Introduction
Decoy receptor 3 (DcR3), a soluble decoy receptor,
is a member of the tumor necrosis factor receptor superfamily
(1). DcR3 can prevent cell death
and may play an important role in the pathogenesis of many
malignancies by binding and inhibiting the function of the Fas
receptor (2). Accumulating evidence
has demonstrated that DcR3 is overexpressed in several cancers
(3,4) and might protect cancer cells against
the cytotoxic and regulatory effects of FasL, LIGHT19 and TNF-like
molecule (5,6). In addition, the expression of DcR3
positively correlated with tumor invasiveness in pancreatic cancer
cells (7). DcR3 also has a
described role as a tumor promoter in renal carcinoma, hepatoma,
gastric carcinoma, and breast cancer (6,8). DcR3
is expected to be an effective target for tumor gene therapy.
However, little is known about the molecular mechanisms underlying
the role of DcR3 in tumor progression and metastasis.
Colon cancer is one of the most common malignancies
around the world, accounting for 8–15% of all cancer deaths
(9). Colon cancer cells can co-opt
signaling pathways stimulated by both exogenous factors and
endogenous genetic agents for growth advantage and evasion of
apoptosis (10). To explore the
effects of DcR3 during tumorigenesis and progression of colon
cancer, we determined the expression of DcR3 in a large number of
colon cancer tissues. Cancerous colon tissues have high DcR3
expression and its expression is correlated with tumor
differentiation, serosal invasion, Duck’s stage and lymph node and
liver metastases. Nevertheless, the precise mechanisms of DcR3 are
not completely clear in progression and metastasis of colon cancer.
Current therapies for colon cancer consist of excision,
chemotherapy and radiotherapy, all of which have toxic or
side-effects. Gene therapy has become a promising strategy for
cancer treatment due to minimal drug resistance, high efficiency
and specificity for drug delivery. In this study, we further
determined the role of DcR3 in colon cancer, and assessed whether
it may serve as a viable therapeutic target.
In recent years, RNA interference (RNAi) has shown
great potential as a gene therapy strategy for cancer. Gene
ablation technology involving short hairpin RNA (shRNA) synthesis
can considerably suppress gene expression (11,12).
RNAi is mediated by small interfering RNAs (siRNAs) that are
produced from double-stranded RNAs (dsRNAs) of exogenous or
endogenous origin by an endonuclease of the ribonuclease-III type
called Dicer. The resulting siRNAs are approximately 21–23
nucleotides (nt) long and are then incorporated into a nuclease
complex, and transported to the targeted RNA for cleavage by the
RNA-inducing silencing complex. shRNA transcribed from a vector are
thought to suppress the expression of targeted genes more
efficiently than synthesized siRNA (13,14).
In this study, we use a plasmid-based polymerase III promoter
system for expressing shRNA to decrease DcR3 levels. We then assess
its effects on the growth and invasive capability of SW480 colon
cancer cells, to provide theoretical basis for DcR3 gene therapy by
RNAi as a treatment strategy for colon cancer.
Materials and methods
Plasmid vector preparation
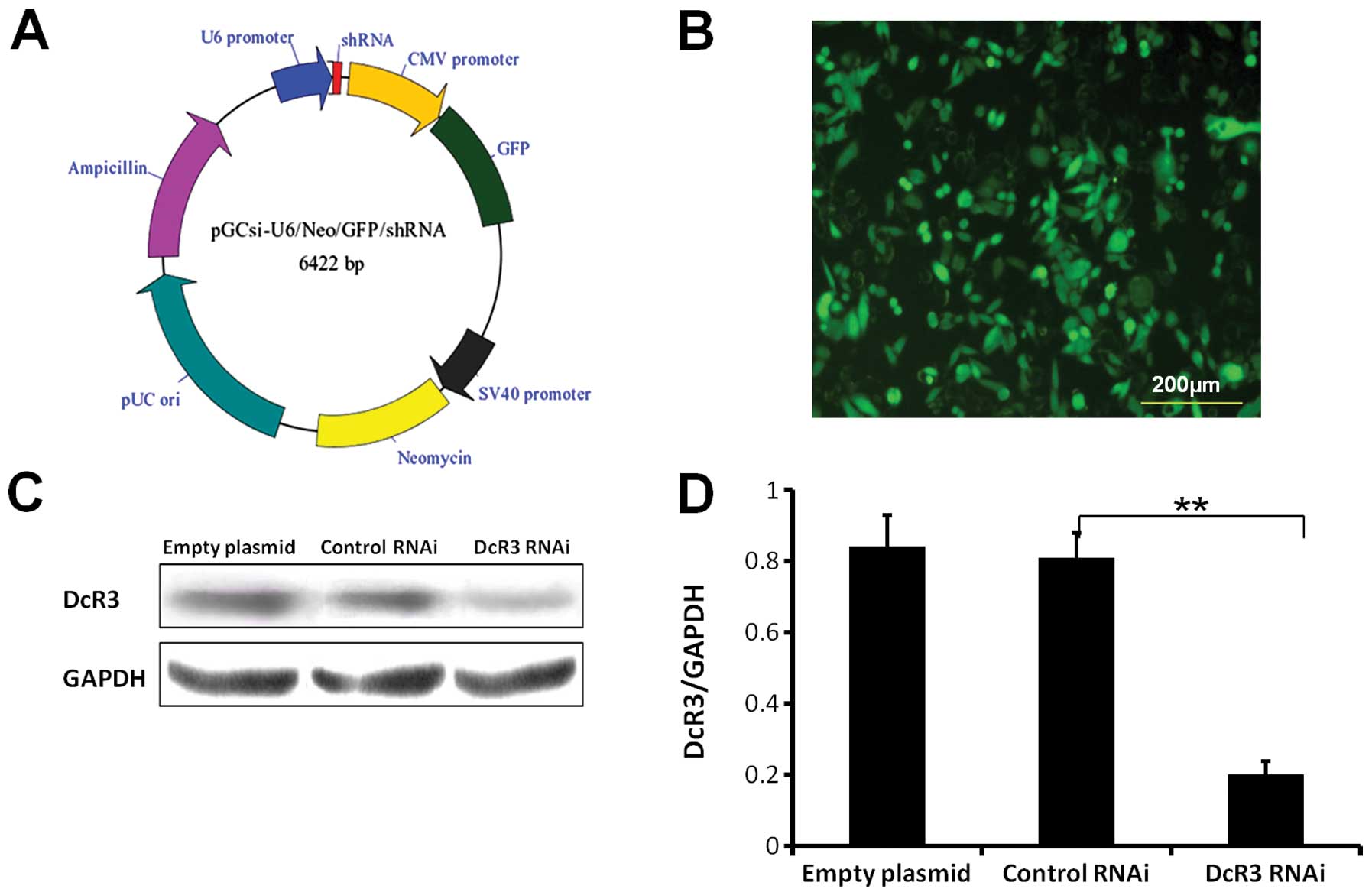
The plasmid vector (pGCSi-U6-Neo-GFP-DcR3shRNA)
carrying DcR3-shRNA was purchased from TransGen Biotech, Co., Ltd.
(China). The sequence of DcR3-shRNA with a stem-loop structure:
CGCTGGTTTCTGCTTGGAGCA- CTCGAG- TGCTCCAAGCAGAAACCAGCG. The plasmid
vector of pGCSi-U6-Neo-GFP-NegConshRNA (as the negative control)
and the empty plasmid were purchased. To amplify the vectors,
cloning was performed. The plasmid vectors were transformed into
the competent E. coli DH5α bacteria followed by ampicillin
selection. The purified plasmid DNAs were tested for identification
by digesting the clones with restriction endonuclease enzyme. The
plasmids were extracted by Fastfiler Endo Free Plasmid Maxiprep kit
(Omega Bio-Tek, USA).
Cell culture and experimental
reagents
SW480 cells were cultured in Leibovitz’s L-15 medium
(Gibco, USA) supplemented with 10% fetal bovine serum (FBS), 100
U/ml penicillin and 100 μg/ml streptomycin, 100% atmospheric air
without additional CO2 at 37°C. The plasmid DNA was
transfected into SW480 cells by Lipofectamine 2000 reagent
(Invitrogen, USA), followed with G418 and flow cytometry
(FACSVantage, BD Biosciences) for selecting stably transfected
cells. Primary antibody of DcR3 (ab8405) was purchased from Abcam
and other antibodies for vascular endothelial growth factor
(VEGF)-C, VEGF-D, MT1-matrix metalloproteinase (MMP) and GAPDH were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Other reagents used in this study, such as anti-mouse-IgG-HRP
and anti-rabbit-IgG-HRP, were purchased from California Biosciences
(USA), Transwell Invasion Chambers were purchased from Corning
(USA).
Plasmid transfection, stably transfected
cell selection
Briefly, the day before transfection,
2×105 of cells were seeded in antibiotic-free L-15
medium into 6-well plates. The cells were transfected with a
mixture of 3 μg plasmids and 10.5 μl of Lipofectamine 2000 reagent
[ratio of plasmid DNA (μg)/Lipofectamine 2000 reagent (μl) was
1:3.5, in our previous study] in 500 μl medium per well. At 6 h
post-transfection, the medium was replaced by complete medium, then
cells were screened by 800 mg/l G418 (the G418 concentration was
adjusted according to cell viability, obtained from our previous
study) and cultured up to 4 weeks. The DcR3-RNAi, negative control,
and empty plasmid stable transfection cells were harvested. The
cells were harvested by trypsinization and then resuspended in PBS
at 4°C. The mean fluorescent intensity of green fluorescent protein
(GFP) from each group was analyzed by flow cytometry (FCM)
(FACSAria; BD Biosciences) through a 488 nm laser for excitation
and the fluorescence emissions at 525 nm for GFP. The GFP positive
cells were sorted. Stable cells of 3 groups were expanded. The
expression levels of DcR3 in the 3 groups were examined by western
blotting to detect the effect of DcR3 knockdown. Other experiments
were performed as indicated.
Cell growth and viability studied by MTT
assay
Cells of 3 groups were seeded with 100 μl medium in
a 96-well plate (5×103 per well), and the MTT assay was
performed after incubation for indicated times. MTT reagent (5
mg/ml) was added to each well, and incubated for 4 h at 37°C. The
resulting formazan crystals were solubilized by the addition of 150
μl DMSO to each well. The optical density at 490 nm was measured
and cell viability was determined by the growth curve for 7 days.
All MTT experiments were performed in triplicate and repeated at
least three times.
Colony formation assay
DcR3 RNAi, empty plasmid, and control RNAi stable
transfected cells were seeded in Leibovitz’s L-15 medium
supplemented with 10% FBS for 14 days prior to crystal violet
staining for colony counting. Cells were then fixed in 4%
paraformaldehyde and incubated in 0.01% crystal violet for 30 min
for colony counting.
Monolayer cell migration assay
Cell migration was modeled using a wound-healing
assay. DcR3 RNAi or empty plasmid/control RNAi stable transfected
SW480 cells were seeded in 6-well plate for 24 h in L-15 medium and
treated as above. The 100% confluent monolayer SW480 cells were
then scraped with a sterile 100 μl pipette tip and cell debris was
washed with PBS. Cells that migrated into the wounded areas were
photographed at the indicated times with an inverted microscope
equipped with a digital camera. The extent of healing was defined
as the ratio of the difference between the original and the
remaining wound areas compared with the original wound area.
Transwell invasion assay
Matrigel Invasion Chambers were hydrated for 4 h
prior to placing the cells into the chambers. Cells from each group
were seeded at 1×105 cells in 200 μl serum-free L-15
medium in the upper chambers of the Transwell and the lower chamber
was filled with 500 μl complete L-15 medium with 10% FBS. The cells
were incubated for 48 h for the invasion assay. The cells remaining
on the upper surface of the filter were removed carefully by cotton
tips, and cells on the underside of the membrane were fixed with 4%
paraformaldehyde. After washing the chambers 5 times by dipping the
chambers in a large beaker filled with dH2O, the cells
remaining on the bottom of the chamber were stained with 0.1%
crystal violet for 30 min. The migrated cells were photographed
under an optical microscope (x400) and counted at 12 different
areas. The experiments were performed in triplicate and the data
were compared with the controls.
Apoptosis and cell cycle analysis
DcR3 RNAi, control RNAi, and empty plasmid cells
were cultured in the same conditions and harvested by
trypsinization. The following steps were carried out in accordance
with the apoptosis detection protocol. Cells were washed once in
PBS, then once in 1X binding buffer, and resuspended in 1X binding
buffer at 5×106/ml. The cell suspension (100 μl) was
stained with 5 μl of fluorochrome-conjugated Annexin V, incubating
15 min at room temperature. Cells were washed and resuspended
before adding 5 μl of propidium iodide (PI). The samples were
stored at 2–8°C in the dark before analyzing by FCM (EPICS XL
Coulter Co., USA) within 30 min. Cells in early-stage apoptosis
(Annexin V positive, viability dye-negative) and late-stage
apoptotic and necrotic cells (Annexin V, viability dye-positive)
were distinguished, and the proportions were measured. The data
were processed using MultiCycle software.
Cell cycle status was determined by staining the
cellular DNA with PI, which was directly proportional to the amount
of DNA within the cell. For cycle detection, cells were treated
with RNase after fixation overnight in 70% ethanol. Cells were
stained with PI for 30 min at 4°C, and single cell suspensions of
the samples were tested by FCM (FACSAria) passing through a 488 nm
laser for excitation and fluorescence emissions were collected at
620 nm for PI. Cell cycle results were analyzed using the
MultiCycle software.
Enzyme-linked immunosorbent assay
(ELISA)
Cells of 3 groups were cultured in complete medium
(with 10% FBS) under the same condition. Medium was refreshed when
the cells reached 70% confluence. Cells were then washed by PBS and
L-15 medium without FBS. The cells were incubated with serum-free
medium for 20 h. Concentrations of MMP9, MMP2, and VEGF in the cell
culture supernatants of SW480 cells were quantified using MMP9,
MMP2 and VEGF ELISA kits (R&D Systems, Inc., USA). Each sample
was analyzed in triplicate and manipulated according to the
manufacturer’s protocol.
Western blotting
Stably transfected cells of 3 groups were lysed in
RIPA buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100,
0.1% SDS, 1% sodium deoxycholate, 5 mM EDTA, 100 mM NaF and 1 mM
Na3VO4] containing a protease inhibitor
cocktail for 30 min on ice, followed by centrifuged for 30 min at
13,200 rpm. Protein concentrations were determined by the BCA
method (Pierce, USA). Equal total proteins were subjected to
electrophoresis by 12% SDS-PAGE gel, followed by transferring onto
a PVDF membrane using a wet transblot system (Bio-Rad, Hercules,
CA, USA). The membranes were blocked for 1 h at room temperature
with 5% nonfat dry milk and incubated overnight at 4°C with
antibodies against DcR3, MT1-MMP, VEGF-C, VEGF-D and GAPDH
(1:1,000). After washing, the membrane was incubated for 1 h with
HRP-conjugated goat anti-rabbit secondary antibody diluted 1:5,000
in PBST. After further washing, the membrane was processed using
SuperSignal West Pico Chemiluminescent Substrate (Pierce). The
membrane was exposed to FujiFilm LAS3000 Image Reader (Fuji,
Japan). The band densities of the western blots were normalized
relative to the relevant GAPDH band density with ImageJ software
(NIH).
Statistical analysis
All experiments were performed three times and the
results were expressed as mean ± SD. Statistical analysis was
performed by SPSS 11.0. t-tests were used to compare the average
values between two data populations. A P-value of less than 0.05
was considered to be statistically significant.
Results
Establishing stably transfected DcR3 RNAi
SW480 cells
The expression level of DcR3 was decreased
significantly in DcR3 RNAi SW480 cells compared to control cells
(P<0.01) (Fig. 1C and D). This
was also accompanied by high expression of GFP (Fig. 1B). Stable knockdown of DcR3 was
still achieved after passaging the cells at least 10 times.
Effects of decreasing DcR3 on the
proliferation and colony formation of SW480 cells
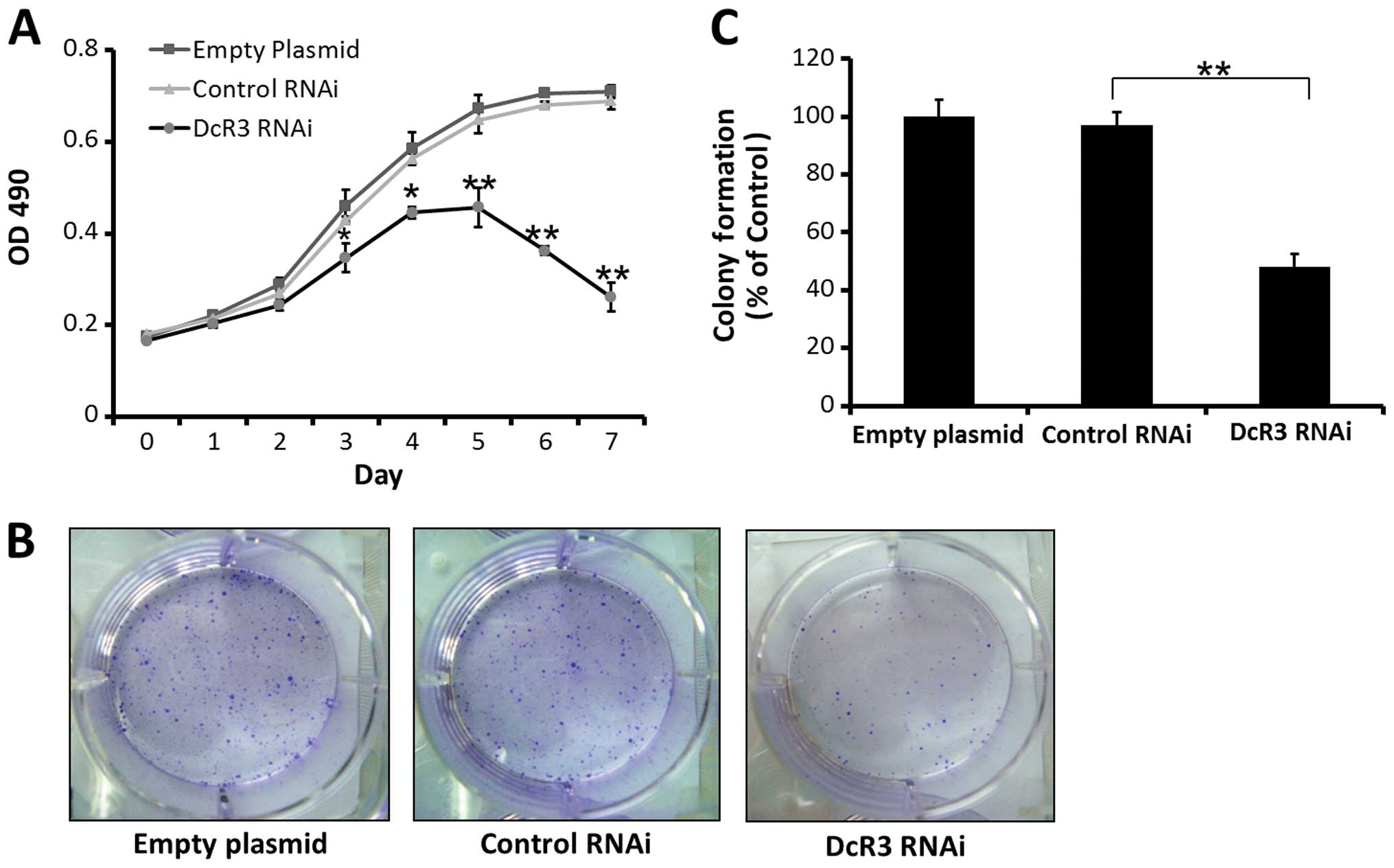
A MTT growth assay showed that inhibiting DcR3
expression can restrain the growth of SW480 cells compared to
control cells, according to the growth curves of 3 groups (Fig. 2A). Inhibiting DcR3 expression also
significantly decrease the number of colony formation compared to
control groups ( Fig. 2B and
C).
Effects of inhibiting DcR3 on the
apoptosis and cell cycle distribution of SW480 cells
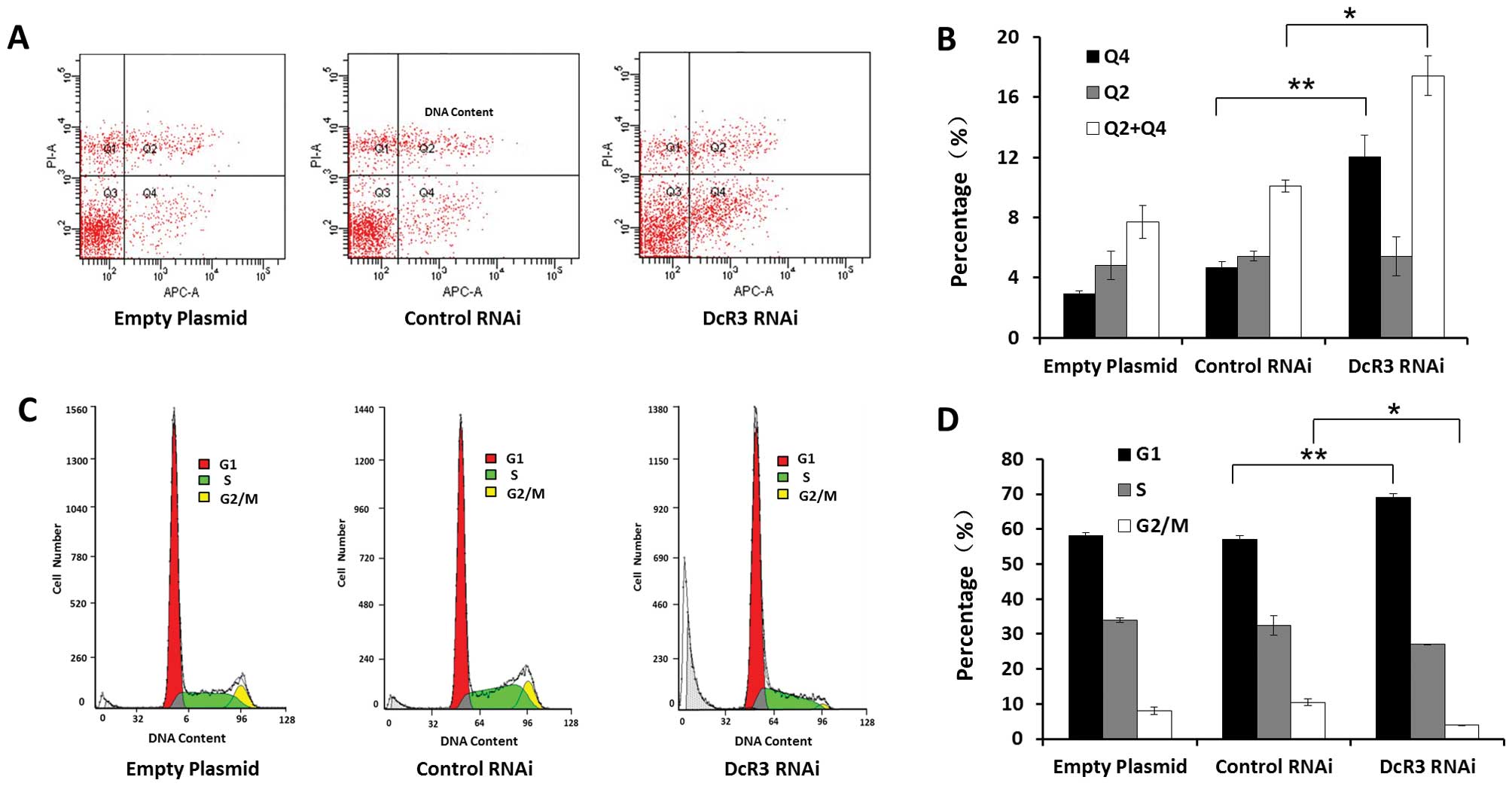
Inhibiting DcR3 by RNAi induced apoptosis and G1
cell cycle arrest of SW480 cells compared to control cells
(Fig. 3). The percentage of
early-stage apoptotic cells was 12.0±1.5%, which was higher than
cells with empty plasmids (2.9±0.2%) or with control RNAi plasmids
(4.7±0.4%) (P<0.01). No observable effects were found on
late-stage apoptotic and necrotic cells (P>0.05) (Fig. 3A and B). The cell cycle analysis
showed a possible G1 phase arrest. The proportion of G1 phase cells
increased, while decreased G2/M phase cells and unchanged S phase
cells were observed. We also found that DcR3 RNAi cells contained a
sub-G1 peak (cell debris) (Fig.
3C). Taken together, these results demonstrate that loss of
DcR3 induced apoptosis and altered the cell cycle distribution of
SW480 cells.
Effects of inhibiting DcR3 on the
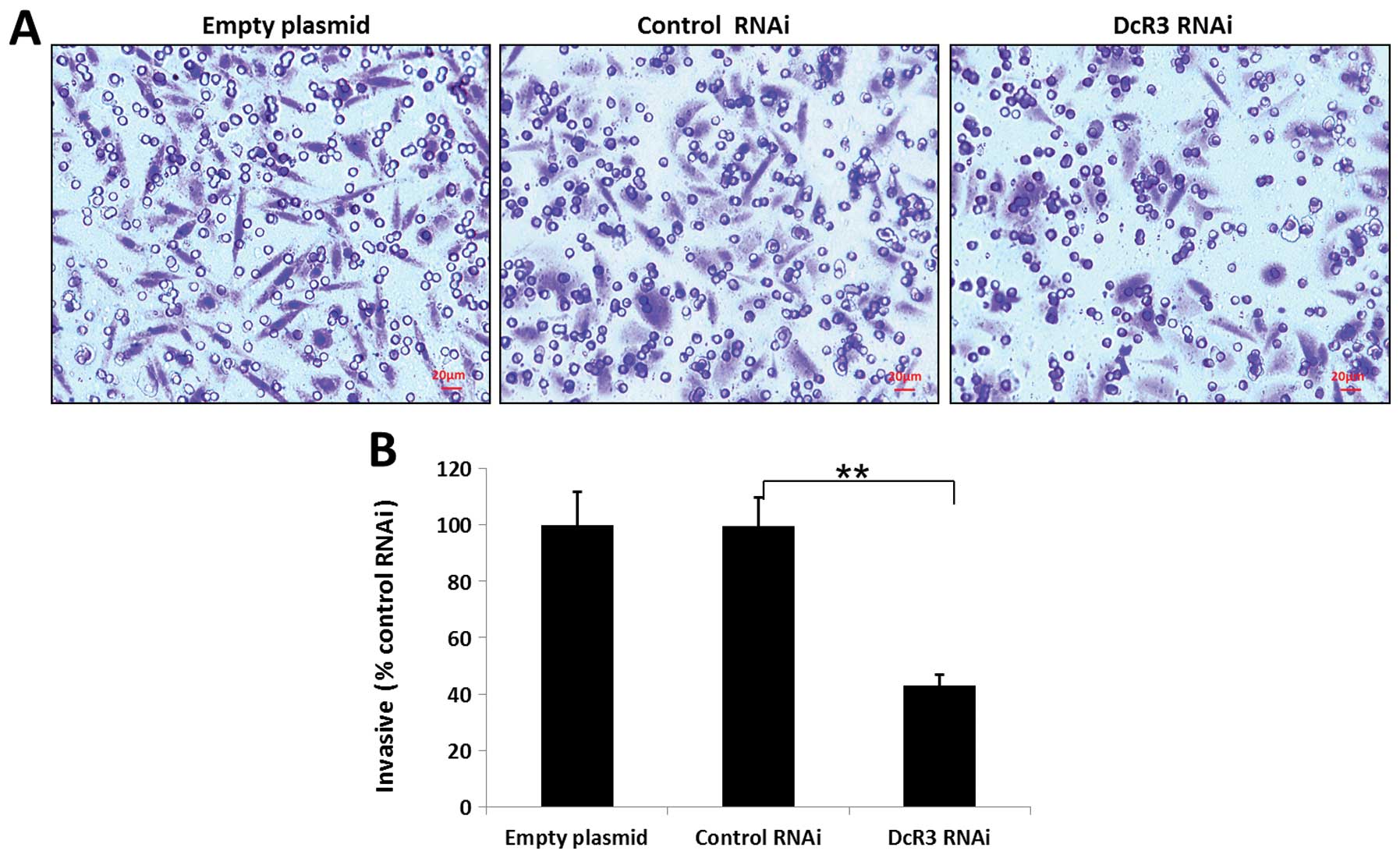
migration and invasiveness of SW480 cells
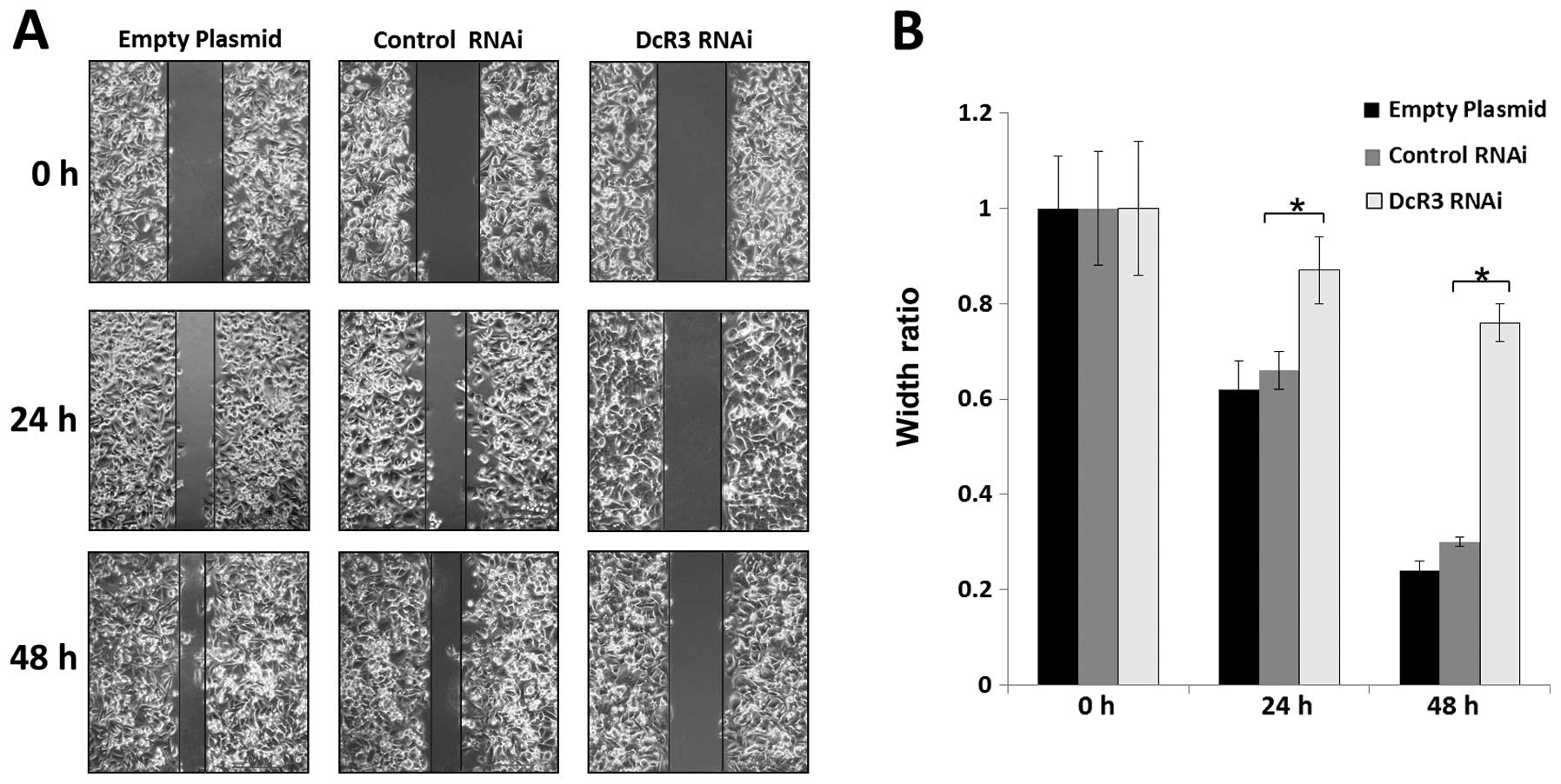
To study whether DcR3 regulates the migration and
invasion, we silenced its expression in SW480 cells and performed
wound healing and Transwell assays. Knockdown of DcR3 was able to
impair the migration (Fig. 4) and
invasiveness (Fig. 5) of SW480
cells (P<0.01).
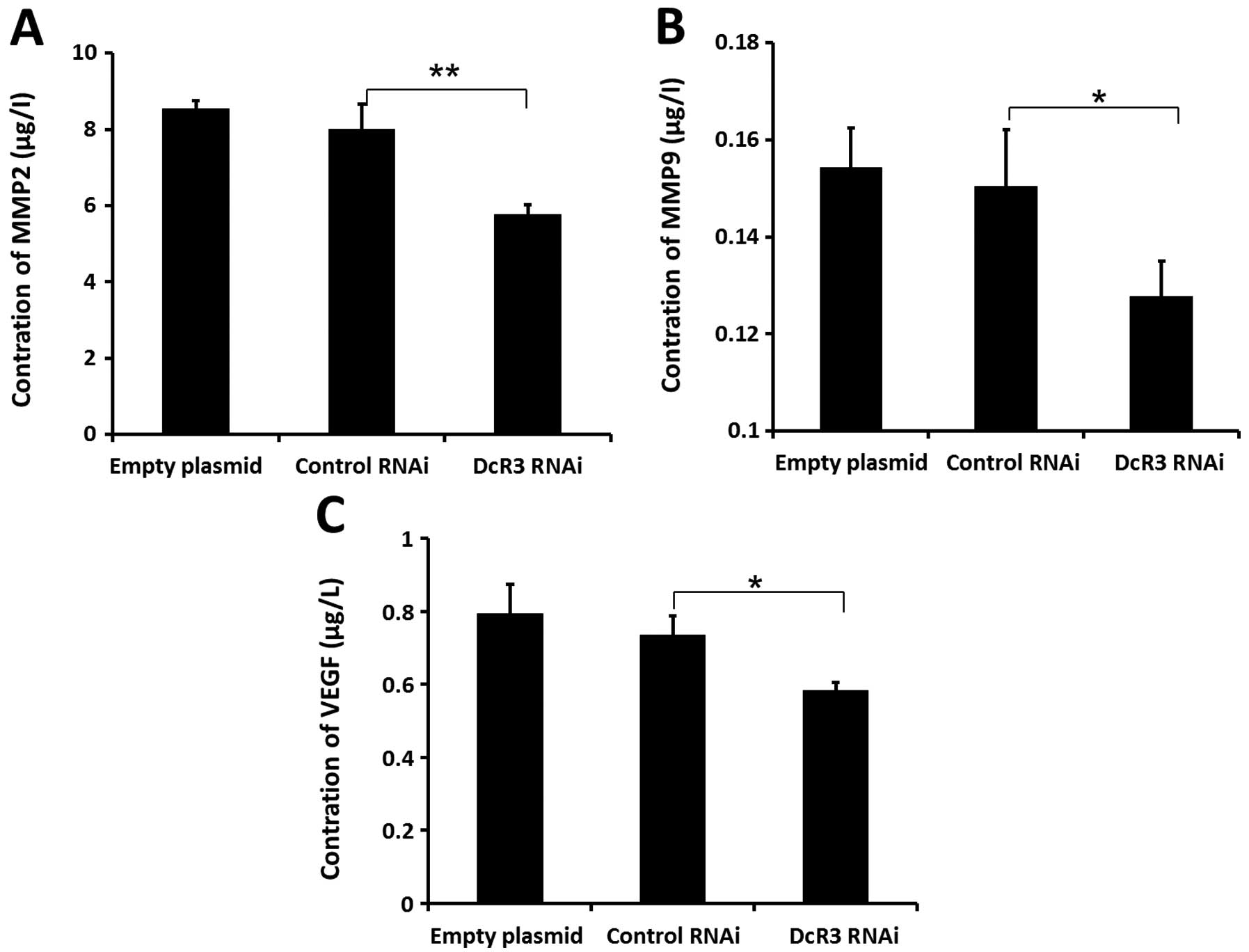
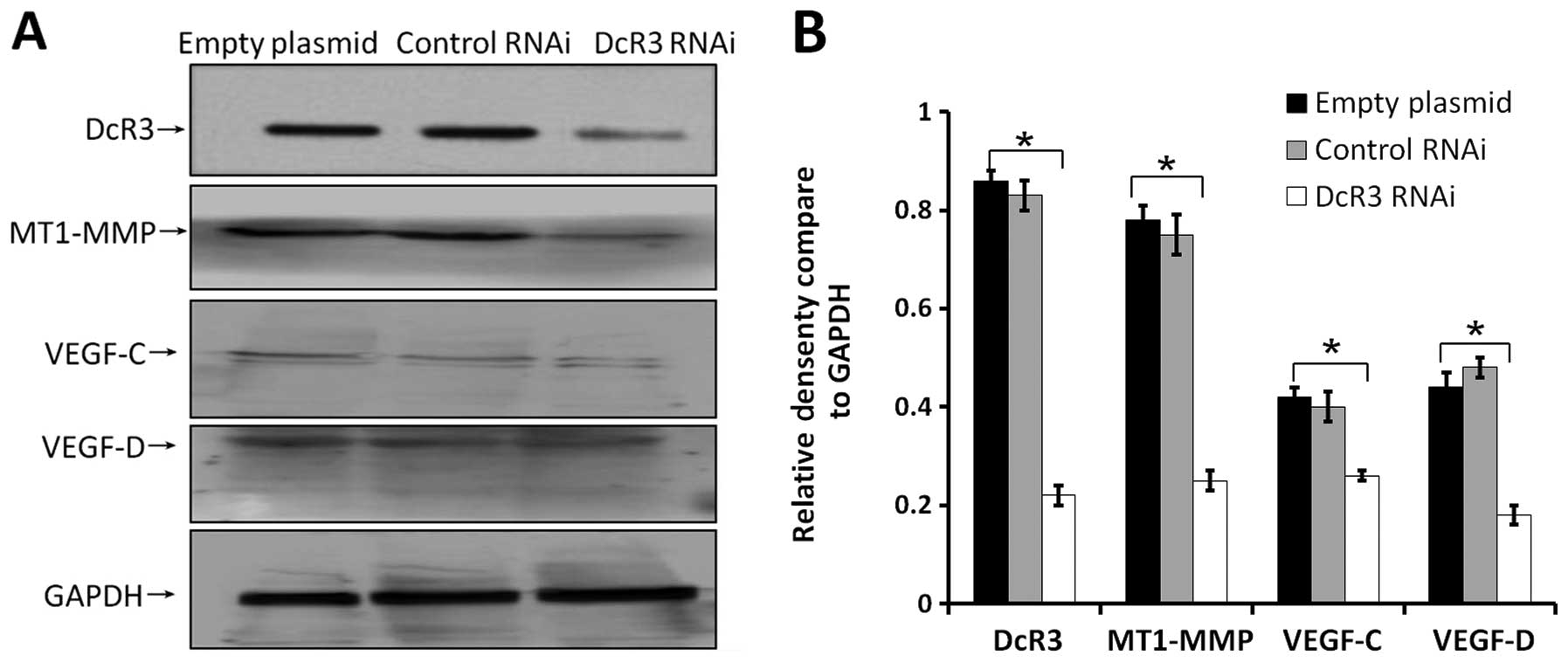
Effects of inhibiting DcR3 on the
pro-metastatic gene expressions
Cancer cell metastasis is regulated by many proteins
in vivo. To fully understand the molecular mechanism of DcR3
during metastasis, we determine the expression levels of some
metastasis-associated proteins in cells of 3 groups. Extracellular
levels of MMP2 (Fig. 6A), MMP9
(Fig. 6B), and VEGF (Fig. 6C) were significantly decreased
compared to control cells. Loss of DcR3 in these cells also led to
decrease endogenous expression of MT1-MMP, VEGF-C, VEGF-D (Fig. 7). These results indicated that the
downregulation of DcR3 expression might inhibit the metastatic
potential of SW480 cells by decreasing MT1-MMP, MMP2/9 and VEGF-C/D
levels.
Discussion
Colon cancer is one of the most common malignancies
in the world. In recent years, the morbidity and mortality of colon
cancer has increased in China (9).
Although significant improvement in diagnosing and treating colon
cancer has been made in recent years, the advanced and metastatic
disease remains a challenge. The 5-year survival rate after
surgical resection in colon cancer patients with liver or lung
metastases is approximately 30–50% (15,16).
More importantly, no curative chemotherapeutic treatment is
presently available for patients with metastatic disease.
DcR3 protein belongs to the tumor necrosis factor
family. Its expression is upregulated in several tumors, and its
function during tumorigenesis has attracted extensive attention.
Several studies have demonstrated that DcR3 can regulate cell
proliferation, apoptosis and immune response by antagonizing FasL-,
LIGH- and TLIA-mediated signals (17–19).
DcR3 is closely related to tumor metastasis. However, the clinical
significance in tumor metastasis of increased DcR3 levels have been
confined to studying its relationship to clinical data (6,8), and
the molecular mechanism of DcR3 on tumor metastasis is rarely
reported. In this study, we inhibited DcR3 expression by RNAi in
SW480 cells with high original expression of the DcR3 gene
(20). Loss of DcR3 inhibited the
proliferation and induced apoptosis of these cells. We further
demonstrated that DcR3 correlated to the growth of colon cancer
cells. Besides, we investigated the change of metastatic ability
and the underlying mechanism of DcR3 on SW480 cells.
As a greatly harmful disease to human health, the
biological processes of colon cancer include tumorigenesis,
progression, invasion and metastasis. The common tumor metastasis
pathways include lymphatic, hematogenous and transcoelomic
metastasises. Lymphatic metastasis is the behavior observed in the
early stage of progression and metastasis of colon cancer. Patients
with colon cancer often succumb to the disease due to multiple
organ failure caused by blood metastasis in its advanced-stage.
Pro-metastatic genes, changes in adhesion molecules, abnormal
angiogenesis, matrix degradation and evasion in immune surveillance
all contribute to the metastatic behavior of cancer cells.
Angiogenesis, basement membrane (BM) and extracellular matrix (ECM)
degradation are two essential steps during metastasis. Currently,
the capability of angiogenesis is considered as an important
character of malignancy (21,22).
Angiogenic regulators promote the growth of vascular endothelial
cells and this is achieved in two ways: autocrine approach by
vascular endothelial cells and paracrine pathway by tumor cells. In
the process of tumor angiogenesis, the latter plays a main role.
VEGF is a potent inducer of a variety of biological effects which
was separated and purified firstly by Leung et al in 1989
(23). VEGF family members regulate
normal physiological and pathological blood vessel growth. In
recent years, studies have shown that macrophages and mast cells
which invade into tumor tissue and tumor cells can secrete high
levels of VEGF to stimulate the proliferation of tumor vascular
endothelial cells through paracrine pathway. VEGF also increases
vascular permeability, being beneficial to the recruitment of
monocytes and fibroblasts, formation of tumor stroma and tumor
cells getting into blood vessels, to enhance the metastatic
potential of cancer cells (24,25).
Studies have shown that the expression of VEGF-C and VEGF-D was
positive in 96 and 90% of colon and rectal cancers, respectively
(26). VEGF-C and VEGF-D can
stimulate the proliferation of lymphatic endothelial cells,
resulting in lymphangiogenesis. VEGF-C and VEGF-D can also enhance
lymphatic metastasis which has been identified as a prognostic
indicator of certain types of tumors. However, different studies by
Yang et al(27) have
reported that the pro-angiogenic role of DcR3 does not depend on
VEGFs. In our study, the silencing of DcR3 gene was associated with
reduced levels of VEGF, VEGF-C and VEGF-D. The invasiveness and
migration ability of SW480 cells were significantly impaired upon
inhibiting DcR3. Knockdown of DcR3 also inhibited growth of
xenograft tumors and decreased the microvessel and lymphatic vessel
densities of tumor tissues. The mechanism by which DcR3 exerts its
effects requires further studies.
Degradation of the extracellular matrix is an
important stage during the metastatic spread of tumor cells to
surrounding tissues. This is mediated by MMPs secreted by tumor
cells. Therefore, MMPs have been associated with poor prognosis in
a variety of cancers. The MMPs family includes 21 kinds of zinc
metal ion-dependent proteolytic enzymes. Some studies have found
that the expression levels of MMP2 and MMP9, which are the key
enzymes in the degradation of BM, were upregulated in a number of
cancers (28). Recent studies have
demonstrated that the MMPs can also directly regulate angiogenesis
through factors such as MMP2, MMP9 and MMP14, in addition to ECM
degradation (29,30). MMP2 is secreted by tumor and stromal
cells through the zymogen form after hydrolysis (31), and able to degrade mucin IV and V,
and collagen. Overexpression of DcR3 in human umbilical vein
endothelial cells promoted cell proliferation, migration, MMP2
expression, tube formation and angiogenesis (27). In our studies, we also demonstrated
a relationship between DcR3 and MMP2 expression. The activity of
MMP2 was regulated by MT1-MMP, which is a type I transmembrane MMP.
MT1-MMP activates MMP2 and MMP3 precursor as well as reconstruct
the ECM (32). Additionally, MMP9
can also degrade ECM and BM located at the surface of tumor cells,
allowing the invasion of these cells to surrounding tissue.
Moreover, MMP9 plays an important role in angiogenesis and survival
of metastatic tumor nodules. Some studies have indicated that MMPs,
such as MMP9 and MT1-MMP, can regulate VEGFs expression. We show
that inhibiting DcR3 also decreased MT1-MMP, MMP2 and MMP9
expression, along with reduced migration and invasiveness of SW480
cells. We will further validate whether the downregulation of DcR3
can reduce enzymatic activity of MMPs.
Our results suggest that DcR3 regulates the growth
and metastasis of colon cancer. DcR3, VEGFs and MMPS factors might
play important roles during tumorigenesis and metastasis of colon
cancer and interact with each other. However, how DcR3 regulates
VEGFs and MMPs expression remains unclear. Our study suggests that
DcR3 might serve as a therapeutic target for colon cancer.
Acknowledgements
The current study was supported by research grants
from the National Natural Science Foundation of China
(30972886).
Abbreviations:
|
DcR3
|
decoy receptor 3
|
|
RNAi
|
RNA interference
|
|
shRNA
|
short hairpin RNA
|
|
dsRNA
|
double-stranded RNA
|
|
VEGFs
|
vascular endothelial growth
factors
|
|
MMPs
|
matrix metalloproteinases
|
|
GFP
|
green fluorescent protein
|
|
FCM
|
flow cytometry
|
|
PI
|
propidium iodide
|
References
|
1
|
Xiong G, Guo H, Ge X, Xu X, Yang X, Yang
K, Jiang Y and Bai Y: Decoy receptor 3 expression in esophageal
squamous cell carcinoma: correlation with tumour invasion and
metastasis. Biomarkers. 16:155–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Colucci S, Brunetti G, Mori G, Oranger A,
Centonze M, Mori C, Cantatore FP, Tamma R, Rizzi R, Liso V, Zallone
A and Grano M: Soluble decoy receptor 3 modulates the survival and
formation of osteoclasts from multiple myeloma bone disease
patients. Leukemia. 23:2139–2146. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Connor JP and Felder M: Ascites from
epithelial ovarian cancer contain high levels of functional decoy
receptor 3 (DcR3) and is associated with platinum resistance.
Gynecol Oncol. 111:330–335. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiong G, Guo H, Wang K, Hu H, Wang D, Xu
X, Guan X, Yang K and Bai Y: Polymorphisms of decoy receptor 3 are
associated with risk of esophageal squamous cell carcinoma in
Chinese Han. Tumour Biol. 31:443–449. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen J, Zhang L and Kim S: Quantification
and detection of DcR3, a decoy receptor in TNFR family. J Immunol
Methods. 285:63–70. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu Y, Han B, Sheng H, Lin M, Moore PA,
Zhang J and Wu J: Clinical significance of detecting elevated serum
DcR3/TR6/M68 in malignant tumor patients. Int J Cancer.
105:724–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsuji S, Hosotani R, Yonehara S, et al:
Endogenous decoy receptor 3 blocks the growth inhibition signals
mediated by Fas ligand in human pancreatic adenocarcinoma. Int J
Cancer. 106:17–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Macher-Goeppinger S, Aulmann S, Wagener N,
Funke B, Tagscherer KE, Haferkamp A, Hohenfellner M, Kim S,
Autschbach F, Schirmacher P and Roth W: Decoy receptor 3 is a
prognostic factor in renal cell cancer. Neoplasia. 10:1049–1056.
2008.PubMed/NCBI
|
|
9
|
Jemal A, Murray T, Ward E, Samuels A,
Tiwari RC, Ghafoor A, Feuer EJ and Thun MJ: Cancer statistics,
2005. CA Cancer J Clin. 55:10–30. 2005. View Article : Google Scholar
|
|
10
|
Lee WS, Kang M, Baek JH, Lee JI and Ha SY:
Clinical impact of tumor-infiltrating lymphocytes for survival in
curatively resected stage IV colon cancer with isolated liver or
lung metastasis. Ann Surg Oncol. 20:697–702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jagani H, Rao JV, Palanimuthu VR,
Hariharapura RC and Gang SA: A nanoformulation of siRNA and its
role in cancer therapy: in vitro and in vivo evaluation. Cell Mol
Biol Lett. 18:120–136. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okamoto S, Amaishi Y, Goto Y, Ikeda H,
Fujiwara H, Kuzushima K, Yasukawa M, Shiku H and Mineno J: A
promising vector for TCR gene therapy: differential effect of
siRNA, 2A peptide, and disulfide bond on the introduced TCR
expression. Mol Ther Nucleic Acids. 1:e632012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baker AM, Cox TR, Bird D, Lang G, Murray
GI, Sun XF, Southall SM, Wilson JR and Erler JT: The role of lysyl
oxidase in SRC-dependent proliferation and metastasis of colorectal
cancer. J Natl Cancer Inst. 103:407–424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gulhati P, Cai Q, Li J, Liu J, Rychahou
PG, Qiu S, Lee EY, Silva SR, Bowen KA, Gao T and Evers BM: Targeted
inhibition of mammalian target of rapamycin signaling inhibits
tumorigenesis of colorectal cancer. Clin Cancer Res. 15:7207–7216.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Binkhathlan Z and Alshamsan A: Emerging
nanodelivery strategies of RNAi molecules for colon cancer therapy:
preclinical developments. Ther Deliv. 3:1117–1130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu B, Zhou H, Hu L, Mu Y and Wu Y:
Involvement of PKCα activation in TF/VIIa/PAR2-induced
proliferation, migration, and survival of colon cancer cell SW620.
Tumour Biol. 34:837–846. 2013.
|
|
17
|
Yu KY, Kwon B, Ni J, Zhai Y, Ebner R and
Kwon BS: A newly identified member of tumor necrosis factor
receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J
Biol Chem. 274:13733–13736. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi G, Mao J, Yu G, Zhang J and Wu J:
Tumor vaccine based on cell surface expression of DcR3/TR6. J
Immunol. 174:4727–4735. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takahama Y, Yamada Y, Emoto K, Fujimoto H,
Takayama T, Ueno M, Uchida H, Hirao S, Mizuno T and Nakajima Y: The
prognostic significance of overexpression of the decoy receptor for
Fas ligand (DcR3) in patients with gastric carcinomas. Gastric
Cancer. 5:61–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bai C, Connolly B, Metzker ML, Hilliard
CA, Liu X, Sandig V, Soderman A, Galloway SM, Liu Q, Austin CP and
Caskey CT: Overexpreasion of M68/DcR3 in human gastrointestinal
tract tumors independent of gene amplificalion and its location in
a four-gene cluster. Proc Natl Acad Sci USA. 97:1230–1235. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Flokman J and D’amore PA: Blood vessel
formation: what is its molecular basis? Cell. 87:1153–1155. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lepelletier Y, Camara-Clayette V, Jin H,
Hermant A, Coulon S, Dussiot M, et al: Prevention of mantle
lymphoma tumor by routing transferrin receptor lysosomal
compartment. Cancer Res. 67:1145–1154. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stacker SA, Caesar C, Baldwin ME, et al:
VEGF-D promotes the metastatic spread of tumor cells via the
lymphatics. Nat Med. 7:186–191. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nagy JA, Vasile E, Feng D, et al: Vascular
permeability factor/vascular endothelial growth factor induces
lymphangiogenesis as well as angiogenesis. J Exp Med.
196:1497–1506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
George ML, Tutton MG, Janssen F, Arnaout
A, Abulafi AM, Eccles SA and Swift RI: VEGF-A, VEGF-C, and VEGF-D
in colorectal cancer progression. Neoplasia. 3:420–427. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang CR, Hsieh SL, Teng CM, Ho FM, Su WL
and Lin WW: Soluble decoy receptor 3 induces angiogenesis by
neutralization of TL1A, a cytokine belonging to tumor necrosis
factor superfamily and exhibiting angiostatic action. Cancer Res.
64:1122–1129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Egeblad M and Werb Z: New functions for
the matrix metallo-proteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar
|
|
29
|
Gao J, Ding F, Liu Q and Yao Y: Knockdown
of MACC1 expression suppressed hepatocellular carcinoma cell
migration and invasion and inhibited expression of MMP2 and MMP9.
Mol Cell Biochem. 376:21–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hotary KB, Allen ED, Brooks PC, Datta NS,
Long MW and Weiss SJ: Membrane type I matrix metalloproteinase
usurps tumor growth control imposed by the three-dimensional
extracellular matrix. Cell. 114:33–45. 2003. View Article : Google Scholar
|
|
31
|
Yamashita K, Upadhay S, Mimori K, Inoue H
and Mori M: Clinical significance of secreted protein acidic and
rich in cystein in esophageal carcinoma and its relation to
carcinoma progression. Cancer. 97:2412–2419. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Koyama H, Iwata H, Kuwabara Y, Iwase H,
Kobayashi S and Fujii Y: Gelatinolytic activity of matrix
metalloproteinase-2 and -9 in oesophageal carcinoma; a study using
in situ zymography. Eur J Cancer. 36:2164–2170. 2000. View Article : Google Scholar : PubMed/NCBI
|