins has been
investigated in the context of organic selenium-induced activation
of the BER pathway. Most reports have proposed a direct role for
p53 in this process via its interaction with BER-associated
proteins, including DNA polymerase β and AP endonuclease (30–33).
In contrast, p53 controls NER by transcriptionally regulating NER
proteins, such as p48XPE and XPC (34). Indeed, we previously demonstrated
that Gadd45a-null cells exhibit a defect in activating the BER
pathway, which is comparable to that previously observed in
p53-null cells. Since isogenic cell lines were used for these
studies, these data suggest that Gadd45a could play a major role in
p53-dependent activation of BER (22). Based on these observations, we
proposed that Gadd45a participates in the BER pathway by
interacting with BER pathway proteins, including PCNA and APE1/Ref1
(22). In fact, Gadd45a was
reported to interact with PCNA and increased NER in response to UV
irradiation (20,35). The data in Fig. 1A and B indicate that the interaction
of Gadd45a with PCNA and APE1/Ref-1 was significantly increased in
p53 wild-type cells treated with organic selenium but not in p53
siRNA-treated cells. These data suggest that organic selenium
modulates the activity of BER via the promotion of an interaction
between p53 downstream genes Gadd45a and PCNA with APE1/Ref-1.
However, the mechanism(s) by which p53 activity affects Gadd45a
expression in organic selenium-treated cells has not yet been
clarified.
Recently, our group suggested that the antioxidant
organic selenium recovers the base damage induced by the alkylating
agent MMS in p53 wild-type cells, as well as the nucleotide damage
induced by UV irradiation (11,36).
In a previous study in which we measured the fraction of unrepaired
AP sites, these pathways were restored more extensively in
wild-type p53 cells that were pretreated with organic selenium than
in untreated cells (18). Treatment
of cells with the alkylating agent MMS primarily induces
N7-methylguanine and 3-methyladenine expression during the BER
process. Then N-methylpurine DNA glycosylase expression is induced,
which generates apurinic sites that are recognized by APE1. APE1
incises the damaged strand immediately at 5′ to the AP site. Here,
we determined that the APE1 enzyme activity was considerably
greater in p53 wild-type cells that were treated with organic
selenium than in mutant p53 cells (Fig.
2A). These data suggest that basal BER activity may be promoted
via the p53-dependent activation of APE1. In addition, the
increased activity induced by SeMet was associated with
Gadd45a-specific binding to APE1 (Fig.
2B). Indeed, APE1/Ref-1 has been reported to mediate the
cysteine reduction of a number of transcription factors, including
c-jun and p53 (37). Furthermore,
APE1 activity induced by the other form of organic selenium,
methylseleninic acid (MSeA), which has shown chemopreventive
properties, was markedly increased in Gadd45a-specific captured
cells (data not shown). These results suggest the existence of a
feedback mechanism whereby organic selenium-mediated activation of
p53 can modulate APE1/Ref-1 localization. However, the localization
of the APE1 protein was not altered in the p53 siRNA-treated cells
(Fig. 3), suggesting that the
activity of the APE1 enzyme may be increased via processes other
than protein shuttling. Our previous study reported that nuclear
APE1 in gadd45a-knockdown cells was released into the cytosol, and
therefore, the mechanism responsible for p53-dependent modulation
of APE activity requires further exploration (22).
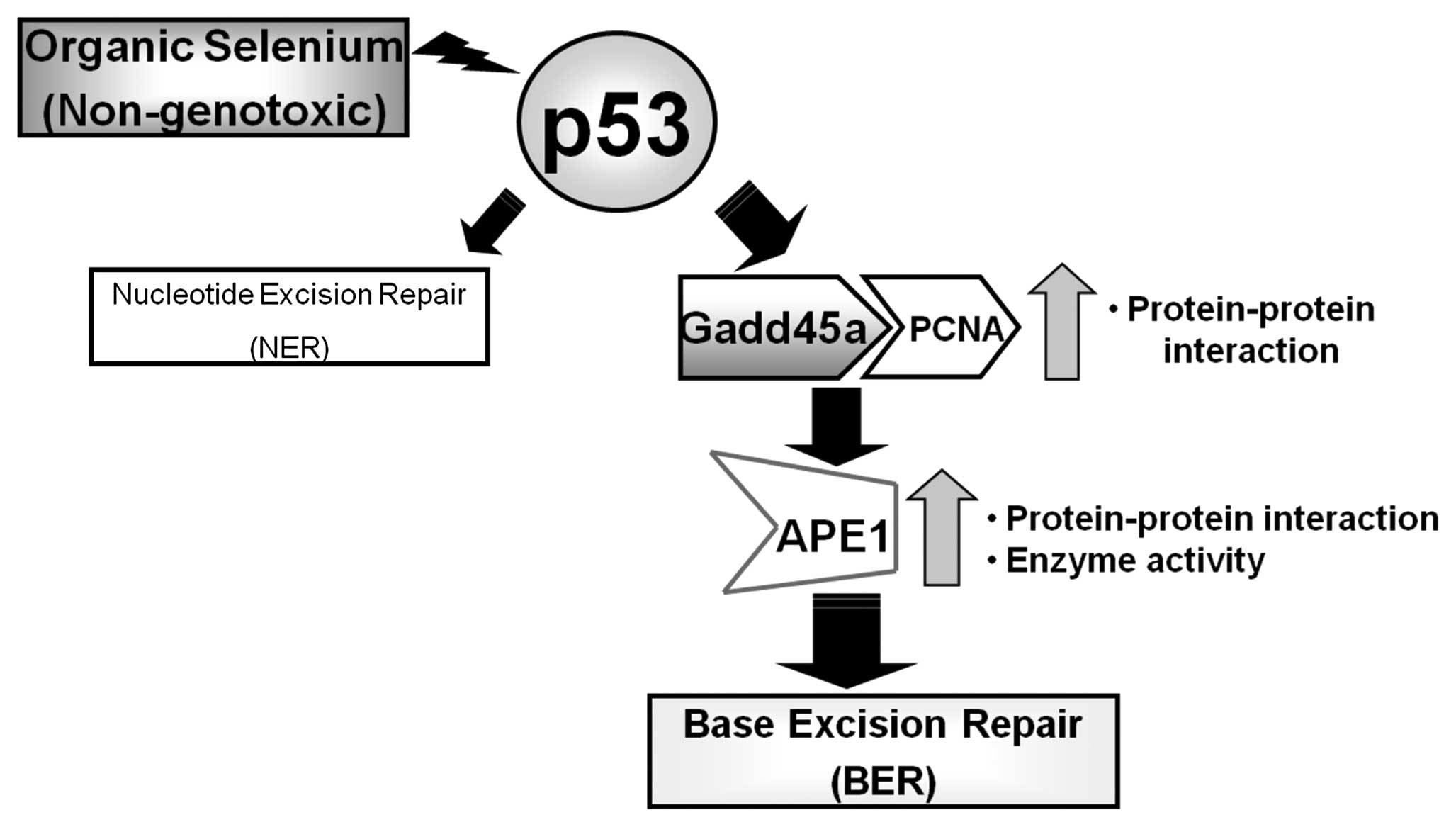
Based on the results of this study, we propose that
the promotion of BER activity by a p53-dependent pathway is
mediated by an interaction between Gadd45a, PCNA and APE1 in the
presence of organic selenium that results in increased APE1
activity (Fig. 4). This study, in
combination with our previous study, which demonstrated that
organic selenium promoted UV-induced NER (11), supports the rationale for targeting
p53-mediated DNA repair as a means of cancer prevention. Our study
supports the possibility of a novel chemopreventive mechanism of
organic selenium in preventing mutagenesis induced by various
oxidative metabolites that cause detrimental DNA base changes in
normal cells.
Acknowledgements
This subject is supported by Basic Science Research
Program through National Research Foundation of Korea (NRF),
Ministry of Education, Science and Technology, Republic of Korea
(2010-0027630) and the Korea Ministry of Environment as ‘The
Ecoinnovation Project’ (412-112-011).
References
|
1
|
Ip C: Prophylaxis of mammary neoplasia by
selenium supplementation in the initiation and promotion phases of
chemical carcinogenesis. Cancer Res. 41:4386–4390. 1981.PubMed/NCBI
|
|
2
|
Ip C, el-Bayoumi K, Upadhyaya P, Ganther
H, Vadhanavikit S and Thompson H: Comparative effect of inorganic
and organic selenocyanate derivatives in mammary cancer
chemoprevention. Carcinogenesis. 15:187–192. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu J, Jiang C, Kaeck M, Ganther H,
Vadhanavikit S, Ip C and Thompson HJ: Cellular and metabolic
effects of triphenylselenonium chloride in a mammary cell culture
model. Carcinogenesis. 16:513–517. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sinha R, Said TK and Medina D: Organic and
inorganic selenium compounds inhibit mouse mammary cell growth in
vitro by different cellular pathways. Cancer Lett. 107:277–284.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Patterson BH and Levander OA: Naturally
occurring selenium compounds in cancer chemoprevention trials: a
workshop summary. Cancer Epidemiol Biomarkers Prev. 6:63–69.
1997.PubMed/NCBI
|
|
6
|
Stewart MS, Spallholtz JE, Neldner KH and
Pence BC: Selenium compounds have disparate abilities to impose
oxidative stress and induce apoptosis. Free Redic Biol Med.
26:42–48. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiao YL, Dawsey SM, Kamangar F, Fan JH,
Abnet CC, Sun XD, Johnson LL, Gail MH, Dong ZW, Yu B, Mark SD and
Taylor PR: Total and cancer mortality after supplementation with
vitamins and minerals: follow-up of the Linxian General Population
Nutrition Intervention Trial. J Natl Cancer Inst. 101:507–518.
2009. View Article : Google Scholar
|
|
8
|
Venkateswaran V, Klotz LH, Ramani M, Sugar
LM, Jacob LE, Nam RK and Fleshner NE: A combination of
micronutrients is beneficial in reducing the incidence of prostate
cancer and increasing survival in the Lady transgenic model. Cancer
Prev Res. 2:473–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clark LC, Combs GF Jr, Turnbull BW, Slate
EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG,
Krongrad A, Lesher JL Jr, Park HK, Sanders BB Jr, Smith CL and
Taylor JR: Effects of selenium supplementation for cancer
prevention in patients with carcinoma of the skin. A randomized
controlled trial. Nutritional Prevention of Cancer Study Group.
JAMA. 276:1957–1963. 1996. View Article : Google Scholar
|
|
10
|
Heinonen OP, Albanes D, Virtamo J, Taylor
PR, Huttunen JK, Hartman AM, Haapakoski J, Malila N, Rautalahti M,
Ripatti S, Mäenpää H, Teerenhovi L, Koss L, Virolainen M and
Edwards BK: Prostate cancer and supplementation with α-tocopherol
and β-carotene: incidence and mortality in a controlled trial. J
Natl Cancer Inst. 90:440–446. 1998.
|
|
11
|
Seo YR, Kelley MR and Smith ML:
Selenomethionine regulation of p53 by a ref1-dependent redox
mechanism. Proc Natl Acad Sci USA. 99:14548–14553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ko LJ and Prives C: p53: puzzle and
paradigm. Genes Dev. 10:1054–1072. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giaccia AJ and Kastan MB: The complexity
of p53 modulation: emerging patterns from divergent signals. Genes
Dev. 12:2973–2983. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Prives C and Hall PA: The p53 pathway. J
Pathol. 187:112–126. 1999. View Article : Google Scholar
|
|
15
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liebermann DA, Hoffman B and Steinman RA:
Molecular controls of growth arrest and apoptosis: p53-dependent
and independent pathways. Oncogene. 11:199–210. 1995.PubMed/NCBI
|
|
17
|
Hwang BJ, Ford JM, Hanawalt PC and Chu G:
Expression of the p48 xeroderma pigmentosum gene is p53-dependent
and is involved in global genomic repair. Proc Natl Acad Sci USA.
96:424–428. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jung HJ, Lee JH and Seo YR: Enhancement of
methyl methanesulfonate-induced base excision repair in the
presence of selenomethionine on p53-dependent pathway. J Med Food.
12:340–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smith ML, Ford JM, Hollander MC, Bortnick
RA, Amundson SA, Seo YR, Deng CX, Hanawalt PC and Fornace AJ Jr:
P53-mediated DNA repair responses to UV-radiation: studies of mouse
cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol.
20:3705–3714. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smith ML, Chen IT, Zhan Q, Bae I, Chen CY,
Gilmer TM, Kastan MB, O’Connor PM and Fornace AJ Jr: Interaction of
the p53-regulated protein Gadd45 with proliferating cell nuclear
antigen. Science. 266:1376–1380. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia L, Zheng L, Lee HW, Bates SE, Federico
L, Shen B and O’Connor TR: Human 3-methyladenine DNA glycosylase:
effect of sequence context on excision, association with PCNA, and
stimulation by AP endonuclease. J Mol Biol. 346:1259–1274. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jung HJ, Kim EH, Mun JY, Park S, Smith ML,
Han SS and Seo YR: Base excision DNA repair defect in
Gadd45a-deficient cells. Oncogene. 26:7517–7525. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kastan MB, Zhan Q, el-Deiry WS, Carrier F,
Jacks T, Walsh WV, Plunkett BS, Vogelstein B and Fornace AJ Jr: A
mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is
defective in ataxia-telangiectasia. Cell. 71:587–597. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smith ML, Chen IT, Zhan Q, O’Connor PM and
Fornace AJ Jr: Involvement of the p53 tumor suppressor in repair of
u.v.-type DNA damage. Oncogene. 10:1053–1059. 1995.PubMed/NCBI
|
|
25
|
Seeberg E, Eide L and Bjørås M: The base
excision repair pathway. Trans Biochem Sci. 20:391–397. 1995.
View Article : Google Scholar
|
|
26
|
Sung JS and Demple B: Roles of base
excision repair subpathways in correcting oxidized abasic sites in
DNA. FEBS J. 273:1620–1629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lindahl T and Andersson A: Rate of chain
breakage at apurinic sites in double-stranded deoxyribonucleic
acid. Biochemistry. 11:3618–3623. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lindahl T and Nyberg B: Rate of
depurination of native deoxyribonucleic acid. Biochemistry.
11:3610–3618. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lippman SM, Klein EA, Goodman PJ, Lucia
MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM,
Hartline JA, Parsons JK, Bearden JD III, Crawford ED, Goodman GE,
Claudio J, Winquist E, Cook ED, Karp DD, Walther P, Lieber MM,
Kristal AR, Darke AK, Arnold KB, Ganz PA, Santella RM, Albanes D,
Taylor PR, Probstfield JL, Jagpal TJ, Crowley JJ, Meyskens FL Jr,
Baker LH and Coltman CA Jr: Effect of selenium and vitamin E on
risk of prostate cancer and other cancers: the Selenium and Vitamin
E Cancer Prevention Trial (SELECT). JAMA. 301:39–51. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gaiddon C, Moorthy NC and Prives C: Ref-1
regulates the transactivation and pro-apoptotic functions of p53 in
vivo. EMBO J. 18:5609–5621. 1999. View Article : Google Scholar
|
|
31
|
Offer H, Zurer I, Banfalvi G, Reha’k M,
Falcovitz A, Milyavsky M, Goldfinger N and Rotter V: p53 modulates
base excision repair activity in a cell cycle-specific manner after
genotoxic stress. Cancer Res. 61:88–96. 2001.PubMed/NCBI
|
|
32
|
Zhou J, Ahn J, Wilson SH and Prives C: A
role for p53 in base excision repair. EMBO J. 20:914–923. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee YS and Chung MH: Base excision repair
synthesis of DNA containing 8-oxoguanine in Escherichia
coli. Exp Mol Med. 35:106–112. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Adimoolam S and Ford JM: p53 and DNA
damage-inducible expression of the xeroderma pigmentosum group C
gene. Proc Natl Acad Sci USA. 99:12985–12990. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vairapandi M, Azam N, Balliet AG, Hoffman
B and Liebermann DA: Characterization of MyD118, Gadd45, and
proliferating cell nuclear antigen (PCNA) interacting domains. PCNA
impedes MyD118 and Gadd45-mediated negative growth control. J Biol
Chem. 275:16810–16819. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Seo YR, Sweeney C and Smith ML:
Selenomethionine induction of DNA repair response in human
fibroblasts. Oncogene. 21:3663–3669. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hanson S, Kim E and Deppert W: Redox
factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting
p53 tetramerization. Oncogene. 24:1641–1647. 2005. View Article : Google Scholar : PubMed/NCBI
|