Introduction
The epidermal growth factor receptor
[(EGFR)/ErbB1/HER1)] is a member of the ErbB tyrosine kinase
family, which consists of ErbB1, ErbB2 (HER2), ErbB3 (HER3) and
ErbB4 (HER4). All receptors of the ErbB family activate and
regulate diverse cellular processes, including proliferation,
survival, adhesion, migration and differentiation (1). Ligand binding potentiates receptor
interaction with either a homologous molecule (homodimerization), a
different ErbB-family receptor, or another cell surface tyrosine
kinase receptor (IGF-1R or c-Met) (heterodimerization) (2–5). Dimer
formation causes autophosphorylation of a tyrosine residue in the
cytoplasmic domain and activates proteins triggering downstream
events. Such proteins include MAPK, PI3K/AKT, phospholipase Cγ, PKC
and STAT. Thus, EGFR proteins indirectly regulate gene expression.
Variants of the genes encoding the EGFR family are oncogenes of
various tumors. Upregulation of EGFR expression in many human
epithelial cancers is associated with advanced tumor stage and an
unfavorable prognosis (6,7). Thus, EGFR is considered to be not only
a useful prognostic biomarker but also a promising therapeutic
target, and specific anti-EGFR monoclonal antibodies (mAbs) to
extracellular domains of the proteins, and tyrosine kinase
inhibitors (TKIs), have been developed and used in cancer
treatment.
Cetuximab is a chimeric IgG1 monoclonal antibody
that binds with high affinity to the extracellular domain of EGFR
(8). The antibody blocks EGFR
activation by preventing tyrosine kinase-mediated phosphorylation
of the protein (9). In addition,
antibody binding may trigger antibody-dependent cellular
cytotoxicity (ADCC); the host immune system may attack cells
covered with antibody bound to EGFR (10,11).
Downstream effects of cetuximab include promotion of apoptosis,
inhibition of cell cycle progression, tumor cell invasion and
angiogenesis.
Cetuximab has been prescribed for patients with
metastatic colorectal cancer (mCRC) (12–16)
and head and neck squamous cell carcinoma (HNSCC) (17–21).
Effects of monotherapy of cetuximab are shown in 13% of recurrent
or metastatic HNSCC and improvement of overall survival benefits is
shown in combination with radiation or other chemotherapy (17,18).
However, use of EGFR inhibitors containing cetuximab is associated
with severe side-effects, a low reaction rate, and recurrence
following treatments. These problems must be solved (21,22).
Thus, it is important to understand not only how cetuximab acts but
also the mechanisms of cetuximab resistance. To date, various
resistance mechanisms of cetuximab have been described. These fall
into two categories. First, resistance may develop via constitutive
activation of growth caused by changes in effectors of the EGFR
signal transduction pathway (23–26).
Second, proliferation may be stimulated by activation of receptors
other than EGFR (27–30). However, the detailed features of
cetuximab remain elusive.
Human cancer tissues are heterogeneous in nature and
become differentiated during expansion of cancer stem cells (CSCs)
(31). CSCs initiate tumorigenesis,
and are involved in tumor recurrence and metastasis (32). Furthermore, data show that CSCs are
highly resistant to anticancer drugs (33–35).
Therefore, analysis of supposedly heterogenous tumor cell masses
containing CSCs is required to identify the molecular mechanisms by
which cetuximab resistance develops.
In the present study, we investigated the
characteristics of cetuximab-resistant oral squamous cell carcinoma
(OSCC) cells. We showed that cetuximab-resistant cells exhibited
stem cell-like properties, and proliferation of such cells in
monolayer culture was EGFR-independent. However, growth became
EGFR-dependent in floatation culture, and cell spheres formed by
cetuximab-resistant stem cell-like cells became
cetuximab-sensitive. Thus, resistance to cetuximab is not only
cell-type-dependent but is also influenced by the microenvironment
in which cells grow.
Materials and methods
Cell culture and reagents
Three human OSCC cell lines, HSC3, HSC4 and SAS
provided by the RIKEN BioResource Center (Ibaraki, Japan), were
used in the present study. The cells were cultured in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal
calf serum (FCS) (both from Life Technologies, Japan) at 37°C in a
humidified atmosphere of 5% (v/v) CO2 in air. Cetuximab
(Erbitux®) was purchased from Merck Serono (Tokyo,
Japan). AG1478 and the EGFR/ErbB2/ErbB4 inhibitor II were from
Calbiochem (Merck Millipore, Billerica, MA, USA). Antibodies used
for western blot analyses and immunofluorescence were from the
following sources; antibodies against EGFR and phosphorylated EGFR
were obtained from Cell Signaling Technology (Denvers, MA, USA);
the antibody against α-tubulin was from Sigma-Aldrich (St. Louis,
MO, USA); and that against Ki-67 from Dako (Tokyo, Japan).
Cell proliferation assay
Human OSCC cells (2×103/well) were plated
in 96-well plates. After 24 h of growth, various reagents were
added at the indicated concentrations and growth continued for a
further 2, 4 or 6 days. All experiments were performed in
triplicate. Cell proliferation was assessed using the CellTiter
96® Non-Radioactive Cell Proliferation Assay (Promega,
Tokyo, Japan).
Aggregation culture and sphere
formation
When aggregation culture was performed,
1×103 cells were seeded into each well of low adhesive
96-well plates (Sumitomo, Tokyo, Japan) and cultured in DMEM
supplemented with 10% (v/v) FCS at 37°C under 5% (v/v)
CO2. To allow sphere formation, 1:1,000 dilution of a
suspension of 1×103 cells was added to the well of
low-adhesive 96-well U-shaped plates in ‘sphere medium’, which was
DMEM/F12 supplemented with 2 mM glutamine, 2% (v/v) B27, 20 ng/ml
EGF, 20 ng/ml bFGF, penicillin, and streptomycin.
Western blotting
OSCC cells were seeded in 60-mm plates at a density
of 2×105/plate. After 2 days of growth, cells were
collected and lysed in RIPA buffer [150 mM NaCl, 10 mM Tris-HCl, pH
8.0, 1% (v/v) Nonidet P-40, 0.5% (w/v) deoxycholic acid, 0.1% (w/v)
SDS, and 5 mM EDTA] containing 1X Halt™ Protease Inhibitor Cocktail
(Thermo Fisher Scientific, Yokohama, Japan). Samples were incubated
at 95°C for 4 min, separated by SDS-PAGE, and electrophoretically
transferred to PVDF membranes (GE Healthcare, Tokyo, Japan).
Non-specific binding was blocked by incubation in 5% (w/v) bovine
serum albumin (BSA) in TBS/Tween-20 (TBS-T) for 1 h at room
temperature. Membranes were probed with antibodies against EGFR and
phosphorylated EGFR in TBS-T overnight at 4°C and then incubated
with HRP-conjugated goat anti-rabbit secondary antibody at a
dilution of 1:5,000. Antibody-antigen complexes were detected by
ECL plus western blotting detection reagent (GE Healthcare).
Immunofluorescence staining
Cultured cells and cell aggregates were fixed in
3.5% (w/v) formaldehyde, permeabilized in 0.2% (v/v) Triton X-100,
and blocked in 2% (w/v) BSA. The primary antibodies were rabbit
anti-EGFR, rabbit anti-phosphorylated EGFR, and mouse anti-Ki-67.
Alexa Fluor 488-conjugated IgG (Life Technologies) was used as the
secondary antibody. After incubation with the antibodies, SlowFade
Gold Antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI;
Invitrogen) was added. The specimens were observed using
fluorescence microscopy.
Results
Cetuximab inhibits proliferation of
AG1478-sensitive HSC3 and HSC4 cells, but not AG1478-resistant SAS
cells
To compare the effect of cetuximab, EGFR inhibitor
(AG1478), and ErbBs inhibitor (EGFR/ErbB2/ErbB4 inhibitor II) on
OSCC cell lines, we first treated HSC3, HSC4 and SAS cells with
increasing concentrations of the drugs for 6 days; proliferation
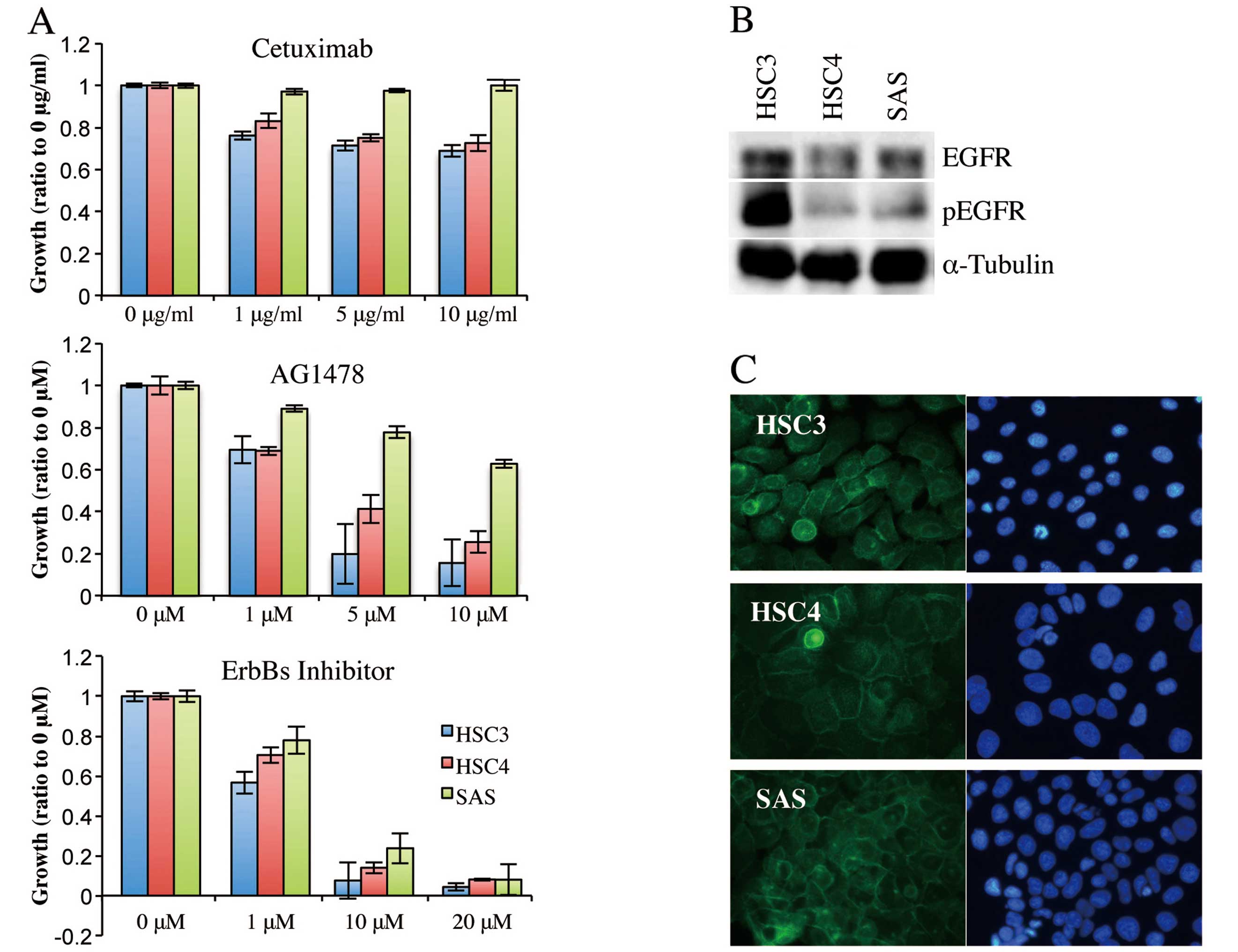
was then assessed by MTT assay (Fig.
1A). Cetuximab at 1 μg/ml reduced the proliferation of HSC3 and
HSC4 cells, but SAS cells proliferated to the extent of untreated
cells even when the cetuximab concentration was 10 μg/ml. Thus,
HSC3 and HSC4 cells were cetuximab-sensitive and SAS cells were
cetuximab-resistant. Proliferation of HSC3 and HSC4 cells was
effectively inhibited, in a concentration-dependent manner, by
AG1478, and proliferation almost ceased when the inhibitor was
added to 5 or 10 μM. AG1478 also reduced proliferation of SAS
cells, but only at higher concentrations; SAS cell proliferation
was not inhibited by AG1478 at 10 μM. Notably, EGFR/ErbB2/ErbB4
inhibitor II strongly inhibited proliferation of all three cell
lines, killing most cells at 10 or 20 μM. These results suggested
that HSC3 and HSC4 proliferation was regulated principally by EGFR,
whereas SAS proliferation was controlled by a receptor of the
ErbB-family other than EGFR.
We next analyzed expression and intracellular
localization of EGFR in these cell lines. Western blotting using
antibodies against EGFR and phosphorylated EGFR showed that EGFR
protein was expressed and phosphorylated in all cell lines,
although the phosphorylated EGFR level was higher in HSC3 cells
than in the other two cell types (Fig.
1B). Immunofluorescence from anti-EGFR antibody was detected in
the cell-cell contact regions of all cell lines, indicating that
EGFR was located in the cell membrane (Fig. 1C). Thus, cetuximab-resistant SAS
cells still expressed EGFR in the cell membrane and EGFR became
phosphorylated when stimulated with a ligand.
Cetuximab-resistant OSCC cell lines
proliferate autonomously in monolayer culture and form spheres in
floating culture
To further characterize the cetuximab resistant OSCC
cells, which are also AG1478 resistant, we compared the
proliferative properties and sphere-formation capacities of the
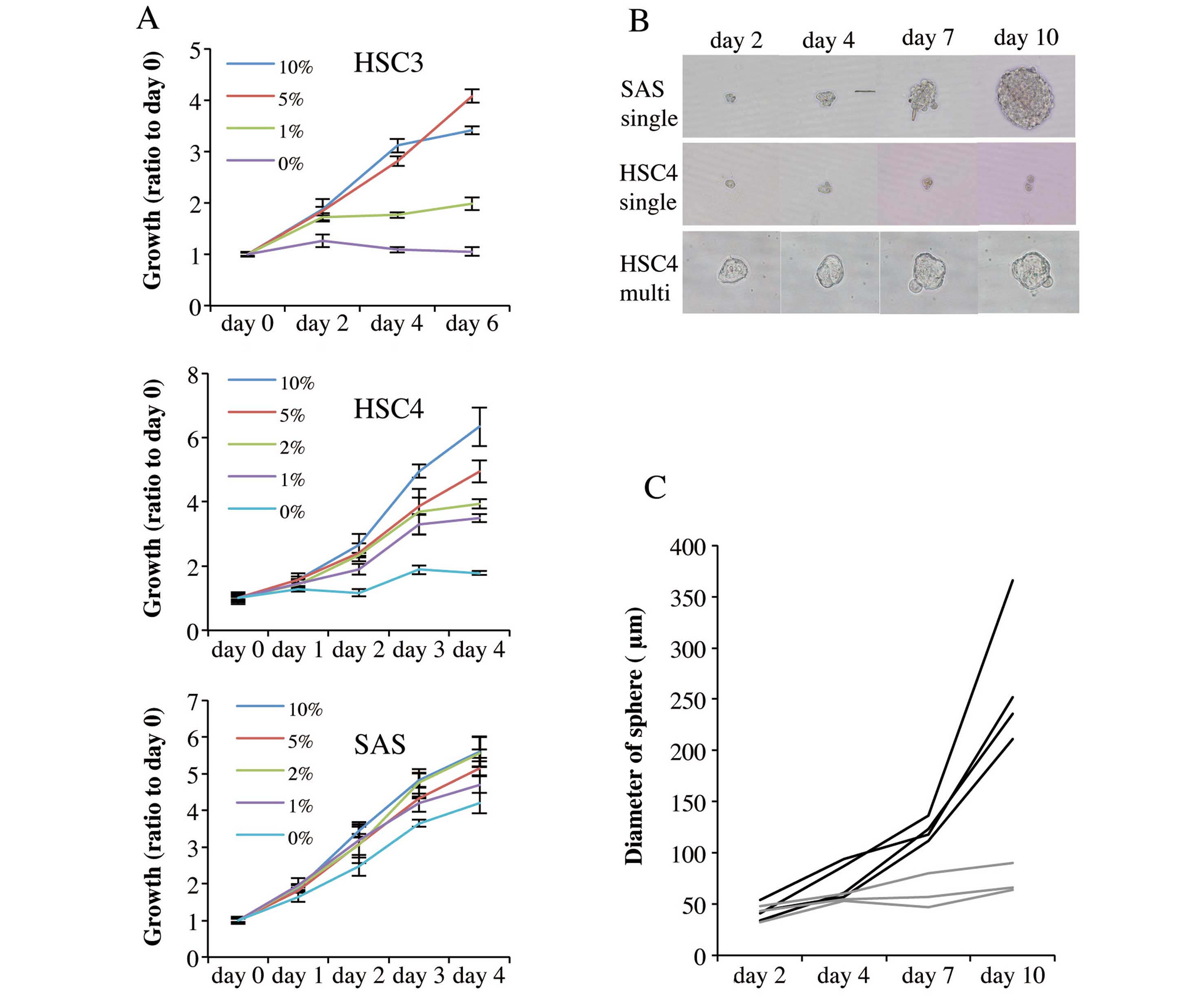
three cell lines. Fig. 2A shows
that proliferation in monolayer culture was promoted by serum in a
concentration-dependent manner. HSC3 and HSC4 cells proliferated
only slightly over 6 and 4 days, respectively of serum-free
culture. Notably, however, SAS cells proliferated strongly in
serum-free medium over 4 days of culture.
We next analyzed the sphere formation capacity of
single cells growing in serum-free sphere formation medium
supplemented with bFGF and EGF (36). Suspensions of 1×103 cells
were diluted so that U-shaped wells of low-adhesion 96-well plates
received single cells in suspension, and the cells were cultured
for 10 days. SAS cells formed spheres from single cells (Fig. 2B), and sphere diameter increased as
culture time rose (Fig. 2B and C).
In contrast, HSC4 cells did not form spheres from either single
(Fig. 2B and C) or multiple cells
(Fig. 2B). Thus,
cetuximab-resistant SAS cells exhibited cancer stem cell-like
characteristics but cetuximab-sensitive cells were incapable of
forming growing aggregates.
Growth of SAS aggregates is regulated by
EGFR and inhibited by cetuximab
HSC4 cells, similar to SAS cells, could form
aggregates containing many cells, but unlike SAS aggregates, the
HSC4 aggregates had poor growth characteristics. To compare the
growth potential of OSCC cell lines under anchorage-independent
culture conditions, 5×103 of the HSC3, HSC4 and SAS
lines were seeded into U-shaped wells of low-adhesive 96-well
plates and cultured in DMEM supplemented with 10% (v/v) FCS.
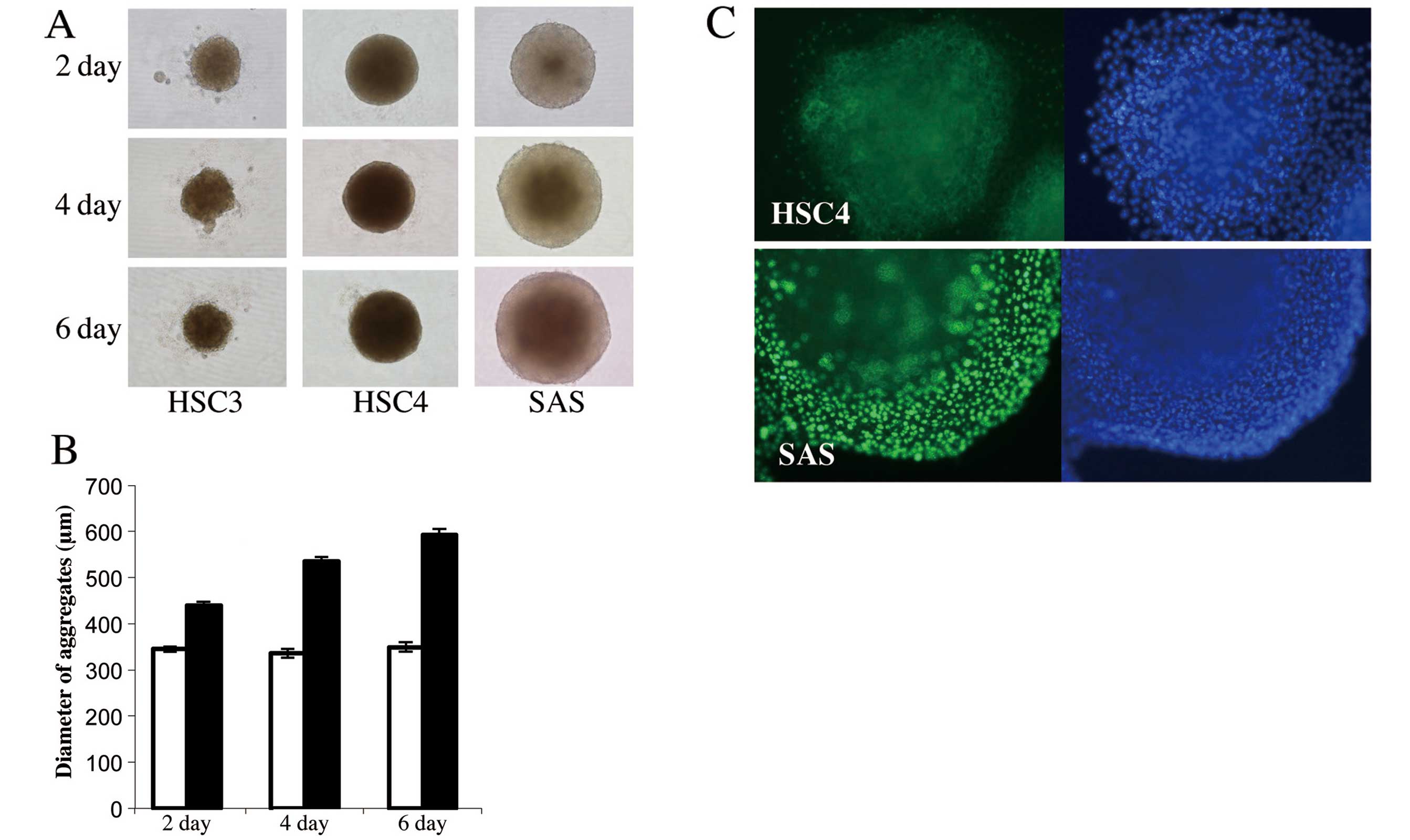
Fig. 3A and B shows that all cell
lines formed aggregates on the first day of culture, but HSC3 and
HSC4 aggregates were smaller than those formed by SAS cells and the
diameters of the former aggregates did not increase thereafter.
HSC4 aggregates were maintained for 6 days, but cell-cell contact
was weak and dissociated cells became obvious in HSC3 aggregates.
Conversely, the SAS aggregates increased in diameter as culture
proceeded, and the aggregates were subjected to immunofluorescence
staining using anti-Ki-67 antibody. Many nuclei of SAS aggregates
stained with this antibody, but Ki-67-positive nuclei were absent
from HSC4 cell aggregates (Fig.
3C), indicating that SAS aggregation reflected proliferation of
aggregated floating cells and that HSC4 could not proliferate in
aggregation culture even in the presence of serum.
To determine whether growth of SAS aggregates was
serum-independent, 5×103 cells were seeded into U-shaped
well of low-adhesive 96-well plates in DMEM with or without 10%
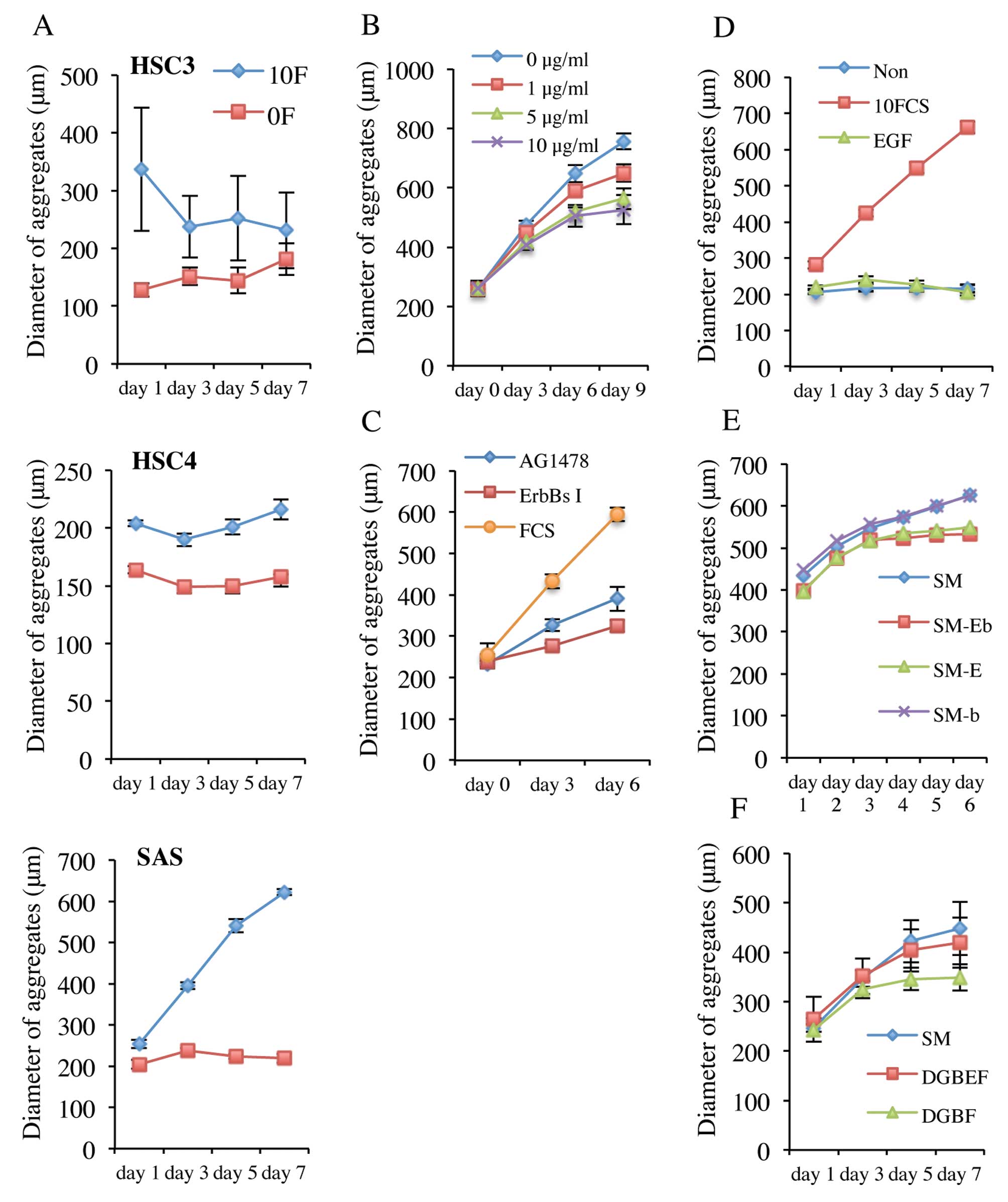
(v/v) FCS. As shown in Fig. 4A, all
cell types formed aggregates in either serum-containing or -free
medium. The HSC3 and HSC4 aggregates formed in serum-containing
medium were larger than those formed in serum-free medium, but SAS
cell aggregates were of similar size in either medium, indicating
that serum influenced the numbers of HSC3 and HSC4 cells that
aggregated, but not the number of SAS cells. The diameter of SAS
aggregates was greater in serum-containing medium, indicating that
anchorage-independent growth of SAS cells was regulated by serum.
Serum-dependent growth of SAS aggregates was reduced not only by an
ErbBs inhibitor but also by cetuximab and AG1478 (Fig. 4B and C), indicating that activation
of the EGFR signal transduction pathway may be associated with
proliferation of SAS aggregates.
However, SAS aggregates did not proliferate when EGF
was added to DMEM (Fig. 4D). On the
other hand, when EGF was removed from sphere medium, proliferation
of SAS aggregates ceased (Fig. 4E).
Furthermore, although SAS aggregates proliferated in DMEM with
added glutamine, B27, EGF and bFGF, removal of EGF caused
proliferation to cease (Fig.
4F).
Discussion
In the present study, we evaluated the proliferative
behavior of oral SCC cell lines including cetuximab-sensitive HSC3
and HSC4 and cetuximab-resistant SAS. Notably, all cell lines
expressed EGFR in the cell membrane and phosphorylated, regardless
of cetuximab sensitivity status.
The monoclonal antibody cetuximab targets the
extracellular domain of EGFR with high specificity and affinity
(37). Cetuximab blocks ligand
binding and thereby inhibits EGFR phosphorylation (9). Thus, cetuximab should inhibit
EGFR-dependent cell proliferation. In the present study,
proliferation of HSC3 and HSC4 cells was strongly associated with
EGFR ligand-EGFR signaling, since proliferation was markedly
reduced by the EGFR kinase inhibitor AG1478. Thus, cetuximab
prevented EGFR phosphorylation, reducing proliferation of HSC3 and
HSC4. These data are consistent with those of a previous report
that EGFR biomarker analysis in non-small cell lung carcinoma
patients showed that those with higher EGFR expression levels
obtained more therapeutic benefit from cetuximab than did patients
with lower EGFR levels (38).
However, we found that SAS proliferation was not affected by
cetuximab, although SAS cells expressed EGFR, which was localized
in the cell membrane and phosphorylated, as did HSC3 and HSC4
cells. A previous report showed that colorectal cancer patients
with EGFR-negative tumors could nonetheless respond to
cetuximab-based therapies (12).
Collectively, the data suggest that EGFR status does not seem to
have predictive value when used to gauge the efficacy of cetuximab
treatment in oral cancer patients.
Although cetuximab is a valuable drug, both
intrinsic resistance to the material and the development of
acquired resistance, are well-recognized problems. Several
mechanisms of resistance to cetuximab have been described. Western
blot and immunofluorescence analysis showed that the expression
level and localization of EGFR in SAS cells were similar to those
of cetuximab-sensitive cell lines, indicating that neither the
expression and activity level of EGFR (39), nor inappropriate cellular
distribution of EGFR (28,40) was probable cause of the drug
resistance shown by SAS cells. However, the fact that
phosphorylated EGFR is expressed by SAS cells suggests that EGFR is
indeed stimulated by a ligand in such cells and plays a role in the
regulation of a function other than growth, which may in turn be
affected by cetuximab. Proliferation of SAS monolayer cultures was
completely inhibited by EGFR/ErbB2/ErbB4 inhibitor II, but only
moderately by AG1478, indicating that SAS proliferation was
regulated principally by a receptor of the ErbB family other than
EGFR. SAS cells also actively proliferated in serum-free culture.
Such autonomous growth of SAS cells was strongly inhibited by
EGFR/ErbB2/ErbB4 inhibitor II and moderately suppressed by AG1478
(data not shown), as was also true of growth in serum-containing
culture. These results suggest that SAS cells express a high level
of ErbB ligands, binding to ErbB receptors other than EGFR, thus
explaining the cetuximab resistance of such cells. It was
previously reported that HB-EGF played an important role in the
development of cetuximab resistance in HNSCC (41).
Sphere formation revealed the stem cell-like
properties of SAS cells. HSC3 and HSC4 cells essentially lacked
such properties, being unable to form spheres from single cells.
This suggests that expression of stem cell-like features is
associated with cetuximab resistance under anchorage-dependent
growth conditions. Spheres were grown in sphere medium supplemented
with bFGF and EGF, but not if EGF was absent. SAS aggregates grew
in DMEM supplemented with 10% (v/v) FCS but HSC3 and HSC4
aggregates did not, although cells of all three tested lines formed
aggregates. SAS aggregates did not grow in serum-free DMEM, rather
requiring serum components including EGF. Aggregated growth was
inhibited by cetuximab and AG1478. Thus, SAS cells lost the ability
to proliferate autonomously in anchorage-independent aggregation
culture; such growth was rather controlled by the EGFR pathway. It
thus appears that the cetuximab sensitivity status of stem
cell-like cancer cells is affected by the cellular
microenvironment.
In summary, cetuximab resistant OSCC cells not only
engaged in EGFR-independent growth in monolayer culture but also
exhibited stem cell-like properties. However, growth was
EGFR-dependent in aggregation culture of the cell, and the cell
aggregate became cetuximab-sensitive. We found that cetuximab
sensitivity is not only cell-type-dependent but is also affected by
the growth microenvironment.
Acknowledgements
This study was supported by funding from the Osaka
University and Osaka Dental University.
References
|
1
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harris RC, Chung E and Coffey RJ: EGF
receptor ligands. Exp Cell Res. 284:2–13. 2003. View Article : Google Scholar
|
|
3
|
Arteaga CL and Baselga J: Tyrosine kinase
inhibitors: why does the current process of clinical development
not apply to them? Cancer Cell. 5:525–531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galer CE, Corey CL, Wang Z, Younes MN,
Gomez-Rivera F, Jasser SA, Ludwig DL, El-Naggar AK, Weber RS and
Myers JN: Dual inhibition of epidermal growth factor receptor and
insulin-like growth factor receptor I: reduction of angiogenesis
and tumor growth in cutaneous squamous cell carcinoma. Head Neck.
33:189–198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C,
Johnson BE, Cantley LC and Jänne PA: MET amplification leads
to gefitinib resistance in lung cancer by activating ERBB3
signaling. Science. 316:1039–1043. 2007. View Article : Google Scholar
|
|
6
|
Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R,
Hammond EH, Fu KK and Milas L: Impact of epidermal growth factor
receptor expression on survival and pattern of relapse in patients
with advanced head and neck carcinoma. Cancer Res. 62:7350–7356.
2002.PubMed/NCBI
|
|
7
|
Chung CH, Zhang Q, Hammond EM, Trotti AM
III, Wang H, Spencer S, Zhang HZ, Cooper J, Jordan R, Rotman MH and
Ang KK: Integrating epidermal growth factor receptor assay with
clinical parameters improves risk classification for relapse and
survival in head-and-neck squamous cell carcinoma. Int J Radiat
Oncol Biol Phys. 81:331–338. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galizia G, Lieto E, De Vita F, Orditura M,
Castellano P, Troiani T, Imperatore V and Ciardiello F: Cetuximab,
a chimeric human mouse anti-epidermal growth factor receptor
monoclonal antibody, in the treatment of human colorectal cancer.
Oncogene. 26:3654–3660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ,
Kussie P and Ferguson KM: Structural basis for inhibition of the
epidermal growth factor receptor by cetuximab. Cancer Cell.
7:301–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
López-Albaitero A and Ferris RL: Immune
activation by epidermal growth factor receptor-specific monoclonal
antibody therapy for head and neck cancer. Arch Otolaryngol Head
Neck Surg. 133:1277–1281. 2007.PubMed/NCBI
|
|
11
|
Kurai J, Chikumi H, Hashimoto K, Yamaguchi
K, Yamasaki A, Sako T, Touge H, Makino H, Takata M, Miyata M,
Nakamoto M, Burioka N and Shimizu E: Antibody-dependent cellular
cytotoxicity mediated by cetuximab against lung cancer cell lines.
Clin Cancer Res. 13:1552–1561. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chung KY, Shia J, Kemeny NE, Shah M,
Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L,
Klimstra DS, Fridman D, Kelsen DP and Saltz LB: Cetuximab shows
activity in colorectal cancer patients with tumors that do not
express the epidermal growth factor receptor by
immunohistochemistry. J Clin Oncol. 23:1803–1810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jonker DJ, O’Callaghan CJ, Karapetis CS,
Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ,
Tebbutt NC, van Hazel G, Wierzbicki R, Langer C and Moore MJ:
Cetuximab for the treatment of colorectal cancer. N Engl J Med.
357:2040–2048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sobrero AF, Maurel J, Fehrenbacher L,
Scheithauer W, Abubakr YA, Lutz MP, Vega-Villegas ME, Eng C,
Steinhauer EU, Prausova J, Lenz HJ, Borg C, Middleton G, Kröning H,
Luppi G, Kisker O, Zubel A, Langer C, Kopit J and Burris HA III:
EPIC: phase III trial of cetuximab plus irinotecan after
fluoropyrimidine and oxaliplatin failure in patients with
metastatic colorectal cancer. J Clin Oncol. 26:2311–2319. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bokemeyer C, Bondarenko I, Makhson A,
Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G,
Stroh C, Loos AH, Zubel A and Koralewski P: Fluorouracil,
leucovorin and oxaliplatin with and without cetuximab in the
first-line treatment of metastatic colorectal cancer. J Clin Oncol.
27:663–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Van Cutsem E, Köhne CH, Hitre E, Zaluski
J, Chang Chien CR, Makhson A, D’Heans G, Pintér T, Lim R, Bodoky G,
Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M,
Nippgen J and Rougier P: Cetuximab and chemotherapy as initial
treatment for metastatic colorectal cancer. N Engl J Med.
360:1408–1417. 2009.PubMed/NCBI
|
|
17
|
Vermorken JB, Mesia R, Rivera F, Remenar
E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol
D, Peyrade F, Benasso M, Vynnychenko I, De Raucourt D, Bokemeyer C,
Schueler A, Amellal N and Hitt R: Platinum-based chemotherapy plus
cetuximab in head and neck cancer. N Engl J Med. 359:1116–1127.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies
MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK and Ang KK:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Herbst RS, Arquette M, Shin DM, Dicke K,
Vokes EE, Azarnia N, Hong WK and Kies MS: Phase II multicenter
study of the epidermal growth factor receptor antibody cetuximab
and cisplatin for recurrent and refractory squamous cell carcinoma
of the head and neck. J Clin Oncol. 23:5578–5587. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baselga J, Trigo JM, Bourhis J, Tortochaux
J, Cortés-Funes H, Hitt R, Gascón P, Amellal N, Harstrick A and
Eckardt A: Phase II multicenter study of the antiepidermal growth
factor receptor monoclonal antibody cetuximab in combination with
platinum-based chemotherapy in patients with platinum-refractory
metastatic and/or recurrent squamous cell carcinoma of the head and
neck. J Clin Oncol. 23:5568–5577. 2005.
|
|
21
|
Burtness B, Goldwasser MA, Flood W, Mattar
B and Forastiere AA; Eastern Cooperative Oncology Group. Phase III
randomized trial of cisplatin plus placebo compared with cisplatin
plus cetuximab in metastatic/recurrent head and neck cancer: an
Eastern Cooperative Oncology Group study. J Clin Oncol.
23:8646–8654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tejani MA, Cohen RB and Mehra T: The
contribution of cetuximab in the treatment of recurrent and/or
metastatic head and neck cancer. Biologics. 4:173–185.
2010.PubMed/NCBI
|
|
23
|
Sok JC, Coppelli FM, Thomas SM, Lange MN,
Xi S, Hunt JL, Freilino ML, Graner NW, Wikstrand CJ, Bigner DD,
Gooding WE, Furnari FB and Grandis JR: Mutant epidermal growth
factor receptor (EGFRvIII) contributes to head and neck cancer
growth and resistance to EGFR targeting. Clin Cancer Res.
12:5064–5073. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen LF, Cohen EE and Grandis JR: New
strategies in head and neck cancer: understanding resistance to
epidermal growth factor receptor inhibitors. Clin Cancer Res.
16:2489–2495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim SM, Kim JS, Kim JH, Yun CO, Kim EM,
Kim HK, Solca F, Choi SY and Cho BC: Acquired resistance to
cetuximab is mediated by increased PTEN instability and leads
cross-resistance to gefitinib in HCC827 NSCLC cells. Cancer Lett.
296:150–159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dunn EF, Iida M, Myers RA, Campbell DA,
Hintz KA, Armstrong EA, Li C and Wheeler DL: Dasatinib sensitizes
KRAS mutant colorectal tumors to cetuximab. Oncogene. 30:561–574.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ciardiello F, Bianco R, Caputo R, Caputo
R, Damiano V, Troiani T, Melisi D, De Vita F, De Placido S, Bianco
AR and Tortora G: Antitumor activity of ZD6474, a vascular
endothelial growth factor receptor tyrosine kinase inhibitor, in
human cancer cells with acquired resistance to antiepidermal growth
factor receptor therapy. Clin Cancer Res. 10:784–793. 2004.
View Article : Google Scholar
|
|
28
|
Wheeler DL, Iida M, Kruser TJ, Nechrebecki
MM, Dunn EF, Armstrong EA, Huang S and Harari PM: Epidermal growth
factor receptor cooperates with Src family kinases in acquired
resistance to cetuximab. Cancer Biol Ther. 8:696–703. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brand TM, Iida M and Wheeler DL: Molecular
mechanisms of resistance to the EGFR monoclonal antibody cetuximab.
Cancer Biol Ther. 11:777–792. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yonesaka K1, Zejnullahu K, Okamoto I,
Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M,
Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun
J, Destro A, Taira K, Takeda K, Okabe T, Swanson J, Itoh H, Takada
M, Lifshits E, Okuno K, Engelman JA, Shivdasani RA, Nishio K,
Fukuoka M, Varella-Garcia M, Nakagawa K and Jänne PA: Activation of
ERBB2 signaling causes resistance to the EGFR-directed therapeutic
antibody cetuximab. Sci Transl Med. 3:99ra862011.PubMed/NCBI
|
|
31
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells - perspectives on current status and future directions:
AACR workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar
|
|
33
|
Todaro M, Alea MP, Di Stefano AB,
Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G,
Medema JP and Stassi G: Colon cancer stem cells dictate tumor
growth and resist cell death by production of interleukin-4. Cell
Stem Cell. 1:389–402. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma S, Lee TK, Zheng BJ, Chan KW and Guan
XY: CD133+ HCC cancer stem cells confer chemoresistance
by preferential expression of the Akt/PKB survival pathway.
Oncogene. 27:1749–1758. 2008.
|
|
35
|
Salmaggi A, Boiardi Ak, Gelati M, Russo A,
Calatozzolo C, Ciusani E, Sciacca FL, Ottolina A, Parati EA, La
Porta C, Alessandri G, Marras C, Croci D and De Rossi M:
Glioblastoma-derived tumorospheres identify a population of tumor
stem-like cells with angiogenic potential and enhanced multidrug
resistance phenotype. Glia. 54:850–860. 2006. View Article : Google Scholar
|
|
36
|
Dontu G, Abdallah WM, Foley JM, Jackson
KW, Clarke MF, Kawamura MJ and Wicha MS: In vitro propagation and
transcriptional profiling of human mammary stem/progenitor cells.
Genes Dev. 17:1253–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thomas SM and Grandis JR: Pharmacokinetic
and pharmacodynamics properties of EGFR inhibitors under clinical
investigation. Cancer Treat Rev. 30:255–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pirker R, Pereira JR, von Pawel J,
Krzakowski M, Ramlau R, Park K, de Marinis F, Eberhardt WE,
Paz-Ares L, Störkel S, Shumacher KM, von Heydebreck A, Celik I and
O’Byrne KJ: EGFR expression as a predictor of survival for
first-line chemotherapy plus cetuximab in patients with advanced
non-small-cell lung cancer: analysis of data from the phase 3 FLEX
study. Lancet Oncol. 13:33–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wheeler DL, Huang S, Kruser TJ,
Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT and
Harari PM: Mechanisms of acquired resistance to cetuximab: role of
HER (ErbB) family members. Oncogene. 27:3944–3956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nevo J, Mattila E, Pellinen T, Yamamoto
DL, Sara H, Iljin K, Kallioniemi O, Bono P, Heikkilä P, Joensuu H,
Wärri A and Ivaska J: Mammary-derived growth inhibitor alters
traffic of EGFR and induces a novel form of cetuximab resistance.
Clin Cancer Res. 15:6570–6581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hatakeyama H, Cheng H, Wirth P, Counsell
A, Marcrom SR, Wood CB, Pohlmann PR, Gilbert J, Murphy B, Yarbrough
WG, Wheeler DL, Harari PM, Guo Y, Shyr Y, Slebos RJ and Chung CH:
Regulation of heparin-binding EGF-like growth factor by miR-212 and
acquired cetuximab-resistance in head and neck squamous cell
carcinoma. PLoS One. 5:e127022010. View Article : Google Scholar : PubMed/NCBI
|