Introduction
Lung cancer is one of the leading causes of
cancer-related death in both men and women worldwide, and
adenocarcinoma is the most predominant histologic subtype in many
parts of the world. Tobacco smoke is clearly the most important
factor associated with the development of lung cancer, accounting
for 80–90% of all cases. Asbestos is another significant inhaled
carcinogen, contributing to the development of ~5–7% of all lung
cancers (1). Many studies on
asbestos-related lung carcinogenesis have analyzed the genotoxic
effects of asbestos; asbestos fibers induce DNA damage, chromosome
aberrations, mitotic disturbances and gene mutations (2). In addition, asbestos fibers can
stimulate a range of other effects including cell proliferation,
chronic inflammation, enhanced gene expression, such as c-fos and
c-jun overexpression, and transformation (3,4).
Despite these studies, the efficacy of asbestos-exposure as a
complete lung carcinogen, independent of tobacco smoke, has not
been demonstrated in humans, since lung cancers of asbestos-exposed
individuals frequently occur in smokers and ex-smokers. The
majority of asbestos-related lung cancers may result from the
combined effects of asbestos and carcinogens in tobacco smoke, with
the possibility of a synergistic relationship first proposed by
Doll (5). Hence, the mechanism of
asbestos-induced lung carcinogenesis still remains unclear.
Both loss of heterozygosity (LOH) and the p53
mutation are genetic alterations. LOH is frequently noted in cancer
cells and is thought to occur through genetic instability at the
chromosomal level. On the other hand, the p53 mutation is a
genetic alteration at the nucleotide level. Mutation in the
p53 tumor suppressor gene is the most frequently observed
gene mutation in cancers. As described below, not only p53
mutations but also LOH spectra differ in different cancer types
associated with different etiologies. Previously we compared the
frequency of LOH on all autosomal chromosomes among non-small cell
lung carcinomas (6,7) as well as p53 mutation patterns
with adenocarcinoma cell morphology (8). The frequency of allelic loss on many
chromosomal arms was commonly higher in squamous cell carcinomas
than in adenocarcinomas. This result suggested that more cumulative
genetic changes are associated with tumorigenesis in squamous cell
carcinomas than contribute to adenocarcinomas, a pattern which may
reflect a difference in the carcinogenic mechanisms responsible for
the two histologies. In addition, we observed high frequencies of
allelic losses on chromosomes 9p, 9q and 13q in squamous cell
carcinomas, the majority of which were from smokers, and higher
frequencies of allelic losses on these arms in adenocarcinomas from
smokers than those from non-smokers. This loss of specific
chromosomes associated with a particular histology is an example of
LOH spectra reflecting etiology. The p53 mutational spectra
differ among cancers of various organs, and its frequency and
mutational spectra can be said to reflect carcinogenic patterns
characteristic of exogenous or endogenous factors and thus may be
helpful for identification of the responsible agents, including,
among others, cigarette smoke, aflatoxin B1 and ultraviolet light.
Hence, the analysis of p53 mutation can provide clues to the
etiology of diverse tumors and to the function of specific regions
of p53 (9,10). The mutation pattern in smokers shows
an excess of G:C to T:A transversions (34.2%), which are relatively
uncommon in non-smokers or passive-smokers (16.6%) (11). These transversions often occur at
codons 157, 158, 245, 248 and 273, experimentally identified as
sites of adduct formation by benzo(a)pyrene, a single polycyclic
aromatic hydrocarbon (PAH)-compound found in cigarette smoke. Other
PAH-compounds also have a similar preference for adduct formation
in these p53 codons (12,13).
In the present study, to elucidate the combined
effects of asbestos-exposure and smoking on development of lung
adenocarcinomas, we used 132 lung adenocarcinomas, for which we
already obtained all detailed smoking histories, comprehensive LOH
data for all autosomal chromosomes (7), and p53 mutation data.
Materials and methods
Patients and sample preparation
A total of 335 cases of lung adenocarcinoma were
surgically removed at the Cancer Institute Hospital (CIH), Tokyo,
Japan, between September 1989 and August 1996. Among the cases,
fresh tumor tissues and corresponding normal lung and detailed
smoking histories were successfully collected from 132 patients,
which were used as materials in this study. Hence, they were
collected semi-randomly without respect to asbestos-exposure
status, and therefore provided a representative population for a
cancer center in Japan. The clinicopathological data for these
samples are summarized in Table I.
We used a differentiation grading that was basically according to
the former version of the Japanese Lung Cancer Society (14), as previously performed (15). Smoking history was surveyed
intensively from patients and their families and presented as
cumulative smoking (CS) in pack-years. The study protocol was
approved by IRB of CIH and informed consent was obtained from all
patients.
 | Table IClinicopathological data of the
patients with lung adenocarcinomas analyzed in this study
(n=132). |
Table I
Clinicopathological data of the
patients with lung adenocarcinomas analyzed in this study
(n=132).
| Clinicopathological
features | No. of patients
(%) |
|---|
| Age (years ±
SD) | 61±11 |
| Gender |
| Male | 74 (56) |
| Female | 58 (44) |
| Cumulative
smoking |
| CS=0 | 54 (41) |
| 0<CS<25 | 18 (14) |
| ≥25 CS | 60 (45) |
| Asbestos
burden |
| AB=0 | 64 (48) |
|
0<AB<1,000 | 28 (21) |
| ≥1,000 AB
<5,000 | 36 (27) |
| ≥5,000 AB | 4 (3) |
| pStage |
| I | 63 (48) |
| II–IV | 69 (52) |
|
Differentiation |
| Well | 35 (27) |
| Moderately | 69 (52) |
| Poorly | 28 (21) |
| Size (mm) |
| <30 | 75 (57) |
| ≥30 | 57 (43) |
Measurement of asbestos-exposure
Asbestos-body burden (AB; in numbers per gram of dry
lung tissue) was measured using paraffin blocks of corresponding
normal lung tissues by a polarizing microscope (16). The detection limit, which means no
AB was found on the measuring filter sample, was ~100 AB/g (dry
lung) and expressed as 0 in this study.
A matrix of smoking-exposure and
asbestos-exposure
To examine the dose-effect relationship of
asbestos-exposure (presented as AB) and smoking-exposure (presented
as CS in pack-years) on lung adenocarcinomas, we classified all
cases into 9 groups based on a matrix of CS in pack-years: CS=0
(n=54, 41%), 0<CS<25 (n=18, 14%), ≥25 CS (n=60, 45%), and AB:
AB=0 (n=64, 48%), 0<AB<1,000 (n=28, 21%), ≥1,000 AB (n=40,
31%). Since the patients were selected consecutively from surgical
tumor files in a general cancer center, only 4 cases (3.0%)
exceeded 5,000 in AB. To investigate the mechanism of
asbestos-induced lung carcinogenesis in a representative population
for a cancer center, not a biased population heavily exposed to
asbestos, we divided the cases between AB <1,000 and ≥1,000
AB.
LOH analysis
For LOH analysis, we performed Southern blotting.
Experimental procedures and probes used were essentially the same
as previously described (6,7). To facilitate the comparison, we used a
fractional allelic loss (FAL) value, defined as: (number of
chromosome arms with LOH)/(number of informative arms) for each
case. Of 132 patients with adenocarcinomas, LOH data were available
for 114 patients.
p53 mutation analysis
Analysis of p53 mutation was performed
essentially as described elsewhere (8). Genomic DNA from fresh tumor samples
was prepared and exons 4–8 and 10 of p53 were analyzed by
polymerase chain reaction and DNA sequencing. Of the 132 patients
with adenocarcinomas, p53 mutation data were available for
123 patients.
Statistical analysis
For statistical analysis, we used the t-test,
Fisher’s exact test, and Chi-square test, as appropriate. The
two-sided significant level was set at p<0.05. Data were
analyzed with the statistical software Stata version 11
(StataCorp., College Station, TX, USA).
Results
LOH frequency of lung adenocarcinomas classified by
CS and AB is shown in Table II and
Fig. 1A. LOH frequency increased
only slightly correlating with the elevation of CS in the AB=0
groups, whereas, in the AB>0 groups, it increased as AB and/or
CS was elevated and was significantly higher in the ≥1,000 AB, ≥25
CS group than in the AB=CS=0 group (p=0.032).
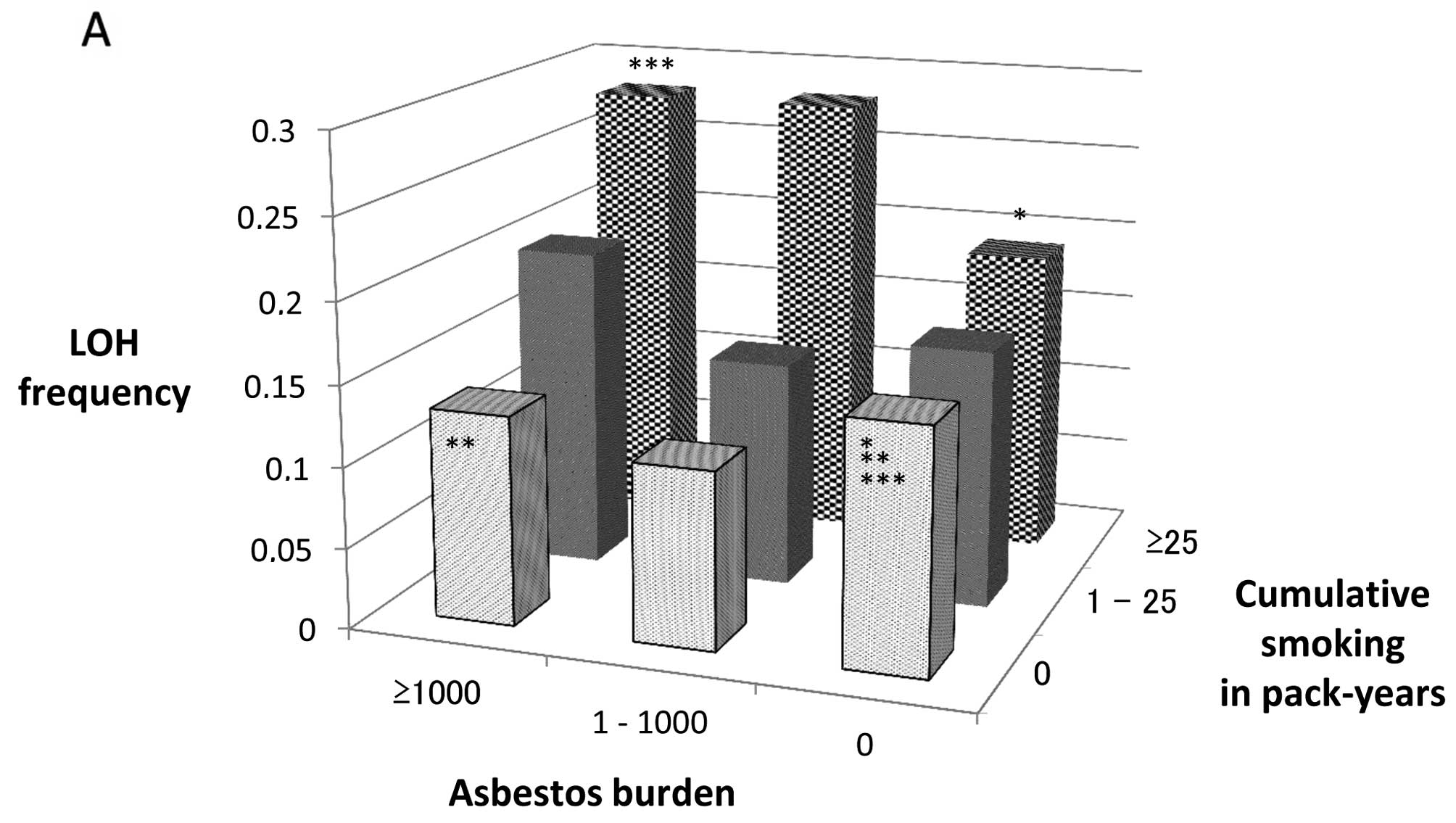 | Figure 1(A) Frequency of the loss of
heterozygosity (LOH) in lung adenocarcinomas classified by
cumulative smoking (CS) in pack-years (CS=0, 0<CS<25, ≥25 CS)
and asbestos burden (AB) (AB=0, 0<AB<1,000, ≥1,000 AB). FAL,
fractional allelic loss. *p=0.30 (AB=0, CS=0 vs. AB=0,
25≤CS); **p=0.69 (AB=0, CS=0 vs. ≥1,000 AB, CS=0);
***p=0.032 (AB=0, CS=0 vs. ≥1,000 AB, ≥25 CS). (B) p53
mutation frequency in lung adenocarcinomas classified by CS in
pack-years (CS=0, 0<CS<25, 25≤CS) and AB (AB=0,
0<AB<1,000, 1,000≤AB). *p=0.14 (AB=0, CS=0 vs.
AB=0, 25≤CS); **p=0.14 (AB=0, CS=0 vs. ≥1,000 AB, CS=0);
***p=0.039 (AB=0, CS=0 vs. ≥1,000 AB, ≥25 CS). |
| Table IIFAL values (± SD) in lung
adenocarcinomas, classified by AB and CS in pack-years. |
Table II
FAL values (± SD) in lung
adenocarcinomas, classified by AB and CS in pack-years.
| AB | |
|---|
|
| |
|---|
| 0 | 1–1,000 | ≥1,000 | Total |
|---|
| CS |
| 0 | 0.15 (±0.13)
(n=20) | 0.11 (±0.13)
(n=16) | 0.13 (±0.16)
(n=13) | 0.13 (±0.12)
(n=49) |
| 1–25 | 0.16 (±0.17)
(n=4) | 0.14 (±0.04)
(n=3) | 0.20 (±0.20)
(n=7) | 0.18 (±0.15)
(n=14) |
| ≥25 | 0.19 (±0.14)
(n=28) | 0.28 (±0.25)
(n=6) | 0.28 (±0.22)
(n=17) | 0.23 (±0.17)
(n=51) |
| Total | 0.17 (±0.12)
(n=52) | 0.15 (±0.16)
(n=25) | 0.21 (±0.18)
(n=37) | 0.18 (±0.15)
(n=114) |
Details of cases with p53 mutations in lung
adenocarcinomas are shown in Table
III and summarized in Table
IV. The p53 mutation rates of pathological stage I and
II–IV lung adenocarcinomas were 32% (18 of 57) and 44% (29 of 66),
respectively, not significantly different by Fisher’s exact test
(p=0.19). p53 mutation frequency of lung adenocarcinomas
classified by CS and AB are depicted in Fig. 1B. p53 mutation frequency was
the lowest in the AB=CS=0 group (18%), increased as AB and/or CS
rose, and was significantly higher in the ≥1,000 AB, ≥25 CS group
(53%) than in the AB=CS=0 group (p=0.039). Tobacco smoke, one of
the most significant exogenous carcinogenic agents has been shown
to frequently cause specific p53 mutations, especially G:C
to T:A transversion (17) at
specific codons described as ‘hotspots’, such as codon 157, 158,
245, 248 and 273 (13). p53
mutations characteristic of smoking, such as G:C to T:A
transversion at the tobacco-specific codons were frequently
observed in the CS>0 groups, whereas non-specific mutations were
often detected in the CS=0, AB>0 groups (Tables III and IV). In the ≥1,000 AB, CS=0 group, there
was only one transversion and no tobacco-specific codons for the
six p53 mutations. In contrast, in the AB=0, ≥25 CS group,
there were five G:C to T:A transversions and five tobacco-specific
codons among 13 p53 mutations. Fig. 2 shows p53 mutation spectra in
lung adenocarcinomas, classified as smokers (A, n=33) or
non-smokers (B, n=14) and asbestos-exposed (C, n=28) or not (D,
n=19). Although p53 mutation spectra varied depending on the
status of smoking history, they showed little difference between
asbestos-exposed or non-exposed. Whereas smokers had frequent G:C
to T:A transversions, which are smoking-associated p53
mutations, non-smokers had frequent G:C to A:T transitions at CpG
sites associated with spontaneous mutations, consistent with
previous reports (9,17).
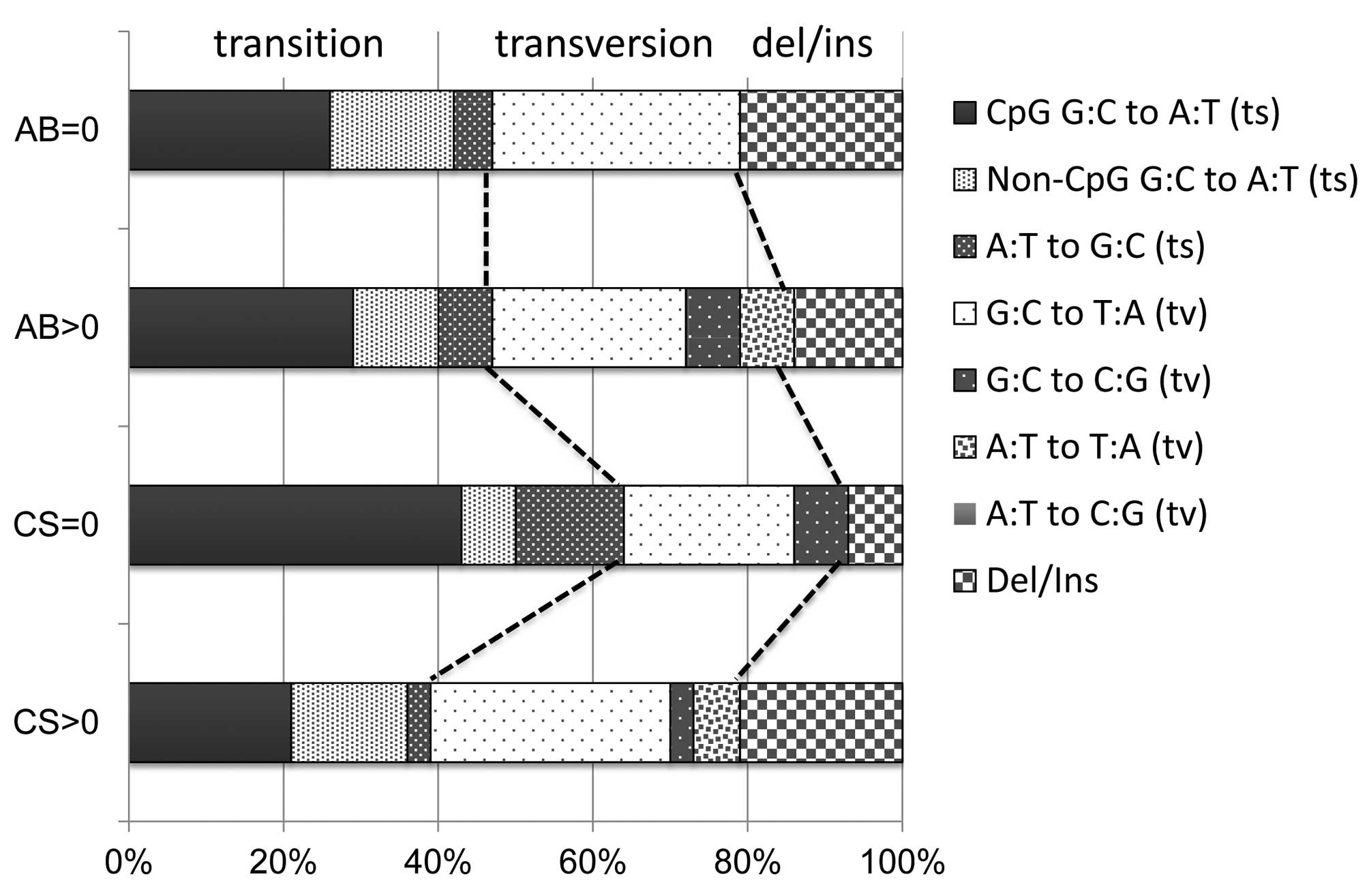 | Figure 2p53 mutation spectra in lung
adenocarcinomas. AB=0 (n=19): lung adenocarcinomas from patients
without AB (0<CS, n=15; CS=0, n=4). AB>0 (n=28): lung
adenocarcinomas from patients with AB (0<CS, n=18; CS=0, n=10).
CS=0 (n=14): lung adenocarcinomas from non-smokers (0<AB, n=10;
AB=0, n=4). CS>0 (n=33): lung adenocarcinomas from smokers and
ex-smokers (0<AB, n=18; AB=0, n=15). CS, cumulative smoking in
pack-years; AB, asbestos burden; ts, transition; tv, transversion;
Del/Ins, deletion/insertion. |
| Table IIIDetails of the cases with p53
mutations in lung adenocarcinomas. |
Table III
Details of the cases with p53
mutations in lung adenocarcinomas.
| Classified by CS
and AB | Case no. | Gender | Age (years) | Diff. | Size (mm) | pStage | AB | CS | FAL | Mut type | Codon | Base change | Amino acid |
|---|
| CS=0 | 33 | F | 49 | Mod | 27 | IIIB | 0 | 0 | 0.13 | ts | 273 | CGT to CAT | Asp→His |
| AB=0 | 70 | F | 26 | W | 43 | IIIB | 0 | 0 | 0.53 | tv | 176 | TGC to TTC | Cys→Phe |
| 73 | M | 44 | P | 35 | IIIA | 0 | 0 | 0.35 | ts | 120 | AAG to AGG | Lys→Arg |
| 74 | F | 70 | W | 20 | IA | 0 | 0 | 0.06 | ts | 248 | CGG to CAG | Arg→Glu |
| CS=0 | 27 | F | 77 | Mod | 38 | IIIB | 187 | 0 | 0.18 | tv | 237 | ATG to ATT | Met→Ile |
|
0<AB<1,000 | 39 | F | 51 | Mod | 24 | IIIB | 214 | 0 | 0.05 | ts | 245 | GGC to AGC | Gly→Ser |
| 47 | F | 65 | W | 24 | IA | 333 | 0 | 0.06 | ts | 335 | CGT to CAT | Arg→His |
| 81 | F | 68 | Mod | 21 | IA | 671 | 0 | 0.05 | tv | 273 | CGT to CTT | Asp→Leu |
| CS=0 | 3 | F | 51 | Mod | 33 | IIIA | 1,715 | 0 | 0.14 | del | 341 | TTC to T---C | Frameshift |
| ≥1,000 AB | 7 | F | 72 | Mod | 60 | IV | 3,939 | 0 | 0.58 | ts | 138 | GCC to GTC | Ala→Val |
| 14 | F | 57 | Mod | 40 | IIIA | 2,305 | 0 | 0.29 | tv | 138 | GCC to CCC | Ala→Pro |
| 84 | F | 63 | Mod | 25 | IIIB | 1,000 | 0 | 0.21 | ts | 282 | CGG to TGG | Arg→Trp |
| 113 | F | 67 | Mod | 32 | IIIB | 1,949 | 0 | 0.13 | ts | 132 | AAG to AGG | Lys→Arg |
| 114 | F | 49 | Mod | 33 | IB | 6,998 | 0 | 0.1 | ts | 213 | CGA to TGA | Arg→Stop |
| 0<CS<25 | 55 | F | 68 | Mod | 42 | IIIB | 0 | 3.8 | 0.17 | ts | 242 | TGC to TAC | Cys→Tyr |
| AB=0 | 126 | M | 66 | Mod | 30 | IA | 0 | 8 | NA | ts | 237 | ATG to ATA | Met→Ile |
| ≥25 CS | 2 | M | 73 | P | 53 | IIIB | 0 | 39.4 | 0.25 | ts | 259 | GAC to AAC | Asp→Ile |
| AB=0 | 12 | M | 69 | Mod | 28 | IA | 0 | 42.3 | 0.04 | del | 113–119 | Del of 19 bp | Frameshift |
| 21 | M | 47 | P | 39 | IIIA | 0 | 32.5 | 0 | tv | 245 | GGC to TGC | Gly→Cys |
| 42 | M | 58 | P | 24 | IIIA | 0 | 80 | 0.36 | ts | 273 | CGT to TGT | Asp→Cys |
| 46 | M | 56 | Mod | 20 | IIIB | 0 | 31 | 0.1 | del | 159 | GCC to---C | Frameshift |
| 54 | M | 54 | Mod | 25 | IIIA | 0 | 48 | 0.38 | tv | 198 | GAA to TAA | Glu→Stop |
| 56 | M | 74 | W | 17 | IA | 0 | 42 | 0.24 | ts | 175 | CGC to CAC | Arg→His |
| 58 | M | 61 | P | 23 | IV | 0 | 80 | 0.33 | tv | 135 | TGC to TTC | Cys→Phe |
| 83 | M | 72 | Mod | 75 | IV | 0 | 126 | 0.44 | del | 274 | GTT to---T | Frameshift |
| 88 | M | 50 | Mod | 48 | IV | 0 | 115.5 | 0.2 | del | 189 | GCC to G---C | Frameshift |
| 96 | M | 54 | Mod | 27 | IA | 0 | 34 | 0.25 | tv | 158 | CGC to CTC | Arg→Leu |
| 102 | M | 56 | W | 16 | IA | 0 | 37.5 | 0.06 | ts | 273 | CGT to TGT | Asp→Cys |
| 116 | M | 50 | P | 60 | IIIA | 0 | 32 | NA | tv | 245 | GGC to TGC | Gly→Cys |
| 0<CS<25 | 17 | M | 58 | W | 27 | IA | 560 | 1.3 | 0.13 | ts | 234 | TAC to TGC | Tyr→Cys |
|
0<AB<1,000 | 128 | F | 69 | P | 60 | IB | 980 | 20 | NA | ts | 245 | GGC to GAC | Gly→Asp |
| ≥25 CS | 11 | M | 64 | Mod | 20 | IA | 333 | 33 | 0.17 | tv | Donor | AGgt to AGtt | Splicing |
|
0<AB<1,000 | 49 | M | 41 | P | 105 | IB | 446 | 37.5 | 0.22 | tv | Acceptor | agG to atG | Splicing |
| 97 | M | 72 | Mod | 16 | IIIB | 929 | 25.5 | 0.26 | ts | 273 | CGT to CAT | Asp→His |
| 131 | M | 51 | P | 28 | IIIA | 339 | 31 | NA | tv | 244 | GGC to TGC | Gly→Cys |
| 0<CS<25 | 23 | M | 59 | Mod | 24 | IA | 1,538 | 24 | 0.12 | tv | 274 | GTT to TTT | Val→Phe |
| ≥1,000 AB | 64 | F | 74 | W | 37 | IIIB | 1,477 | 12 | 0.11 | tv | 209 | AGA to TGA | Arg→Stop |
| 86 | M | 49 | Mod | 23 | IIA | 2,039 | 1 | 0.45 | tv | 238 | TGT to AGT | Cys→Ser |
| ≥25 CS | 16 | M | 72 | P | 35 | IIIA | 2,490 | 40 | 0.31 | ts | 158 | CGC to CAC | Arg→His |
| ≥1,000 AB | 53 | M | 60 | P | 28 | IIA | 1,750 | 40 | 0.64 | ts | 158 | CGC to CAC | Arg→His |
| 85 | M | 65 | Mod | 28 | IA | 2,337 | 45 | 0.55 | ts | 275 | TGT to TAT | Cys→Tyr |
| 103 | M | 67 | Mod | 28 | IIIB | 1,293 | 30.6 | 0.2 | ins | 305–306 | Ins of 23 bp | Frameshift |
| 104 | M | 74 | Mod | 32 | IB | 2,378 | 53 | 0.05 | ts | Donor | AGgt to AGat | Splicing |
| 105 | M | 50 | Mod | 24 | IA | 2,212 | 58 | 0.29 | del | 179–185 | Del of 18 bp | Frameshift |
| 109 | M | 47 | P | 64 | IIIA | 3,207 | 81 | 0.64 | tv | 158 | CGC to CCC | Arg→Pro |
| 110 | M | 55 | Mod | 15 | IA | 3,881 | 35 | 0 | ins | 46 | Ins of 16 bp | Frameshift |
| 115 | M | 71 | Mod | 20 | IIIA | 5,308 | 48 | 0.46 | tv | 157 | GTC to TTC | Val→Phe |
| Table IVp53 mutational spectra in lung
adenocarcinomas, classified by AB and CS in pack-years. |
Table IV
p53 mutational spectra in lung
adenocarcinomas, classified by AB and CS in pack-years.
| | | Transition | | |
|---|
| | |
| Transversion | |
|---|
| | | CpG | Non-CpG | | |
| |
|---|
| Classified by CS
and AB | No. | With p53
mutation (%) | G:C to A:T | G:C to A:T | A:T to G:C | Total (%) | G:C to T:A | G:C to C:G | A:T to T:A | A:T to C:G | Total (%) | Del/Ins (%) |
|---|
| All cases | 123 | 47 (38) | 13 | 6 | 3 | 22 (47) | 13 | 2 | 2 | 0 | 17 (36) | 8 (17) |
| CS=0 | 49 | 14 (28) | 6 | 1 | 2 | 9 (64) | 3 | 1 | 0 | 0 | 4 (29) | 1 (7) |
| AB=0 | 22 | 4 (18) | 2 | 0 | 1 | 3 (75) | 1 | 0 | 0 | 0 | 1 (25) | 0 (0) |
|
0<AB<1,000 | 13 | 4 (31) | 2 | 0 | 0 | 2 (50) | 2 | 0 | 0 | 0 | 2 (50) | 0 (0) |
| ≥1,000 AB | 14 | 6 (43) | 2 | 1 | 1 | 4 (67) | 0 | 1 | 0 | 0 | 1 (17) | 1 (17) |
| 0<CS<25 | 17 | 7 (41) | 1 | 2 | 1 | 4 (57) | 1 | 0 | 2 | 0 | 3 (43) | 0 (0) |
| AB=0 | 6 | 2 (33) | 0 | 2 | 0 | 2 (100) | 0 | 0 | 0 | 0 | 0 (0) | 0 (0) |
|
0<AB<1,000 | 3 | 2 (67) | 1 | 0 | 1 | 2 (100) | 0 | 0 | 0 | 0 | 0 (0) | 0 (0) |
| ≥1,000 AB | 8 | 3 (38) | 0 | 0 | 0 | 0 (0) | 1 | 0 | 2 | 0 | 3 (100) | 0 (0) |
| ≥25 CS | 57 | 26 (46) | 6 | 3 | 0 | 9 (35) | 9 | 1 | 0 | 0 | 10 (38) | 7 (27) |
| AB=0 | 33 | 13 (39) | 3 | 1 | 0 | 4 (31) | 5 | 0 | 0 | 0 | 5 (38) | 4 (31) |
|
0<AB<1,000 | 7 | 4 (57) | 1 | 0 | 0 | 1 (25) | 3 | 0 | 0 | 0 | 3 (75) | 0 (0) |
| ≥1,000 AB | 17 | 9 (53) | 2 | 2 | 0 | 4 (44) | 1 | 1 | 0 | 0 | 2 (22) | 3 (33) |
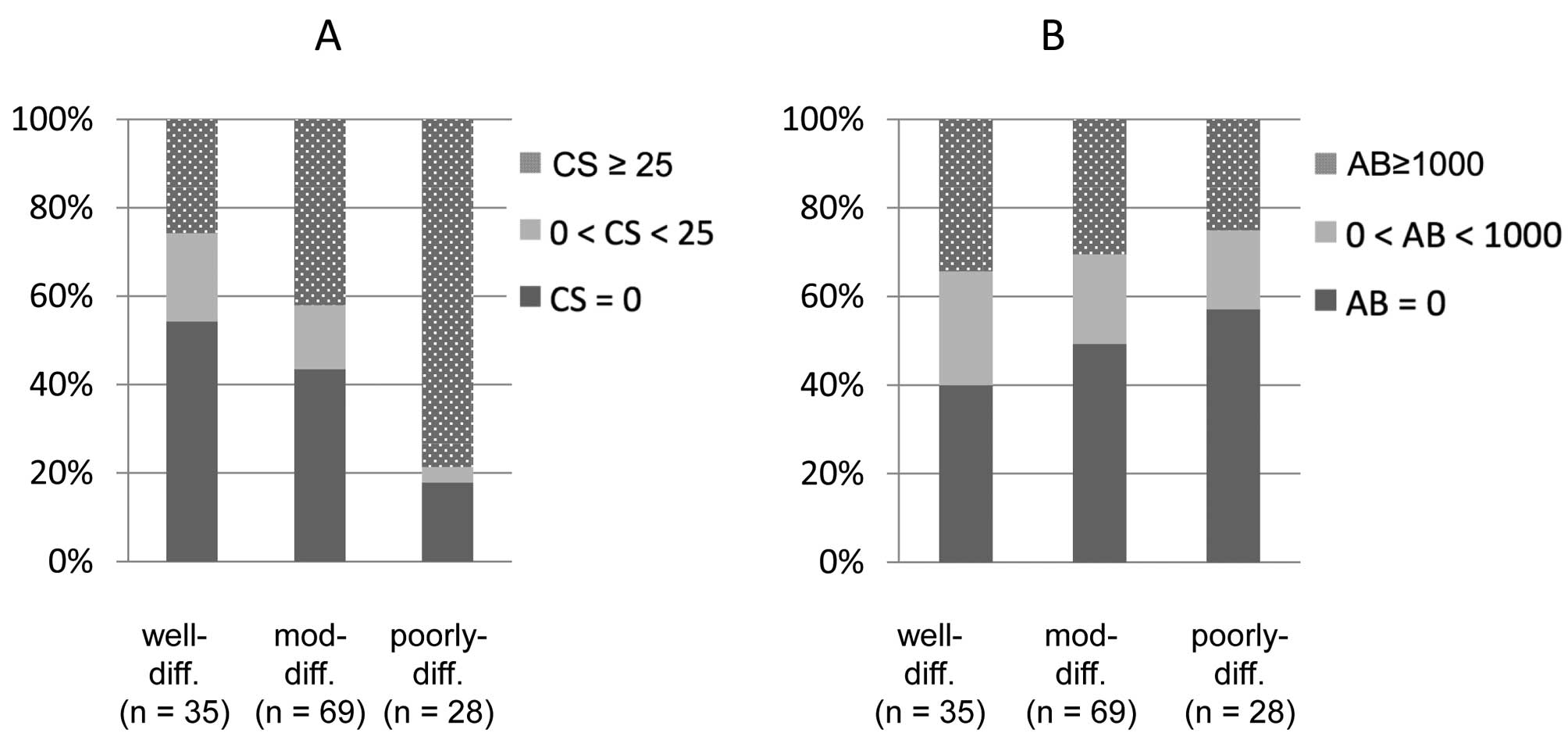
With respect to tumor differentiation grade, a
heavier smoking habit was associated with less-differentiated
adenocarcinomas (Fig. 3A, p=0.0010,
Chi-square test), in line with a previous study (18). On the other hand, there was no
correlation between asbestos deposition and the differentiation
grade (Fig. 3B, p=0.75).
Discussion
Both tobacco smoke and asbestos fibers are
significant inhaled carcinogens which contribute significantly to
lung adenocarcinoma development. We previously revealed that
chromosome instability and LOH, rather than minisatellite and
microsatellite instability, play major roles in the development of
lung adenocarcinomas (19). The LOH
and p53 spectra provide clues concerning the etiology and
nature of carcinogenesis. To elucidate the carcinogenic mechanisms
of two different inhaled carcinogens, asbestos and cigarette smoke,
we investigated LOH on all autosomal chromosomes and measured
asbestos burden (AB; asbestos body per gram of dry lung tissue)
using corresponding normal lung tissue and investigated p53
mutation employing fresh tumor samples.
The p53 mutational spectra may be helpful for
identification of the origins of the mutations that give rise to
human cancers. For example, aflatoxin B1-associated hepatocellular
carcinomas frequently have the specific p53 mutations: G:C
to T:A transversions at the 3rd base of codon 249, AGG to AGT (Arg
to Ser) (20). Another example of a
clearly characteristic ‘finger-print’ mutation in p53 is the
CC to TT double mutation in skin cancer (21). Exposure to UV light, a physical
mutagen, produces distinctive pyrimidine dimers that, if
unrepaired, can produce tandem mutations, most characteristically
CC to TT transitions. Similar to these, the p53 mutational
spectra can provide clues to the etiology of cancers.
The possible role of asbestos-exposure in the
genesis of p53 mutations in lung cancers is less well
understood. Husgafvel-Pursiainen et al investigated
p53 mutation of 105 lung cancers from smokers, comprising 53
squamous cell carcinomas, 39 adenocarcinomas and other 13
carcinomas, focusing on the presence or absence of
asbestos-exposure (22). They found
p53 mutations in 39% of asbestos-exposed patients with lung
cancer while the percentage was 54% in patients not exposed to
asbestos, indicating that the p53 mutations were less common
among the cases with occupational asbestos-exposure than in the
non-exposed cases. These results have not been verified yet by
another study, and need additional examinations of smoking
status.
In adenocarcinoma without asbestos-exposure or
smoking-exposure, the p53 mutation rate was the lowest. It
increased in correlation with the elevation of asbestos-exposure
and/or smoking-exposure. Adenocarcinomas associated with frequent
smoking have characteristic p53 mutations, especially G:C to
T:A transversions (17), at
specific ‘hotspot’ codons (13).
However, adenocarcinomas associated only with asbestos-exposure had
non-specific p53 mutations, such as transitions which are
thought to be caused by endogenous mechanisms associated with
spontaneous events (9,17). Asbestos may work in a promoter-like
manner. Production of reactive oxygen species and/or induction of
tissue regeneration may be relevant.
Adenocarcinomas have different etiologies from
squamous cell carcinomas, which can be reflected also in terms of
LOH. As we revealed, LOH frequency was higher in squamous cell
carcinomas than in adenocarcinomas (6,7).
Poorly differentiated adenocarcinomas, which are often noted in
smokers such as squamous cell carcinomas, have higher LOH frequency
than differentiated adenocarcinomas, which have a relatively weaker
association with smoking (23).
Smoking induces complicated genetic changes in lung cancers.
One of the most intriguing recent discoveries in the
field of lung cancer research is the identification of new driver
mutations in lung adenocarcinomas, such as EGFR mutations
(24,25) and ALK fusion (26). Both lung cancers with EGFR
mutations or ALK translocations are characterized by
negative or light smoking history. Lung cancers in non-smokers are
considered to be less genetically complex than those in smokers and
therefore they often have distinct characteristics developing on
simple gene mutations for maintenance and survival. Consequently,
patients with tumors harboring such simple oncogenic mutations
represent good candidates who may stand to benefit from
molecular-targeted drugs. To date, two-thirds of Japanese
adenocarcinomas and a little more than half of Caucasian
adenocarcinomas have mutually exclusive oncogenic mutations or
other genetic alterations including EGFR, KRAS,
MET, ALK and HER2 (27). Asbestos-associated alterations in
chromosomal regions, such as 19p13 (28), 9q33.1 (29) and 2p16 (30) have been identified. Whereas the
smoking status has a significant association with driver mutations
in lung adenocarcinomas, the relationship with asbestos-exposure
remains unclear.
In adenocarcinomas without asbestos-exposure, the
LOH frequency increased only slightly, correlating with the
elevation in smoking-exposure. On the other hand, in
adenocarcinomas with asbestos-exposure, the LOH frequency increased
as asbestos-exposure and/or smoking-exposure was elevated. This
suggests that asbestos-exposure in concert with smoking-exposure
increases LOH frequency.
In the present study, lung adenocarcinomas, for
which asbestos-exposure and smoking-exposure data could be
obtained, were examined for LOH and the p53 mutation.
Combined effects of asbestos and cigarette smoke were suggested by
these analyses. Asbestos-exposure alone did not increase the LOH
frequency but increased non-specific p53 mutations. These
findings suggest that the major carcinogenic mechanism of asbestos
in lung adenocarcinomas may be as a promoter, contributing to the
genotoxic effect of cigarette smoke. Since this study was based on
a general cancer center’s experience, the limited sample size does
not permit consideration that the result is conclusive. Further
investigation with a large sample size is required to establish the
mechanism of asbestos-induced lung carcinogenesis.
Acknowledgements
The authors thank Ms. Miyuki Kogure, Mr. Motoyoshi
Iwakoshi, Ms. Tomoyo Kakita, and Ms. Shizue Kurimori for their
technical assistance, and Ms. Yuki Takano and Ms. Yumiko Toriyama
for secretarial assistance. Parts of this study were supported
financially by Grants-in-Aid for Scientific Research from the
Ministry of Education, Culture, Sports, Science and Technology,
from the Japan Society for the Promotion of Science including
Grant-in-Aid for Young Scientists (B), and by grants from the
Ministry of Health, Labour and Welfare, the Smoking Research
Foundation, and the Vehicle Racing Commemorative Foundation.
References
|
1
|
LaDou J: The asbestos cancer epidemic.
Environ Health Perspect. 112:285–290. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jaurand MC: Mechanisms of fiber-induced
genotoxicity. Environ Health Perspect. 105(Suppl 5): 1073–1084.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heintz NH, Janssen YM and Mossman BT:
Persistent induction of c-fos and c-jun expression by
asbestos. Proc Natl Acad Sci USA. 90:3299–3303. 1993.PubMed/NCBI
|
|
4
|
Timblin CR, Janssen YW and Mossman BT:
Transcriptional activation of the proto-oncogene c-jun by
asbestos and H2O2is directly related to
increased proliferation and transformation of tracheal epithelial
cells. Cancer Res. 55:2723–2726. 1995.PubMed/NCBI
|
|
5
|
Doll R: Mortality from lung cancer in
asbestos workers. Br J Ind Med. 12:81–86. 1955.
|
|
6
|
Tsuchiya E, Nakamura Y, Weng SY, et al:
Allelotype of non-small cell lung carcinoma - comparison between
loss of heterozygosity in squamous cell carcinoma and
adenocarcinoma. Cancer Res. 52:2478–2481. 1992.PubMed/NCBI
|
|
7
|
Sato S, Nakamura Y and Tsuchiya E:
Difference of allelotype between squamous cell carcinoma and
adenocarcinoma of the lung. Cancer Res. 54:5652–5655.
1994.PubMed/NCBI
|
|
8
|
Hashimoto T, Tokuchi Y, Hayashi M, et al:
Different subtypes of human lung adenocarcinoma caused by different
etiological factors. Evidence from p53 mutational spectra. Am J
Pathol. 157:2133–2141. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vähäkangas K: TP53 mutations in workers
exposed to occupational carcinogens. Hum Mutat. 21:240–251.
2003.PubMed/NCBI
|
|
11
|
Petitjean A, Mathe E, Kato S, et al:
Impact of mutant p53 functional properties on TP53 mutation
patterns and tumor phenotype: lessons from recent developments in
the IARC TP53 database. Hum Mutat. 28:622–629. 2007.PubMed/NCBI
|
|
12
|
Denissenko MF, Pao A, Tang M and Pfeifer
GP: Preferential formation of benzo[a]pyrene adducts at lung
cancer mutational hotspots in P53. Science. 274:430–432.
1996.
|
|
13
|
Smith LE, Denissenko MF, Bennett WP, et
al: Targeting of lung cancer mutational hotspots by polycyclic
aromatic hydrocarbons. J Natl Cancer Inst. 92:803–811. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Japan Lung Cancer Society. General Rules
for Clinical and Pathologic Record of Lung Cancer. 5th edition.
Kanahara, Tokyo: 1999, (In Japanese).
|
|
15
|
Inamura K, Satoh Y, Okumura S, et al:
Pulmonary adenocarcinomas with enteric differentiation: histologic
and immunohistochemical characteristics compared with metastatic
colorectal cancers and usual pulmonary adenocarcinomas. Am J Surg
Pathol. 29:660–665. 2005. View Article : Google Scholar
|
|
16
|
Kohyama N and Suzuki Y: Analysis of
asbestos fibers in lung parenchyma, pleural plaques, and
mesothelioma tissues of North American insulation workers. Ann NY
Acad Sci. 643:27–52. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Greenblatt MS, Bennett WP, Hollstein M and
Harris CC: Mutations in the p53 tumor suppressor gene: clues
to cancer etiology and molecular pathogenesis. Cancer Res.
54:4855–4878. 1994.PubMed/NCBI
|
|
18
|
Nakachi K, Hayashi S, Kawajiri K and Imai
K: Association of cigarette smoking and CYP1A1 polymorphisms
with adenocarcinoma of the lung by grades of differentiation.
Carcinogenesis. 16:2209–2213. 1995.PubMed/NCBI
|
|
19
|
Ninomiya H, Nomura K, Satoh Y, et al:
Genetic instability in lung cancer: concurrent analysis of
chromosomal, mini- and microsatellite instability and loss of
heterozygosity. Br J Cancer. 94:1485–1491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang
NJ and Harris CC: Mutational hotspot in the p53 gene in human
hepatocellular carcinomas. Nature. 350:427–428. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brash DE, Rudolph JA, Simon JA, et al: A
role for sunlight in skin cancer: UV-induced p53 mutations in
squamous cell carcinoma. Proc Natl Acad Sci USA. 88:10124–10128.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Husgafvel-Pursiainen K, Karjalainen A,
Kannio A, et al: Lung cancer and past occupational exposure to
asbestos. Role of p53 and K-ras mutations. Am J
Respir Cell Mol Biol. 20:667–674. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishikawa Y, Furuta R, Miyoshi T, et al:
Loss of heterozygosity and the smoking index increase with decrease
in differentiation of lung adenocarcinomas: etiologic implications.
Cancer Lett. 187:47–51. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paez JG, Jänne PA, Lee JC, et al:
EGFR mutations in lung cancer: correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar
|
|
26
|
Soda M, Choi YL, Enomoto M, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature. 448:561–566. 2007.PubMed/NCBI
|
|
27
|
Inamura K and Ishikawa Y: Lung Cancer.
Asian Pacific Organization for Cancer Prevention Cancer Report
2010. Tuncer MA: New Hope in Health Foundation; Turkey: pp.
202–204. 2010
|
|
28
|
Ruosaari ST, Nymark PE, Aavikko MM, et al:
Aberrations of chromosome 19 in asbestos-associated lung cancer and
in asbestos-induced micronuclei of bronchial epithelial cells in
vitro. Carcinogenesis. 29:913–917. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nymark P, Kettunen E, Aavikko M, et al:
Molecular alterations at 9q33.1 and polyploidy in asbestos-related
lung cancer. Clin Cancer Res. 15:468–475. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kettunen E, Aavikko M, Nymark P, et al:
DNA copy number loss and allelic imbalance at 2p16 in lung cancer
associated with asbestos exposure. Br J Cancer. 100:1336–1342.
2009. View Article : Google Scholar : PubMed/NCBI
|