Introduction
Bladder cancer (BCa) ranks sixth in cancer incidence
in the United States, and it is the fourth most common cancer in
men with a male dominance (male to female ratio 4:1) (1). In 2011, there were 10,670 estimated
male deaths from BCa and it was the eighth cause of cancer-related
mortality among males (1).
Therefore, males are more susceptible to BCa than females. It is
proposed that the gender difference in BCa is linked to sex
hormones and their receptors (2,3).
However, the exact mechanism of how sex hormone receptor(s) affect
BCa development and progression remains unclear. Miyamoto et
al(4) demonstrated the possible
involvement of androgens and androgen receptor (AR) in BCa
development. Using the carcinogen of
N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN) to induce BCa in
wild-type male and female mice, Miyamoto et al(4) found male mice lacking AR (ARKO) (with
little androgen and deleted AR) failed to develop BCa, yet almost
all wild-type male mice developed BCa, suggesting AR might play key
roles during BCa development. The study also found 50% of BBN
treated castrated mice (with little androgen yet existence of AR)
still developed BCa, indicating AR in these castrated mice might
still be able to promote BCa development, indicating that other
factor(s) could affect AR activity and facilitate the development
of BCa at the castration level of androgens (4). Other reports also indicated that in a
BBN-induced rat model, surgical or medical castration could only
reduce (and not completely eliminate) the number of rats with BCa
(5).
AR is a transcriptional factor that may need
androgens to transactivate its target genes via direct binding on
androgen response element (ARE). Accumulating evidence indicates AR
could also be transactivated via rapid non-androgen intracellular
signals, including c-Src, the downstream MAPK signals, G-protein
coupled receptor and downstream calcium signals (6). However, almost all these
non-androgens-induced AR transactivations were found in in
vitro cell lines without strong in vivo evidence. The
indication that non-androgens could induce BCa development via
transactivation of AR might therefore represent promising in
vivo evidence that warrants further characterization.
Among several potential candidates, we first decided
to assay the EGF, which is excreted in high concentrations in the
urine and stimulates urothelial cell growth, and acts through EGFR
to promote the development of various types of cancer, including
urothelial tumors (7). EGF was
shown to increase BCa cell growth and invasion activity (8,9). EGFR
belongs to the human epidermal receptor (HER) family of receptor
tyrosine kinases that contains 4 receptors; HER1 (EGFR, erb-B1),
HER2 (neu, erb-B2), HER3 (erb-B3), HER4 (erb-B4) and is a 170-kDa
membrane-spanning glycoprotein with an extracellular ligand-binding
domain, a transmembrane domain and an intracellular cytoplasmic
domain with tyrosine kinase activity (10). The overexpression of EGFR has been
linked to several malignant features and prognosis in superficial
BCa (11), tumor proliferation
(12) and the development of
secondary recurrences (13).
In prostate cancer cells, EGF was shown to
transactivate AR via signaling involving the MAPK to TIF2/GRIP1
(14). Therefore, we hypothesized
that EGF might be able to potentiate AR transcriptional activity
that may enhance BCa development.
Materials and methods
Cell culture and chemicals
Human urothelial carcinoma cell line J82, and human
embryonic kidney cell line 293T (all obtained from the American
Type Culture Collection, Manassas, VA, USA) were maintained in
appropriate medium (DMEM for 293T and MEM for J82) supplemented
with 10% fetal bovine serum (FBS) at 37°C in a humidified
atmosphere of 5% CO2. Cells were cultured in phenol
red-free medium supplemented with 5% charcoal-stripped FBS at least
18 h before experimental treatment. We purchased
dihydrotestosterone (DHT) and EGF from Sigma. Casodex
(anti-androgen), LY 294002 and PD168393 were purchased from Enzo
Life Sciences (Farmingdale, NY, USA). PD98059 was purchased from
Gibco (Frederick, MD, USA).
Luciferase reporter gene assay
Bladder cancer cells at a density of 50–60%
confluence in 24-well plates were co-transfected with 250 ng of
ARE-luc reporter plasmid DNA and 2.5 ng of PRL-TK-luc plasmid DNA,
using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). After 24
h of transfection, the cells were treated with EGF in the presence
or absence of ligands (DHT) for 24 h. Cells were then harvested,
lysed and assayed for luciferase activity, which was determined
using a Dual-Luciferase Reporter Assay kit (Promega, Madison, WI,
USA) and luminometer (TD-20/20; Turner Biosystems, Sunnyvale, CA,
USA).
AR lentiviral cDNA overexpression
To establish AR overexpression cells, we used
lentiviral vectors containing AR cDNA (15). In brief, a full-length wild-type
human AR cDNA was subcloned into pWPI plasmid (Addgene, Cambridge,
MA, USA), and the lentivirus-based vector (pWPI-AR/pWPI-control)
with pMD2.G packaging and psPAX2 envelope plasmids
(lentivirus:packaging:envelope=2:1:1) was co-transfected into 293T
cells, using lipofectamine reagent. After 48 h of transfection, the
target cells (J82) were cultured in the presence of viral
supernatant containing 8 μg/ml polybrene (Millipore, Billerica, MA,
USA) for 6 h. Flow cytometry was used to evaluate the population of
cells overexpressing AR (J82-hAR) or vector only (J82-pWPI).
Colony formation assay
For colony formation assay, 103 cells
were subcultured into 60-mm tissue culture dishes and incubated for
10–14 days with designated treatments, with Charcoal/Dextran
treated fetal bovine serum (CD FBS) medium and the medium was
changed at days 4 and 8. Cells were then washed twice with PBS and
stained with 0.1% crystal violet for 15 min before counting under a
light microscope. Clusters of at least 50 cells were counted as
colonies.
Wound healing migration assay
Cells were seeded onto a 12-well tissue culture
plate and were grown to confluence. Cell monolayers were then
wounded by sterile pipette tips (200 μl) that generated a gap.
Wounded monolayers were then washed 3 times with PBS to remove cell
debris and incubated in 10% CD FBS medium with designated
treatments for 24 h to allow cells to migrate. Images of cells were
then captured using a microscope equipped with a camera. The
relative migration folds were determined by the area covered by
migrating cells of test cells in the wounded area compared to that
of control cells, analyzed with NIS-Elements BR (Nikon)
software.
Transwell invasion assay
Cell invasion through a three-dimensional
extracellular matrix was assessed by a Matrigel invasion assay
using BD Matrigel coated-Transwell with 8.0 μm filter membranes.
Cells resuspended in 200 μl of serum free medium were plated onto
each filter with designated treatments, and 750 μl of DMEM
containing 10% CD FBS was added into the lower compartment of
invasion chambers. The cells in the top well with invasive capacity
could migrate through the Matrigel layer and 8.0 μm pores. After 24
h, filters were washed in 4% paraformaldehyde and stained with 1%
crystal violet. Cells on the upper surface of the filters were
removed with cotton swabs. Cells that had invaded to the lower
surface of the filter were counted under the microscope.
Statistical analysis
All experimental data are reported as means ± SD
(standard error). Statistical analysis was performed by t-test. A
P-value of <0.05 was considered to indicate a statistically
significant result.
Results
EGF increases AR-mediated BCa cell
growth
To examine if EGF can function through AR to
influence BCa progression, we first performed the colony formation
assay to investigate the effects of EGF on BCa cell growth with or
without the addition of AR. Therefore, we added AR in J82 cells by
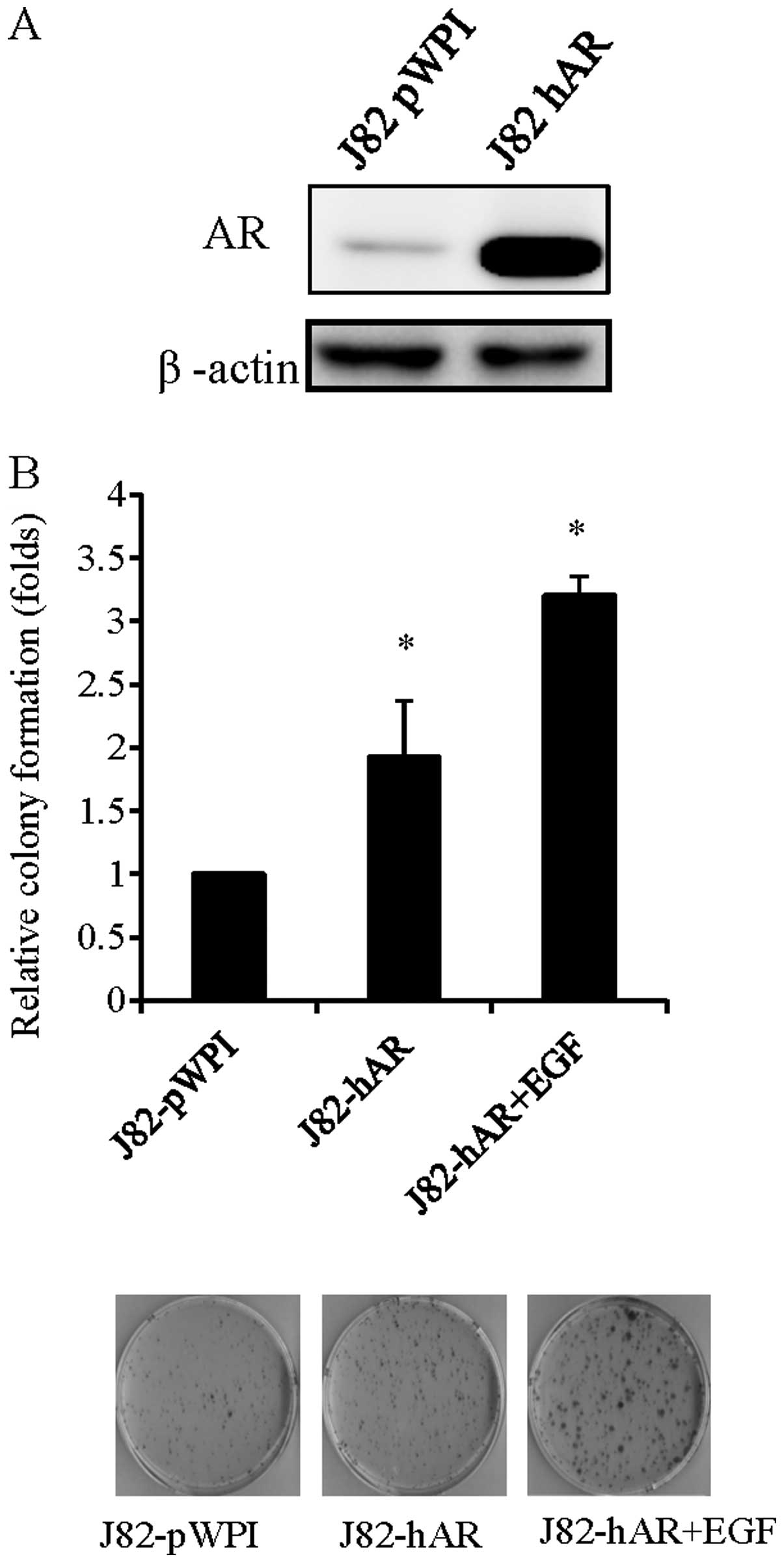
lentiviral infection and confirmed the addition by western blot
analysis (Fig. 1A). The addition of
AR in J82 cells increased colony formation number as compared to
the cells infected with virus carrying parent expression vector
(J82-pWPI) (Fig. 1B). Notably,
addition of EGF in J82-hAR cells further enhanced BCa cell growth,
suggesting that EGF could function through AR to affect BCa cell
growth.
EGF increases AR-mediated BCa cell
migration
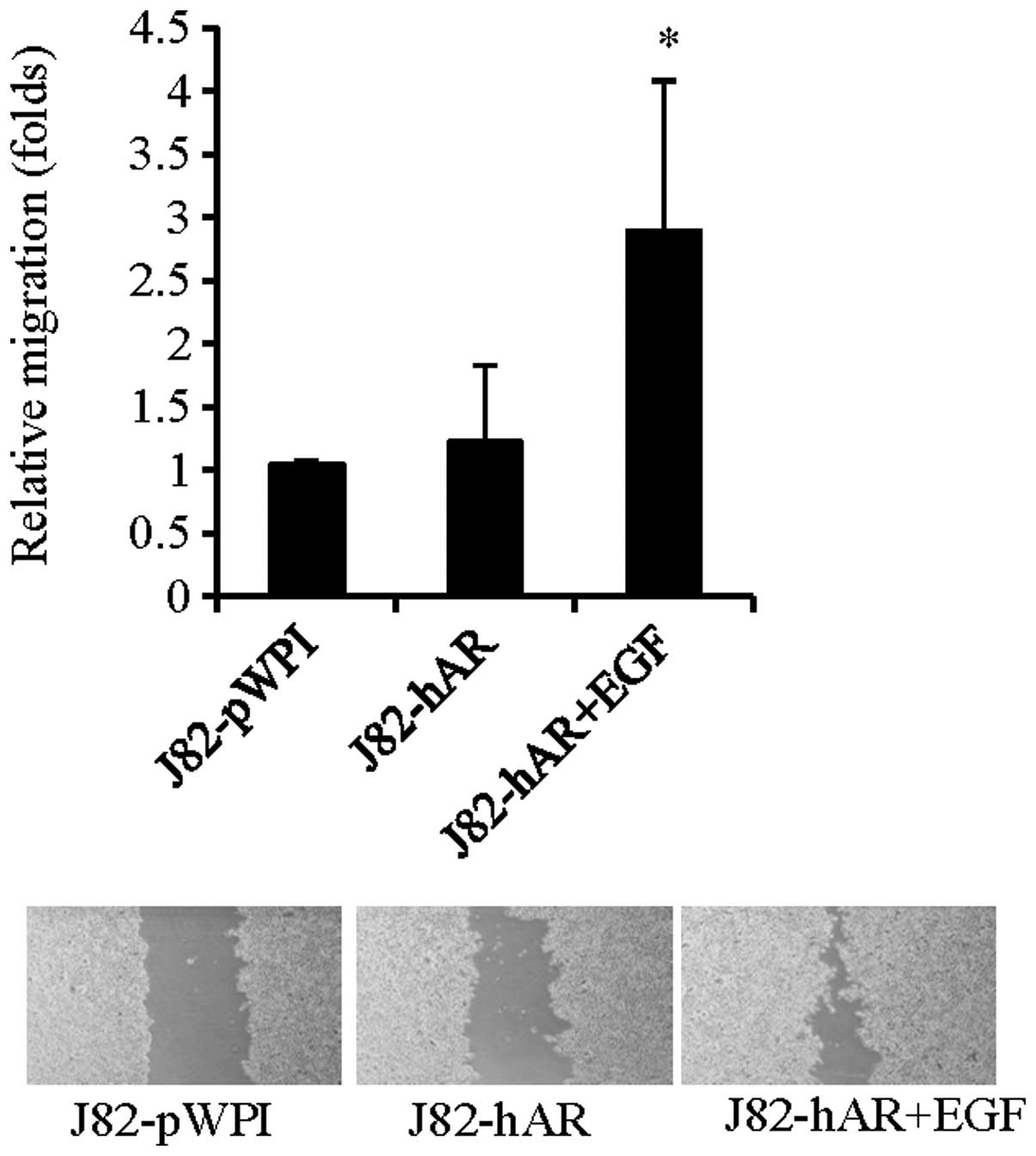
To further investigate if EGF could also influence
BCa cell migration via modulation of AR function, we then performed
wound healing migration assay to investigate the effects of EGF on
BCa cell migration. As shown in Fig.
2, addition of EGF in J82-hAR cells increased cell migration,
suggesting that EGF could also enhance BCa cell migration via
modulation of AR function.
EGF increases AR-mediated BCa cell
invasion
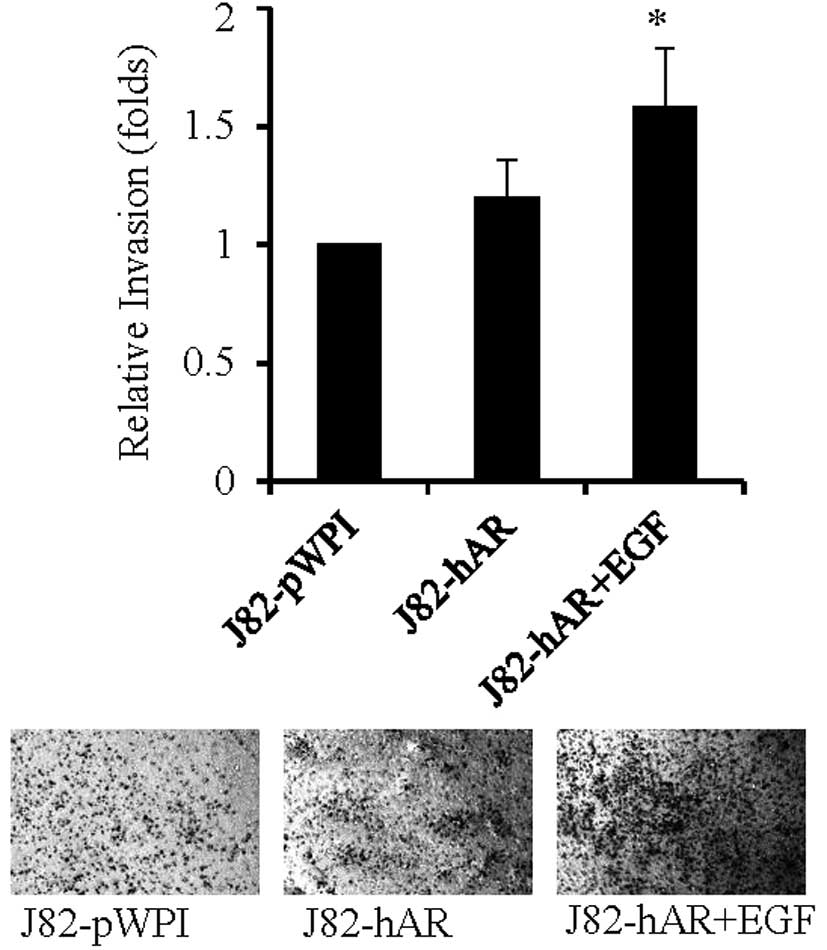
To further investigate if EGF could also influence
BCa cell invasion via modulation of AR function, we then performed
Transwell invasion assay to investigate the effects of EGF on BCa
cell invasion. As shown in Fig. 3,
addition of EGF in J82-hAR cells increased the number of cells
invading through Matrigel-coated Transwell filters, suggesting that
EGF could also enhance BCa cell invasion via modulation of AR
function.
EGF potentiates AR transcriptional
activity
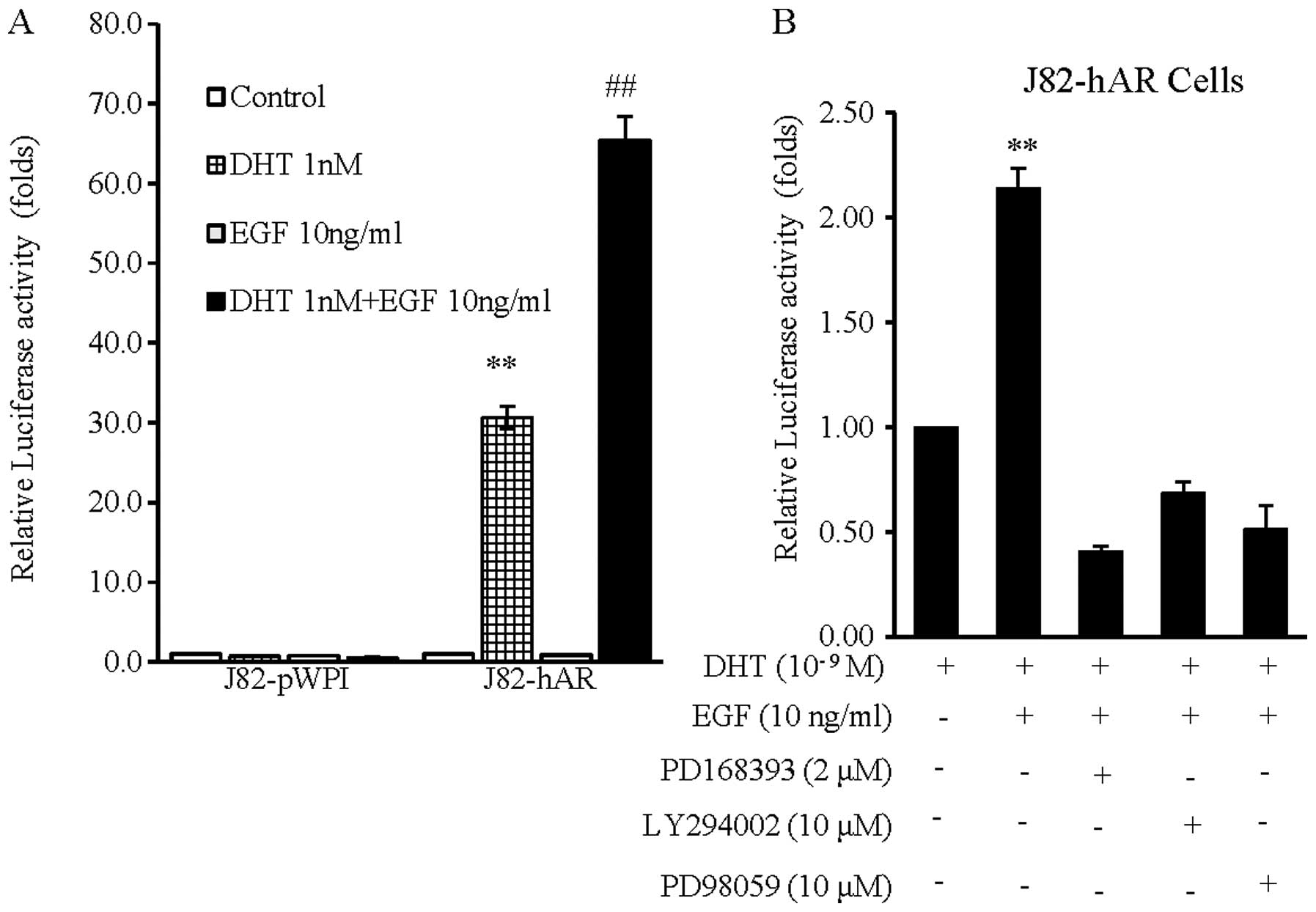
We then investigated the potential mechanism of how
EGF could function through AR signaling to enhance BCa cell growth,
migration and invasion. Using the luciferase reporter assays with
androgen-inducible promoter with four AREs in BCa J82 cells, we
first proved addition of the very low concentration of 1 nM DHT
(which is the DHT concentration in prostate after patients received
androgen deprivation therapy with surgical/chemical castration)
could still induce AR transcriptional activity in J82-hAR cells,
and not in J82-pWPI cells (Fig.
4A). Notably, addition of EGF (10 ng/ml) alone in J82-hAR cells
induced little AR transcriptional activity, but addition of EGF (10
ng/ml) and 1 nM DHT led to significantly further enhanced AR
transcriptional activity (Fig. 4A and
B), suggesting EGF was able to enhance AR transcriptional
activity in the presence of the castration level of DHT.
To elucidate the mechanism of how EGF could enhance
AR transactivation, we assessed the effects of PD168393 (a specific
EGFR inhibitor), LY294002 [a potent inhibitor of phosphoinositide
3-kinases (PI3Ks)] and PD98059 (a potent and selective inhibitor of
MEK kinase) on EGF-enhanced AR transactivation in BCa AR-positive
J82-hAR cells. The results showed that EGF-induced AR
transcriptional activity (Fig. 4B,
lane 2 vs. 1) was suppressed significantly by PD168393 (Fig. 4B, lane 3 vs. 2), Ly294002 (Fig. 4B, lane 4 vs. 2) and PD98059
(Fig. 4B, lane 5 vs. 2). These
results suggest that EGF enhances AR transactivation through the
modulation of EGFR and PI3K or MAPK/ERK signaling pathways.
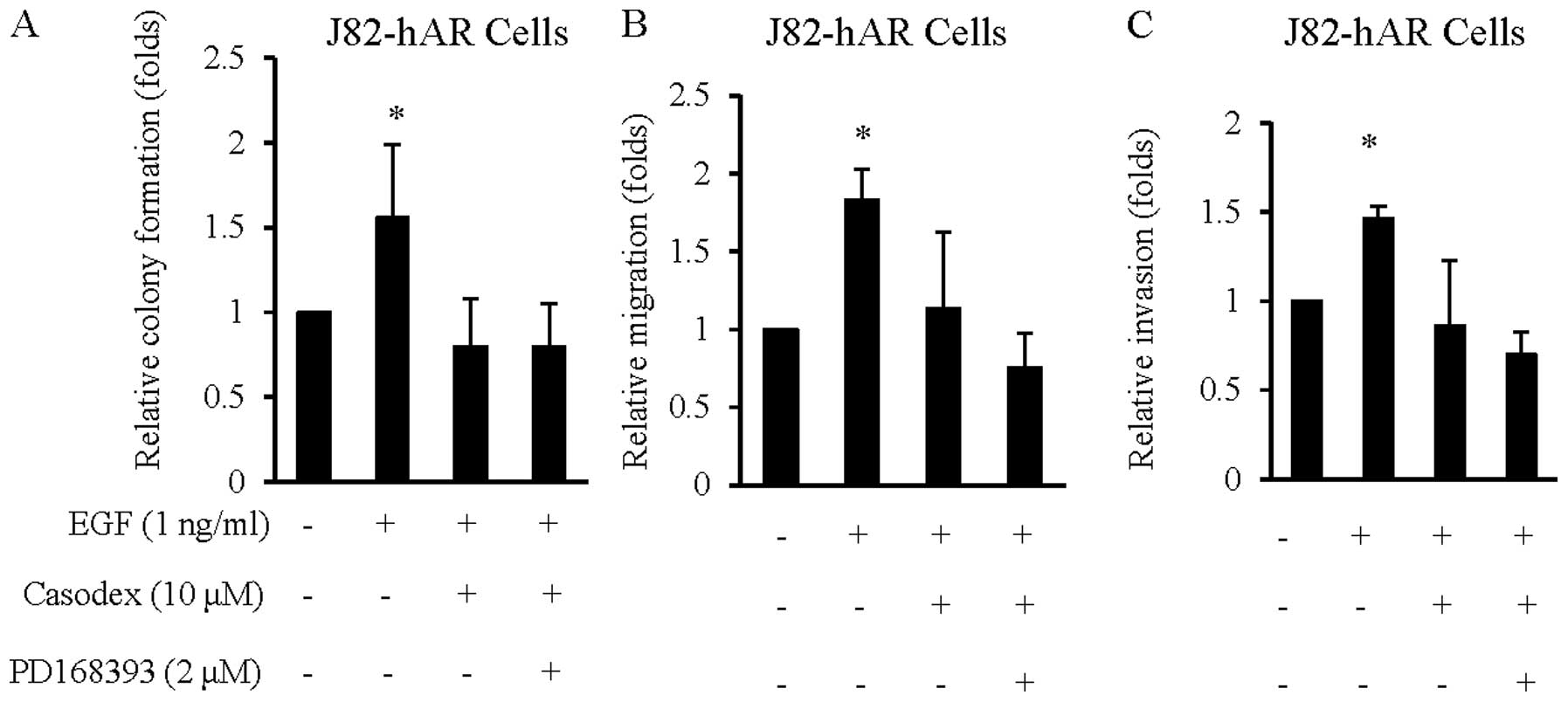
Targeting AR blocks the effect of EGF in
BCa cell growth, migration and invasion
Results from Figs.
1–4 suggest that EGF could
enhance AR transcriptional activity at the castration level of
androgen that resulted in increasing BCa cell growth, migration and
invasion. We then examined if anti-androgen could alter these EGF
effects on BCa cell growth, migration and invasion. As shown in
Fig. 5, addition of the
anti-androgen, Casodex suppressed EGF effects on BCa cell growth
(Fig. 5A), migration (Fig. 5B) and invasion (Fig. 5C). The increased suppression effects
by EGFR inhibitor of PD168393 on AR function after addition of
Casodex (Fig. 5A–C) further
suggested AR might play a key role in the effects of EGF on BCa
progression and metastasis.
Discussion
In the present study, we showed that EGF could act
through AR to further promote BCa progression by increasing cell
growth, migration and invasion. Overexpression of EGFR in
urothelial tumors may be associated with cancer progression
(16,17). Zheng et al(18) demonstrated that androgen could
enhance EGFR expression that might then lead to activation of AKT
and ERK. Similar results were also found in prostate cancer showing
that androgen could promote EGFR expression (19). In addition, Migliaccio et
al(20) also revealed that AR
might play an essential role in activating EGFR signaling in the
AR-positive prostate cancer LNCaP cells. Finally, Craft et
al(21) reported that EGFR
signal could amplify AR signaling to facilitate prostate cancer
relapse following hormone therapy. Taken together, these reports
suggest that a positive regulation loop exists between AR and EGFR
in either prostate cancer or BCa.
The interaction between EGF and AR in BCa has not
been well investigated, but in prostate cancer, this interaction
has been documented in a number of studies (22). EGF in prostatic tissue has a
mitogenic role in the prostate gland (23). Compared to other growth factors,
such as IGF-I and KGF, EGF has fewer effects on the transcriptional
activity of the androgen-responsive reporter gene when compared to
IGF-I and KGF in prostate cancer cells (14). Further studies indicated that EGF
induced activation of the MAPK pathway and the interaction with the
PKA pathway to regulate proliferation of prostate cancer cells
(24,25). In addition, EGF was demonstrated to
induce DNA synthesis, cytoskeletal changes and Src activation in
LNCaP cells through triggering rapid association of Src with AR and
ERb in LNCaP cells, suggesting AR played a key role in EGF
signaling by forming a complex with Src to induce EGFR tyrosine
phosphorylation (20). Furthermore,
EGF signaling can act through MAPK to increase TIF2/GRIP1
coactivation of AR transactivation in recurrent prostate cancer
(26). EGF requires the presence of
androgen to initiate the nuclear translocation of AR induced
ligand-independent transcriptional activity suggesting EGF causes
an over-activated AR (27). In the
present study, we further proved the cross-talk between AR and EGF
signaling pathways, which may have significant implications for the
understanding of BCa development and progression.
Our previous in vivo studies (4,18) and
our present in vitro study provided important finding that
EGF may function through AR signals to influence the BCa cell
growth, migration and invasion. However, there may be a reciprocal
positive regulation between these two key factors as well. From our
results, we propose a possible molecular mechanism to delineate how
EGF affects BCa progression via AR, by which EGF binds to EGFR to
activate ERK and AKT to act on its downstream effectors and
potentiate AR effects on BCa progression. The implication of this
finding is that we identified the link between EGF and AR signals.
EGFR inhibitors were proposed to treat urothelial cancer, but in
vitro data using BCa cell lines showed that some cells failed
to have such antiproliferative effect of EGFR inhibitors,
suggesting cells survive from molecular target agent attack by
activating downstream signals (28). The clinical trials of molecular
targeting agents, including EGFR antagonist, have been applied in
transitional urothelial carcinoma and muscular invaded BCa. There
is currently no effective EGFR antagonist single administration for
urothelial cancer trials. However, increasingly more combinational
therapy trials including EGFR antagonists are ongoing in phase II
trials with urothelial cancer patients. Therefore, combinational
therapies targeting EGFR and AR may provide a better therapeutic
strategy to suppress BCa progression. We previously tested the
combinational therapy by targeting AR and other factors in liver
cancer therapy (15,29,30).
The data in the present study may provide a novel therapeutic
strategy of combining targeting EGFR and AR in urothelial cancer
therapy.
Acknowledgements
The present study was supported by the NIH grant
(CA155477), China Medical University grants (CMU99-N2-06-2 and CMU
DMR-100-166), the Buddhist Tzu Chi General Hospital grant (TTCRD
101-10), the National Science Council grant (NSC
101-2314-B-027-MY3), and the Taiwan Department of Health Clinical
Trial and Research Center of Excellence grant
DOH99-TD-B-111-004.
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shariat SF, Sfakianos JP, Droller MJ,
Karakiewicz PI, Meryn S and Bochner BH: The effect of age and
gender on bladder cancer: a critical review of the literature. BJU
Int. 105:300–308. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fajkovic H, Halpern JA, Cha EK, et al:
Impact of gender on bladder cancer incidence, staging, and
prognosis. World J Urol. 29:457–463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miyamoto H, Yang Z, Chen YT, et al:
Promotion of bladder cancer development and progression by androgen
receptor signals. J Natl Cancer Inst. 99:558–568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Imada S, Akaza H, Ami Y, Koiso K, Ideyama
Y and Takenaka T: Promoting effects and mechanisms of action of
androgen in bladder carcinogenesis in male rats. Eur Urol.
31:360–364. 1997.PubMed/NCBI
|
|
6
|
Heinlein CA and Chang C: The roles of
androgen receptors and androgen-binding proteins in nongenomic
androgen actions. Mol Endocrinol. 16:2181–2187. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grivas PD, Day M and Hussain M: Urothelial
carcinomas: a focus on human epidermal receptors signaling. Am J
Transl Res. 3:362–373. 2011.PubMed/NCBI
|
|
8
|
Kanno N, Nonomura N, Miki T, et al:
Effects of epidermal growth factor on the invasion activity of the
bladder cancer cell line. J Urol. 159:586–590. 1998. View Article : Google Scholar
|
|
9
|
Kawamata H, Azuma M, Kameyama S, Nan L and
Oyasu R: Effect of epidermal growth factor/transforming growth
factor alpha and transforming growth factor beta 1 on growth in
vitro of rat urinary bladder carcinoma cells. Cell Growth Differ.
3:819–825. 1992.PubMed/NCBI
|
|
10
|
Rowinsky EK: The erbB family: targets for
therapeutic development against cancer and therapeutic strategies
using monoclonal antibodies and tyrosine kinase inhibitors. Annu
Rev Med. 55:433–457. 2004. View Article : Google Scholar
|
|
11
|
Lipponen P and Eskelinen M: Expression of
epidermal growth factor receptor in bladder cancer as related to
established prognostic factors, oncoprotein (c-erbB-2, p53)
expression and long-term prognosis. Br J Cancer. 69:1120–1125.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sauter G, Haley J, Chew K, et al:
Epidermal-growth-factor-receptor expression is associated with
rapid tumor proliferation in bladder cancer. Int J Cancer.
57:508–514. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chow NH, Chan SH, Tzai TS, Ho CL and Liu
HS: Expression profiles of ErbB family receptors and prognosis in
primary transitional cell carcinoma of the urinary bladder. Clin
Cancer Res. 7:1957–1962. 2001.PubMed/NCBI
|
|
14
|
Culig Z, Hobisch A, Cronauer MV, et al:
Androgen receptor activation in prostatic tumor cell lines by
insulin-like growth factor-I, keratinocyte growth factor, and
epidermal growth factor. Cancer Res. 54:5474–5478. 1994.
|
|
15
|
Ma WL, Hsu CL, Yeh CC, et al: Hepatic
androgen receptor suppresses hepatocellular carcinoma metastasis
through modulation of cell migration and anoikis. Hepatology.
56:176–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nguyen PL, Swanson PE, Jaszcz W, et al:
Expression of epidermal growth factor receptor in invasive
transitional cell carcinoma of the urinary bladder. A multivariate
survival analysis. Am J Clin Pathol. 101:166–176. 1994.PubMed/NCBI
|
|
17
|
Chow NH, Tzai TS, Lin SN, Chan SH and Tang
MJ: Reappraisal of the biological role of epidermal growth factor
receptor in transitional cell carcinoma. Eur Urol. 24:140–143.
1993.PubMed/NCBI
|
|
18
|
Zheng Y, Izumi K, Yao JL and Miyamoto H:
Dihydrotestosterone upregulates the expression of epidermal growth
factor receptor and ERBB2 in androgen receptor-positive bladder
cancer cells. Endocr Relat Cancer. 18:451–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brass AL, Barnard J, Patai BL, Salvi D and
Rukstalis DB: Androgen up-regulates epidermal growth factor
receptor expression and binding affinity in PC3 cell lines
expressing the human androgen receptor. Cancer Res. 55:3197–3203.
1995.PubMed/NCBI
|
|
20
|
Migliaccio A, Di Domenico M, Castoria G,
et al: Steroid receptor regulation of epidermal growth factor
signaling through Src in breast and prostate cancer cells: steroid
antagonist action. Cancer Res. 65:10585–10593. 2005. View Article : Google Scholar
|
|
21
|
Craft N, Shostak Y, Carey M and Sawyers
CL: A mechanism for hormone-independent prostate cancer through
modulation of androgen receptor signaling by the HER-2/neu tyrosine
kinase. Nat Med. 5:280–285. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Culig Z, Hobisch A, Cronauer MV, et al:
Regulation of prostatic growth and function by peptide growth
factors. Prostate. 28:392–405. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Heinlein CA and Chang C: Androgen receptor
in prostate cancer. Endocr Rev. 25:276–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Putz T, Culig Z, Eder IE, et al: Epidermal
growth factor (EGF) receptor blockade inhibits the action of EGF,
insulin-like growth factor I, and a protein kinase A activator on
the mitogen-activated protein kinase pathway in prostate cancer
cell lines. Cancer Res. 59:227–233. 1999.
|
|
25
|
Chen T, Cho RW, Stork PJ and Weber MJ:
Elevation of cyclic adenosine 3′,5′-monophosphate potentiates
activation of mitogen-activated protein kinase by growth factors in
LNCaP prostate cancer cells. Cancer Res. 59:213–218. 1999.
|
|
26
|
Gregory CW, Fei X, Ponguta LA, et al:
Epidermal growth factor increases coactivation of the androgen
receptor in recurrent prostate cancer. J Biol Chem. 279:7119–7130.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Orio F, Terouanne B, Georget V, et al:
Potential action of IGF-1 and EGF on androgen receptor nuclear
transfer and transactivation in normal and cancer human prostate
cell lines. Mol Cell Endocrinol. 198:105–114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kassouf W, Dinney CPN, Brown G, et al:
Uncoupling between epidermal growth factor receptor and downstream
signals defines resistance to the antiproliferative effect of
Gefitinib in bladder cancer cells. Cancer Res. 65:10524–10535.
2005. View Article : Google Scholar
|
|
29
|
Ma WL, Hsu CL, Wu MH, et al: Androgen
receptor is a new potential therapeutic target for the treatment of
hepatocellular carcinoma. Gastroenterology. 135:947–955. 955.e1–5.
2008.PubMed/NCBI
|
|
30
|
Wu JT, Han BM, Yu SQ, Wang HP and Xia SJ:
Androgen receptor is a potential therapeutic target for bladder
cancer. Urology. 75:820–827. 2010. View Article : Google Scholar : PubMed/NCBI
|