Introduction
Colorectal cancer is one of the leading causes of
mortality and ranks among the three most common cancers in
developed countries (1,2). More than one million new cases of
colorectal cancer are diagnosed every year (3). Multiple lines of evidence suggest that
bioactive compounds present in various edible and medicinal plants
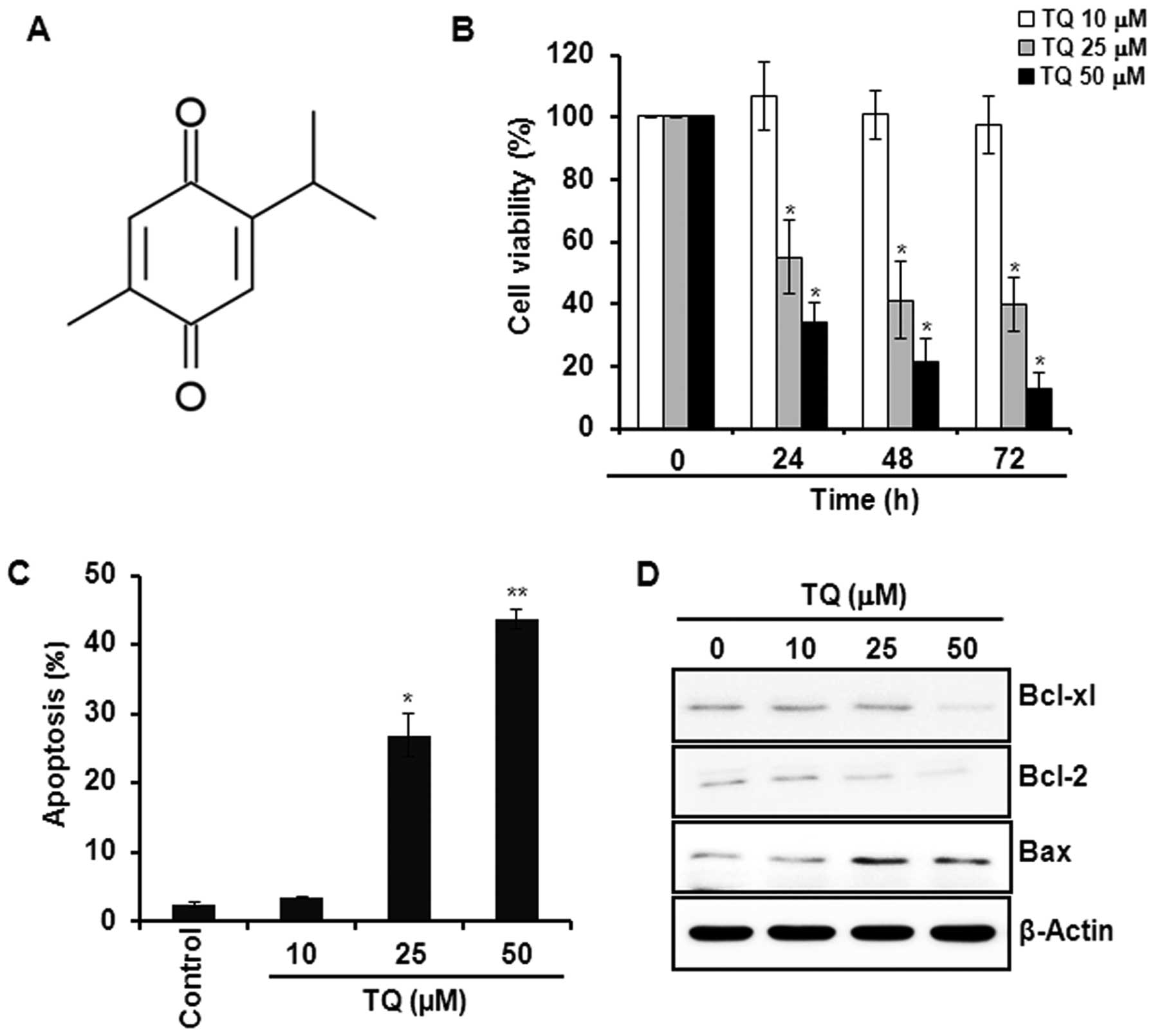
can prevent colon carcinogenesis (4). Thymoquinone (TQ; Fig. 1A), a dietary phytochemical, is the
major bioactive constituent present in black seed oil (Nigella
sativa), which is widely consumed as a condiment and has long
been used in Ayurvedic medicine. TQ has been reported to exert
antioxidative, anti-inflammatory and anticancer effects (5–7).
Several studies have reported that TQ inhibits cell proliferation,
induces apoptosis and impedes the in vivo growth of
xenograft tumors of various human cancer cells including those of
the stomach (8,9), colorectal (10,11),
lungs (12), breast (5) and prostate (13). In a recent study, TQ was shown to
inhibit 1,2-dimethylhydrazine-induced initiation and promotion of
colon carcinogenesis in Wister rats (14). However, the molecular basis of the
anticancer effects of TQ in colon cancer has yet to be fully
elucidated.
Evasion from apoptosis is one of the hallmarks of
cancer (15). Tumor cells bypass
the apoptotic process following two biochemical pathways (16). The intrinsic or
mitochondria-mediated pathway of apoptosis involves the
depolarization of mitochondrial membrane, release of cytochrome
c, sequential activation of caspase-9, -7 and -3, and the
cleavage of poly-(ADP-ribose) polymerase (PARP). The extrinsic
pathway, on the other hand, is mediated through the activation of
cell membrane-bound death receptors, followed by the activation of
pro-caspase-8, which then execute cell death by triggering the
activity of caspase-3 (16,17). According to the intrinsic mechanism
of apoptosis induction, the mitochondrial membrane potential is
largely regulated by relative abundance of different B-cell
lymphoma (Bcl) family proteins. Whereas localization of Bcl-2 and
Bcl-xl proteins stabilizes mitochondrial membrane integrity,
downregulation of Bcl-2 and concomitant overexpression and
subsequent translocation of proapoptotic protein Bax to the
mitochondria lead to mitochondrial membrane depolarization and
release of cytochrome c, which triggers the activation
caspase cascade, thereby inducing cell death (16,17).
Bcl-2 and Bcl-xl are oncoproteins that are
frequently overexpressed in many cancers (16). One of the transcriptional regulators
of Bcl-2 family proteins is signal transducer and activator of
transcription-3 (STAT3), which is a latent transcription factor
that normally resides in the cytoplasm (18). In response to diverse growth
stimulatory signals, STAT3 gets activated through phosphorylation
by upstream kinases, such as Janus-activated kinase-2 (JAK2)
(19) and Src tyrosine kinase
(20) followed by STAT3
dimerization and nuclear localization. The oncogenic signal
transduction pathway mediated through phosphorylation of epidermal
growth factor receptor (EGFR) tyrosine kinases also transmits
activating signals to STAT3 (21,22).
While transient activation of STAT3 is associated with the growth
and development of various organs, constitutive activation of STAT3
has been implicated in carcinogenesis (23). The activation of STAT3 not only
assists tumor cells to evade apoptosis but also promotes cell
proliferation by transactivating the genes encoding various cell
survival proteins, such as cyclins, c-Myc and survivin (24,25).
The blockade of inappropriate activation of STAT3 signaling
inhibits cell proliferation and induces apoptosis. Thus, STAT3
appears as a molecular target of many anticancer agents. Herein, we
report that TQ induces apoptosis in HCT116 cells through the
inhibition of STAT3 signaling pathway by blocking the
phosphorylation of EGFR tyrosine kinase via modulation of JAK2 and
Src kinases.
Materials and methods
Materials
TQ (purity 99%), z-VAD-fmk and β-actin antibody were
purchased from Sigma-Aldrich (St. Louis, MO, USA). AG490 and PP2
were purchased from Cayman Chemical Co. (Ann Arbor, MI, USA).
Gefitinib was purchased from LC Laboratories (Woburn, MA, USA) and
antibodies against cleaved caspase-9, -3, and -7, PARP, Bcl-2,
Bcl-xl, Bax, STAT3, p-STAT3 (Y705), JAK2, p-JAK2, Src, p-Src, EGFR,
p-EGFR (Y1173), cyclin-D1, -D2, and survivin were obtained from
Cell Signaling Technology Inc. (Beverly, MA, USA). Primary
antibodies against each of c-Myc, p27, p21, lamin A and horseradish
peroxidase-conjugated secondary antibodies were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals
were of analytical or highest purity grade available.
Cell culture and treatment
HCT116 cells were obtained from American Type
Culture Collection (ATCC) and maintained in RPMI-1640 medium
supplemented with 10% fetal bovine serum and antibiotics (100 U/ml
penicillin and 100 μg/ml streptomycin) at 37°C in a humidified
incubator containing 5% CO2 and 95% air. In all the
experiments, cells were seeded at 2×105 cells/ml and
treated with TQ at 50–60% confluence. All chemicals were dissolved
in ethanol.
Cell proliferation assay
The anti-proliferative effect of TQ against HCT116
cells was measured by using a solution of tetrazolium compound
[3-(4,5-dimethylthiazol-2-yl)-5-
(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner
salt (MTS) (Promega, Madison, WI, USA). Briefly, cells
(2×103) were incubated in triplicate in a 96-well plate
in the presence or absence of TQ in a final volume of 0.1 ml for
different time intervals at 37°C. Thereafter, 20 μl of MTS solution
was added to each well and incubated for 60 min. The number of
viable cells was measured in a 96-well plate at an optical density
of 492 nm on a microplate reader (Tecan Trading AG, Männedorf,
Switzerland). Cell viability was described as the relative
percentage of control.
Annexin V staining
Annexin V staining was performed using FITC-Annexin
V staining kit (BD Biosciences, San Jose, CA, USA) following the
manufacturer’s instructions. Briefly, TQ-treated cells were washed
with PBS and resuspended in binding buffer containing Annexin V and
propidium iodide (PI). Fluorescence intensity was measured using
flow cytometry (BD Biosciences).
Western blot analysis
Cells were harvested and lysed with RIPA buffer to
obtain whole cell lysate. Collected protein samples were quantified
by using bicinchoninic acid protein assay kit (Pierce
Biotechnology, Rockford, IL, USA). The cytosolic and nuclear
extracts were prepared by using NE-PER Nuclear and Cytoplasmic
Extraction Reagent kit (Thermo Scientific, Rockford, IL, USA)
according to the procedure described previously (6). Nuclear proteins were collected and
stored at −70°C after determination of protein concentration by
using Bradford Reagent (Bio-Rad Laboratories, Hercules, CA, USA).
The protein samples were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and immunoblot analysis
was performed according to the protocol described previously
(6). Immunoblot membranes were
incubated with SuperSignal Pico Chemiluminescent substrate or Dura
Luminol substrate (Thermo Scientific), according to the
manufacturer’s instructions, and visualized with ImageQuant™ LAS
4000 (Fujifilm Life Science, Tokyo, Japan).
Caspase-3 activity assay
The activity of caspase-3 was detected using the
Caspase-3 Colorimetric Activity Assay kit (Millipore, Billerica,
MA, USA). The assay was performed in 96-well plates by incubating
cell lysates (50 μg) in 100 μl reaction buffer containing caspase-3
substrate Ac-DEVD-pNA at 37°C for 2 h and 30 min according to the
manufacturer’s protocol.
STAT3-Luciferase reporter gene assay
Cells were seeded into 12-well plates at a density
of 5×104 cells/well prior to transfection. Cells were
transfected with p-STAT3-TA-luc (Clontech, Palo Alto, CA, USA) or
control vector using Genefectin transfection reagent (Genetrone
Biotech, Seoul, Korea). After 24 h of transfection, cells were
treated with TQ for an additional 24 h and cell lysis was carried
out with 1X reporter lysis buffer. After mixing the cell lysates
with luciferase substrate (Promega), the luciferase activity was
measured by using luminometer (Tecan Trading AG). The
β-galactosidase assay was carried out according to the supplier’s
instructions (Promega Enzyme Assay System) for normalizing the
luciferase activity and the results were expressed as fold
transactivation.
Statistical analysis
When necessary, data are expressed as mean ± SD of
at least three independent experiments, and statistical analysis
for single comparison was performed using the Student’s t-test. A
p-value <0.05 was considered to indicate a statistically
significant difference.
Results
TQ induces apoptosis in HCT116 cells
We initially examined the effect of TQ on the
viability of HCT116 cells. Incubation of cells with TQ (10, 25 or
50 μM) reduced the cell viability in a time- and
concentration-dependent manner (Fig.
1B). Annexin V staining of cells treated with the indicated
concentrations of TQ showed that the compound induced apoptosis in
a concentration-dependent manner (Fig.
1C). Moreover, TQ reduced the expression of anti-apoptotic
proteins Bcl-2 and Bcl-xl, while the compound increased expression
of pro-apoptotic protein Bax in HCT116 cells (Fig. 1D).
TQ-induced apoptosis is mediated through
caspase activation
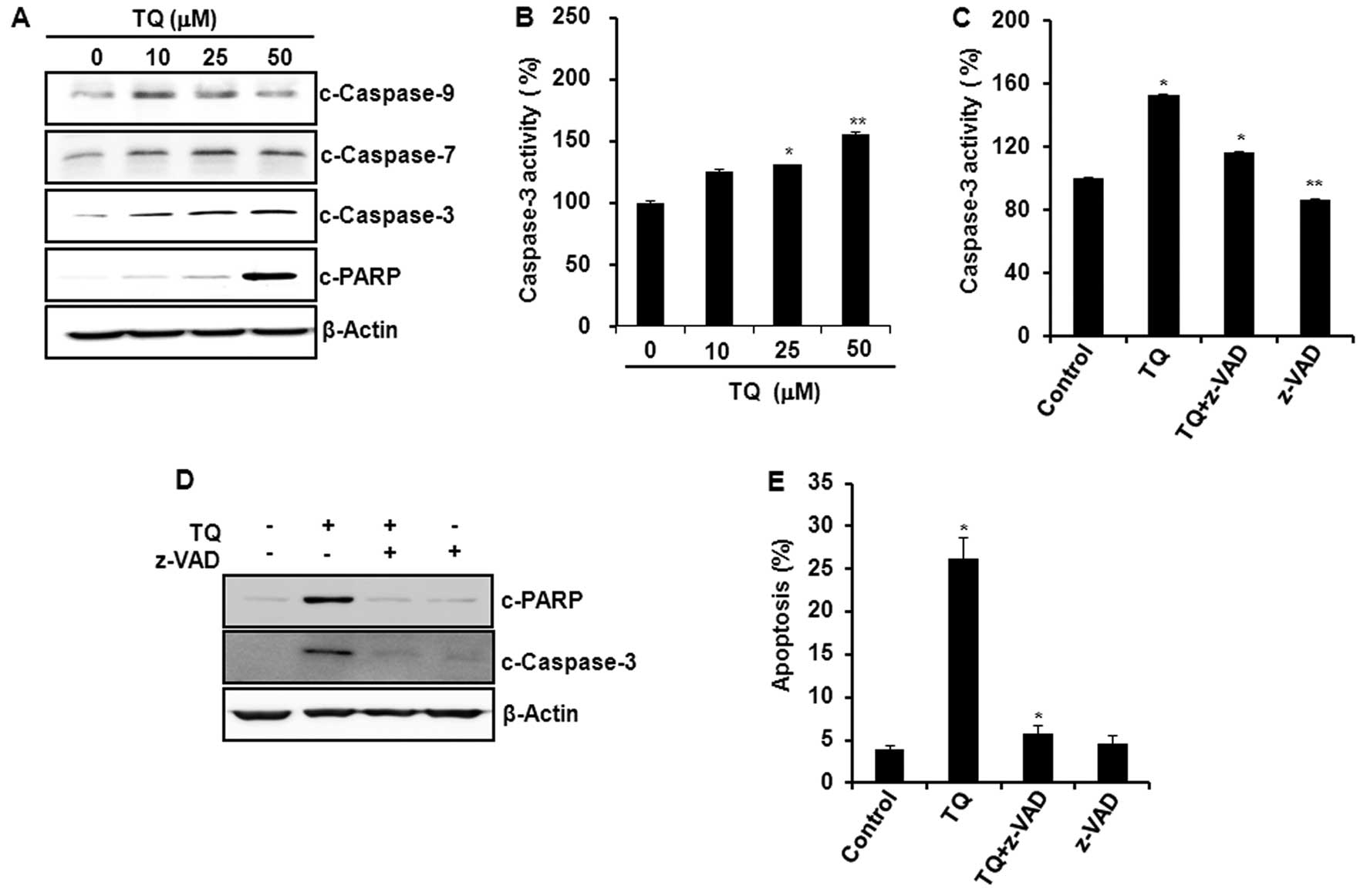
Treatment of HCT116 cells with TQ induced the
cleavage of caspase-9, -7, and -3, and PARP (Fig. 2A) and increased the caspase-3
activity (Fig. 2B). Pretreatment of
cells with a pan-caspase inhibitor z-VAD-fmk abrogated
TQ-induced caspase-3 activity (Fig.
2C), and the cleavage of caspase-3 and PARP (Fig. 2D). Moreover, the blockade of
caspase-3 activation resulted in the abrogation of TQ-induced
apoptosis as assessed by Annexin V staining (Fig. 2E).
TQ attenuates the constitutive activation
of STAT3 and expression of its target gene products in HCT116
cells
Since STAT3 is constitutively active in colon cancer
and plays a key role in cell proliferation through transcriptional
activation of pro-survival genes (23), we examined the effect of TQ on STAT3
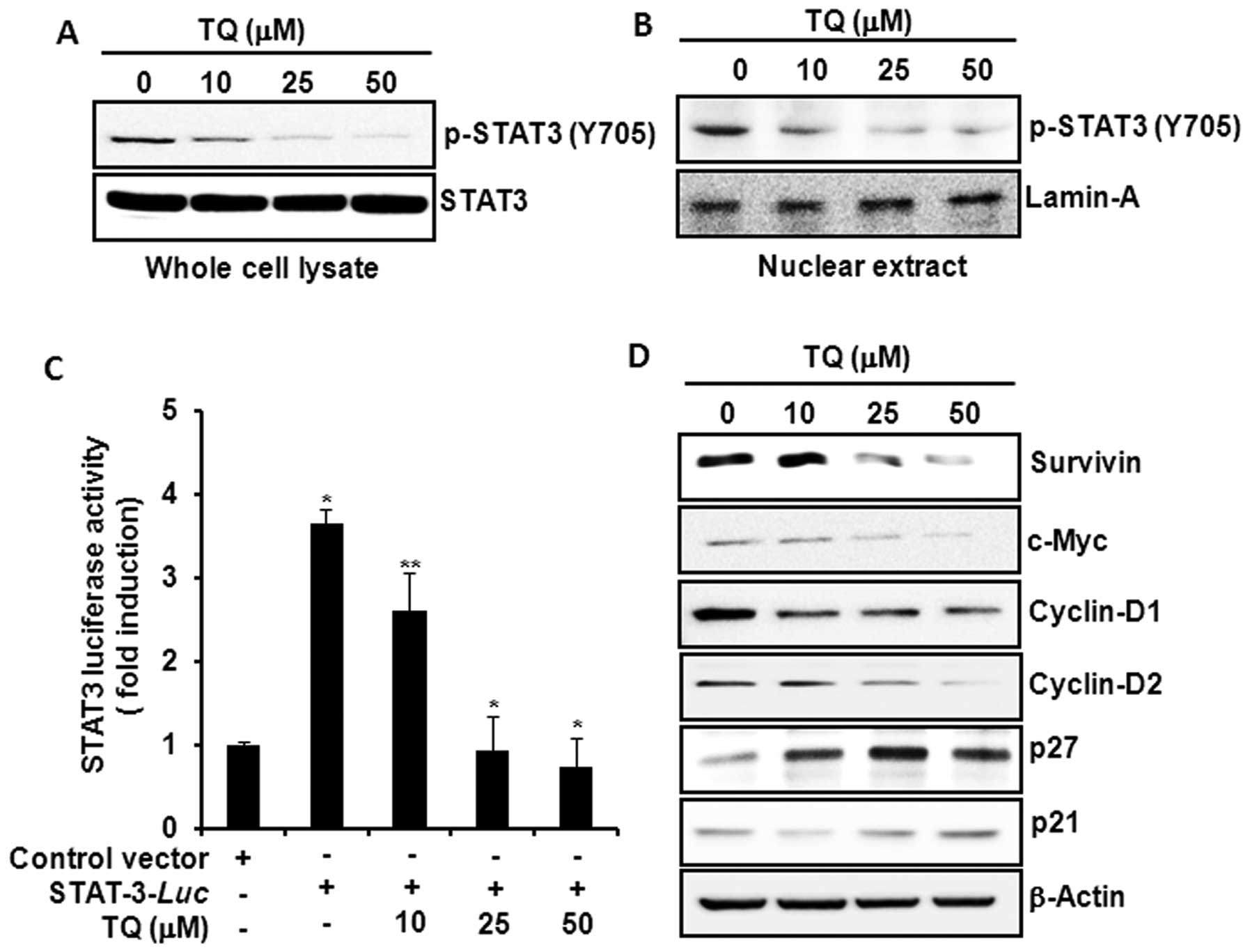
activation in HCT116 cells. Incubation of cells with TQ inhibited
constitutive phosphorylation of STAT3 at tyrosine-705 residue
(Fig. 3A) and decreased the nuclear
localization of p-STAT3 (Fig. 3B).
TQ also reduced the STAT3 reporter gene activity in HCT116 cells
transiently transfected with STAT3-luc vector (Fig. 3C). Moreover, TQ attenuated the
expression of STAT3 target gene products, such as survivin, c-Myc,
cyclin-D1, -D2, and increased the expression levels of the cell
cycle regulatory proteins p27 and p21 (Fig. 3D).
TQ inhibits STAT3 activation by blocking
the phosphorylation of EGFR (Y1173), JAK2 and Src kinases in HCT116
cells
Several upstream kinases, such as EGFR tyrosine
kinase (21,22), JAK2 (19), and Src (20), are known to regulate STAT3
activation. We, therefore, examined the effect of TQ on the
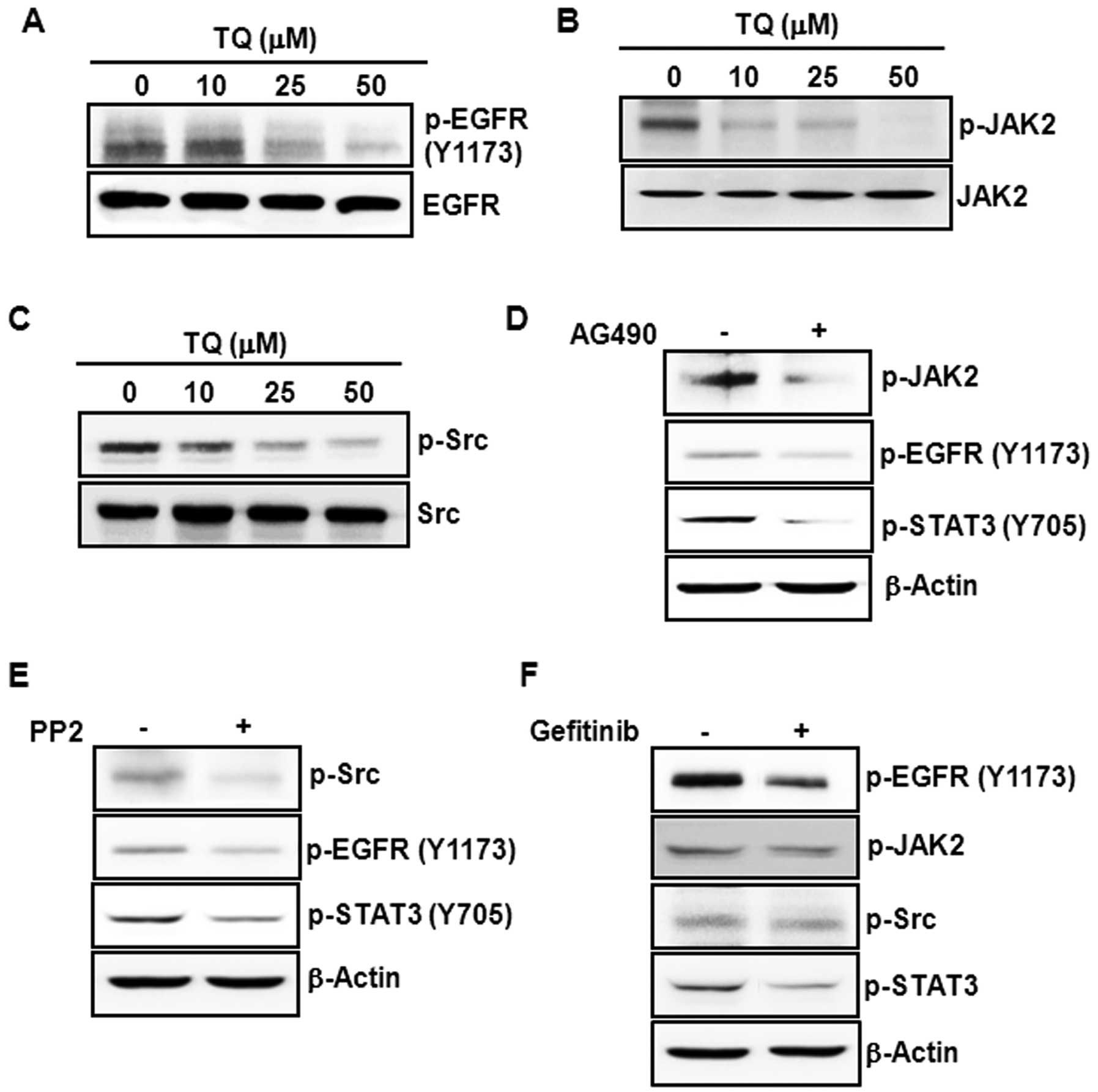
constitutive activation of these kinases in HCT116 cells. Treatment
with TQ markedly diminished the phosphorylation of EGFR at
tyrosine-1173 residue (Fig. 4A),
JAK2 (Fig. 4B) and Src kinase
(Fig. 4C). While JAK2 can directly
phosphorylate STAT3, it has been reported that JAK2 (26) and Src (27) function as upstream kinases to EGFR,
which transmits activating signals to STAT3 (28). Pretreatment of cells with AG490, a
JAK2 inhibitor, attenuated the phosphorylation of EGFR and STAT3
(Fig. 4D). Moreover,
pharmacological inhibition of Src kinase by treatment of cells with
PP2 diminished the tyrosine phosphorylation of EGFR and STAT3 in
HCT116 cells (Fig. 4E). Treatment
of cells with gefitinib, an EGFR antagonist, also negated STAT3
phosphorylation without affecting the levels of phosphorylated JAK2
and Src (Fig. 4F).
Discussion
Despite the marked progress in the development of
anticancer therapeutics, cancer remains as a major public health
concern worldwide. It has been estimated that the incidence of and
mortality from cancer will double in the next 50 years (29). There is now a growing interest in
searching for anticancer agents from natural sources, especially
those present in edible medicinal plants. Thymoquinone (TQ), the
major bioactive constituent of black seed essential oil, has been
reported to possess diverse pharmacological activities (5,30).
However, the molecular mechanisms of the anticancer activity of TQ
have not been fully elucidated. One of the hallmarks of cancer is
the ability of tumor cells to evade the apoptosis process and many
anticancer therapies have been shown to induce apoptosis
selectively in cancer cells.
TQ has been reported to inhibit proliferation and
induce apoptosis in human colon cancer cells without exhibiting
cytotoxicity to normal human intestinal FHs74Int cells (31). According to this study, TQ-induced
cytotoxicity in DLD-1 colon cancer cells was associated with the
generation of reactive oxygen species, activation of extracellular
signal-regulated kinase and c-Jun-N-terminal kinase, and the
cleavage of caspase-7 and -3. Moreover, TQ induced apoptosis and G1
phase cell cycle arrest in HCT116 colon cancer cells by increasing
the expression of a cell cycle inhibitory protein p21 with
concomitant inhibition of anti-apoptotic protein Bcl-2 (10) and the inactivation of stress
response pathway sensor CHEK1 (32)
in a p53-dependent manner. In accordance with these previous
reports, our study revealed that TQ decreased the cell viability
and induced apoptosis in HCT116 cells. Herein, we provide new
mechanistic findings that TQ induced apoptosis in HCT116 cells
through the downregulation of constitutive activation of STAT3
signaling via inhibition of EGFR tyrosine kinase by blocking
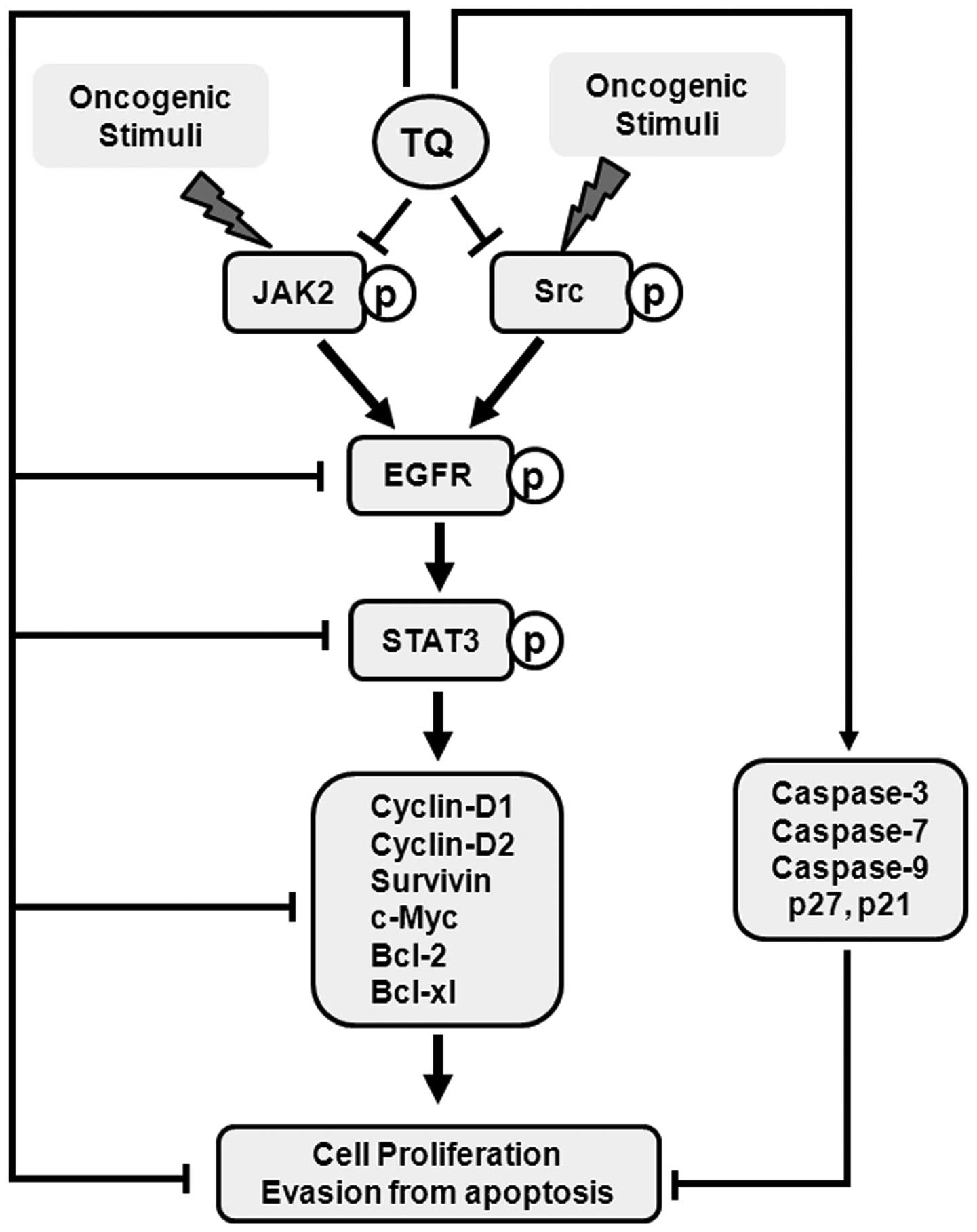
phosphorylation of upstream JAK2 and Src kinases (Fig. 5).
The induction of apoptosis by TQ was associated with
decreased expression of anti-apoptotic proteins Bcl-2 and Bcl-xl,
and the elevated expression of proapoptotic protein Bax. Moreover,
TQ treatment caused the cleavage of caspase-9, -7 and -3, and
induced the caspase-3 activity followed by the cleavage of PARP.
Pretreatment of cells with a pan-caspase inhibitor z-VAD-fmk
abrogated TQ-induced apoptosis by blocking the caspase-3 activity
and the cleavage of caspase-3 and PARP. Since caspase-9 and -7 are
initiator caspases in mitochondria-mediated death signaling
(16), the activation of caspase-3
and the cleavage of PARP suggest that TQ may induce apoptosis
through the activation of the intrinsic pathway. Contrary to our
findings, TQ failed to induce the caspase activity and treatment
with z-VAD-fmk was unable to block TQ-induced apoptosis in human
prostate cancer (PC3) cells (33),
suggesting that the mechanisms of apoptosis induction in cancer
cells by TQ are variable depending on the cell type. A recent study
reported that TQ increased the expression of death receptor-5 (DR5)
in neuroblastoma cells, but the upregulation of DR5 was not
associated with TQ-induced apoptosis (34). Whether TQ-induced apoptosis in
HCT116 cells is associated with the activation of DR-mediated
pathway has yet to be investigated.
It has been reported that Bcl-2 is overexpressed in
colon cancer (35) and the
Bcl-2-mediated inhibition of apoptosis restores the tumorigenicity
of spontaneously regressed colon tumors in vivo (36). The inhibition of Bcl-2 and Bcl-xl
expression and the increase in Bax protein level, thus, provide a
mechanistic basis of apoptosis induction by TQ. The expression of
anti-apoptotic proteins Bcl-2 and Bcl-xl is transcriptionally
regulated by STAT3 (25), which is
overexpressed in colon cancer (24,37).
Persistent STAT3 activation increases the proliferation of colon
cancer cells in culture and enhances the growth of colon cancer
cell xenograft tumors in nude mice (38). Moreover, the inhibition of STAT3
signaling induces apoptosis and cell cycle arrest in colon
carcinoma cells (39).
We, therefore, examined the effect of TQ on STAT3
activation in HCT116 cells and, for the first time, we found that
TQ suppressed the constitutive activation of STAT3 signaling in
human colon cancer HCT116 cells. Our findings that TQ inhibited the
constitutive phosphorylation of STAT3 at tyrosine-705 residue and
decreased the STAT3 reporter gene activity in HCT116 cells are in
good co-relation with previous studies reporting that TQ inhibited
proliferation and induced apoptosis in multiple myeloma cells by
blocking the activation of STAT3 (40,41).
It has been reported that treatment with a STAT3 decoy
oligonucleotide diminished the expression of Bcl-2 and activated
caspase-3, thereby resulting in the induction of apoptosis in
ovarian cancer cell xenograft tumors (42). Thus, TQ-induced apoptosis in HCT116
cells is mediated, at least in part, by the blockade of STAT3
activation. This was further supported by the findings that TQ
inhibited the expression of D-series cyclins, c-Myc and survivin,
which are STAT3 target gene products (25,43).
While survivin inhibits apoptosis in cancer cells
(44), cyclins (45) and c-Myc (46) promote tumor cell proliferation.
Thus, the reduced expression of survivin, c-Myc and D-series
cyclins by TQ is associated with the induction of apoptosis by this
compound. Cyclins form active complexes with cyclin-dependent
kinases, which are inhibited by cell cycle inhibitory proteins p27
and p21 (47). The elevated
expression of p27 and p21 by TQ suggests that the compound can
block cell cycle progression. In fact, TQ has been shown to induce
G1 phase cell cycle arrest through the induction of p53 and p21 in
HCT116 cells (10).
The STAT3 activation mechanism involves the
phosphorylation of its tyrosine-705 and serine-727 residues
followed by the formation of STAT3 dimer, which translocates to the
nucleus and interacts with the promoter region of target genes
(24). Although we have found that
TQ can inhibit STAT3 tyrosine phosphorylation, the possible
inhibitory effect of TQ on STAT3 serine phosphorylation merits
further investigation. Several upstream kinases including EGFR
tyrosine kinase (22), JAK2
(19) and Src (20) have been reported to activate STAT3.
In the present study, we reported the novel finding that TQ
treatment attenuated the constitutive phosphorylation of EGFR at
tyrosine-1173 residue, JAK2 and Src kinases in HCT116 cells. We
confirmed the roles of these kinases in STAT3 activation in HCT116
cells. Treatment of cells with specific pharmacological inhibitors
of JAK2 and Src diminished the phosphorylation of EGFR and STAT3,
while the blockade of EGFR by gefitinib inhibited STAT3
phosphorylation at tyrosine-705 residue without affecting that of
JAK2 and Src. These findings suggest that JAK2 and Src regulate
STAT3 activation via phosphorylation of EGFR in HCT116 cells.
Our findings are in accordance with a previous study
by Nourazarian et al (48),
who demonstrated that combined EGFR and c-Src antisense
deoxyoligonucleotides inhibited the proliferation of human colon
cancer (HT29) cells. It has been reported that the blockade of Src
kinase by PP2 attenuates EGFR phosphorylation at both tyrosine-845
and tyrosine-1173 residues in cervical cancer cells. According to
this study, the phosphorylation of EGFR at tyrosine-1173, but not
tyrosine-845, is involved in the proliferation of cervical cancer
cells (27). The distinct roles of
different EGFR tyrosine phosphorylation in colon cancer cell
proliferation have not yet been clarified and the effect of TQ on
EGFR phosphorylation at other tyrosine residues warrants further
examination. The activation of Src also phosphorylates STAT3 at
tyrosine-705 residue and contributes to colon cancer cell
proliferation (20). Besides Src,
the upstream kinase JAK2 has been shown to transactivate EGFR as
the treatment of cells with JAK2 inhibitor AG490 attenuated EGFR
phosphorylation in human epidermoid carcinoma cells (26). In agreement with this study, we
found that JAK2 inhibition by AG490 diminished EGFR phosphorylation
in HCT116 cells and TQ attenuated the activation of both JAK2 and
EGFR. Since combined inhibition of EGFR and STAT3 have been shown
to be more effective in suppressing growth of various cancer cells
(26,49), the ability of TQ to inhibit both
EGFR and STAT3, hence, appears as a mechanistic basis of its
antitumor effects in colon cancer cells.
Acknowledgements
The present study was funded by the Settlement
Research Grant of Keimyung University in 2011 allocated to
Kyung-Soo Chun.
References
|
1
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
3
|
Merika E, Saif MW, Katz A, Syrigos K and
Morse M: Review. Colon cancer vaccines: an update. In Vivo.
24:607–628. 2010.PubMed/NCBI
|
|
4
|
Chung MY, Lim TG and Lee KW: Molecular
mechanisms of chemopreventive phytochemicals against
gastroenterological cancer development. World J Gastroenterol.
19:984–993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Banerjee S, Padhye S, Azmi A, et al:
Review on molecular and therapeutic potential of thymoquinone in
cancer. Nutr Cancer. 62:938–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kundu J, Kim DH, Kundu JK and Chun KS:
Thymoquinone induces heme oxygenase-1 expression in HaCaT cells via
Nrf2/ARE activation: Akt and AMPKα as upstream targets. Food Chem
Toxicol. 65:18–26. 2014.PubMed/NCBI
|
|
7
|
Kundu JK, Liu L, Shin JW and Surh YJ:
Thymoquinone inhibits phorbol ester-induced activation of NF-κB and
expression of COX-2, and induces expression of cytoprotective
enzymes in mouse skin in vivo. Biochem Biophys Res Commun.
438:721–727. 2013.PubMed/NCBI
|
|
8
|
Lei X, Lv X, Liu M, et al: Thymoquinone
inhibits growth and augments 5-fluorouracil-induced apoptosis in
gastric cancer cells both in vitro and in vivo. Biochem Biophys Res
Commun. 417:864–868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yi T, Cho SG, Yi Z, et al: Thymoquinone
inhibits tumor angiogenesis and tumor growth through suppressing
AKT and extracellular signal-regulated kinase signaling pathways.
Mol Cancer Ther. 7:1789–1796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gali-Muhtasib H, Diab-Assaf M, Boltze C,
et al: Thymoquinone extracted from black seed triggers apoptotic
cell death in human colorectal cancer cells via a p53-dependent
mechanism. Int J Oncol. 25:857–866. 2004.PubMed/NCBI
|
|
11
|
Gali-Muhtasib H, Ocker M, Kuester D, et
al: Thymoquinone reduces mouse colon tumor cell invasion and
inhibits tumor growth in murine colon cancer models. J Cell Mol
Med. 12:330–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jafri SH, Glass J, Shi R, Zhang S, Prince
M and Kleiner-Hancock H: Thymoquinone and cisplatin as a
therapeutic combination in lung cancer: In vitro and in vivo. J Exp
Clin Cancer Res. 29:872010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaseb AO, Chinnakannu K, Chen D, et al:
Androgen receptor and E2F-1 targeted thymoquinone therapy for
hormone-refractory prostate cancer. Cancer Res. 67:7782–7788. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jrah-Harzallah H, Ben-Hadj-Khalifa S,
Almawi WY, Maaloul A, Houas Z and Mahjoub T: Effect of thymoquinone
on 1,2-dimethyl-hydrazine-induced oxidative stress during
initiation and promotion of colon carcinogenesis. Eur J Cancer.
49:1127–1135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Burz C, Berindan-Neagoe I, Balacescu O and
Irimie A: Apoptosis in cancer: key molecular signaling pathways and
therapy targets. Acta Oncol. 48:811–821. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martin SJ and Green DR: Protease
activation during apoptosis: death by a thousand cuts? Cell.
82:349–352. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ihle JN: The Stat family in cytokine
signaling. Curr Opin Cell Biol. 13:211–217. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Slattery ML, Lundgreen A, Kadlubar SA,
Bondurant KL and Wolff RK: JAK/STAT/SOCS-signaling pathway and
colon and rectal cancer. Mol Carcinog. 52:155–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Laird AD, Li G, Moss KG, et al: Src family
kinase activity is required for signal tranducer and activator of
transcription 3 and focal adhesion kinase phosphorylation and
vascular endothelial growth factor signaling in vivo and for
anchorage-dependent and -independent growth of human tumor cells.
Mol Cancer Ther. 2:461–469. 2003.
|
|
21
|
Berclaz G, Altermatt HJ, Rohrbach V,
Siragusa A, Dreher E and Smith PD: EGFR dependent expression of
STAT3 (but not STAT1) in breast cancer. Int J Oncol. 19:1155–1160.
2001.PubMed/NCBI
|
|
22
|
Chan KS, Carbajal S, Kiguchi K, Clifford
J, Sano S and DiGiovanni J: Epidermal growth factor
receptor-mediated activation of Stat3 during multistage skin
carcinogenesis. Cancer Res. 64:2382–2389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, et al: Signal transducer and activator of transcription-3,
inflammation, and cancer: how intimate is the relationship? Ann NY
Acad Sci. 1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnston PA and Grandis JR: STAT3
signaling: anticancer strategies and challenges. Mol Interv.
11:18–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Masuda M, Suzui M, Yasumatu R, et al:
Constitutive activation of signal transducers and activators of
transcription 3 correlates with cyclin D1 overexpression and may
provide a novel prognostic marker in head and neck squamous cell
carcinoma. Cancer Res. 62:3351–3355. 2002.PubMed/NCBI
|
|
26
|
Dowlati A, Nethery D and Kern JA: Combined
inhibition of epidermal growth factor receptor and JAK/STAT
pathways results in greater growth inhibition in vitro than single
agent therapy. Mol Cancer Ther. 3:459–463. 2004.PubMed/NCBI
|
|
27
|
Kong L, Deng Z, Shen H and Zhang Y: Src
family kinase inhibitor PP2 efficiently inhibits cervical cancer
cell proliferation through down-regulating phospho-Src-Y416 and
phospho-EGFR-Y1173. Mol Cell Biochem. 348:11–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kopetz S: Targeting SRC and epidermal
growth factor receptor in colorectal cancer: rationale and progress
into the clinic. Gastrointest Cancer Res. 1:S37–S41.
2007.PubMed/NCBI
|
|
29
|
Mann JR, Backlund MG and DuBois RN:
Mechanisms of disease: Inflammatory mediators and cancer
prevention. Nat Clin Pract Oncol. 2:202–210. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Woo CC, Kumar AP, Sethi G and Tan KH:
Thymoquinone: potential cure for inflammatory disorders and cancer.
Biochem Pharmacol. 83:443–451. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
El-Najjar N, Chatila M, Moukadem H, et al:
Reactive oxygen species mediate thymoquinone-induced apoptosis and
activate ERK and JNK signaling. Apoptosis. 15:183–195. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gali-Muhtasib H, Kuester D, Mawrin C, et
al: Thymoquinone triggers inactivation of the stress response
pathway sensor CHEK1 and contributes to apoptosis in colorectal
cancer cells. Cancer Res. 68:5609–5618. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koka PS, Mondal D, Schultz M, Abdel-Mageed
AB and Agrawal KC: Studies on molecular mechanisms of growth
inhibitory effects of thymoquinone against prostate cancer cells:
role of reactive oxygen species. Exp Biol Med. 235:751–760. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hussain AR, Uddin S, Ahmed M, Al-Dayel F,
Bavi PP and Al-Kuraya KS: Phosphorylated IκBα predicts poor
prognosis in activated B-cell lymphoma and its inhibition with
thymoquinone induces apoptosis via ROS release. PLoS One.
8:e605402013.
|
|
35
|
Valassiadou KE, Stefanaki K, Tzardi M, et
al: Immunohistochemical expression of p53, bcl-2, mdm2 and waf1/p21
proteins in colorectal adenocarcinomas. Anticancer Res.
17:2571–2576. 1997.PubMed/NCBI
|
|
36
|
Bonnotte B, Favre N, Moutet M, et al:
Bcl-2-mediated inhibition of apoptosis prevents immunogenicity and
restores tumorigenicity of spontaneously regressive tumors. J
Immunol. 161:1433–1438. 1998.PubMed/NCBI
|
|
37
|
Dobi E, Monnien F, Kim S, et al: Impact of
STAT3 phosphorylation on the clinical effectiveness of
anti-EGFR-based therapy in patients with metastatic colorectal
cancer. Clin Colorectal Cancer. 12:28–36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Corvinus FM, Orth C, Moriggl R, et al:
Persistent STAT3 activation in colon cancer is associated with
enhanced cell proliferation and tumor growth. Neoplasia. 7:545–555.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin Q, Lai R, Chirieac LR, et al:
Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and
cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis
and cell cycle arrest of colon carcinoma cells. Am J Pathol.
167:969–980. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Badr G, Mohany M and Abu-Tarboush F:
Thymoquinone decreases F-actin polymerization and the proliferation
of human multiple myeloma cells by suppressing STAT3
phosphorylation and Bcl2/Bcl-XL expression. Lipids Health Dis.
10:236–243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li F, Rajendran P and Sethi G:
Thymoquinone inhibits proliferation, induces apoptosis and
chemosensitizes human multiple myeloma cells through suppression of
signal transducer and activator of transcription 3 activation
pathway. Br J Pharmacol. 161:541–554. 2010. View Article : Google Scholar
|
|
42
|
Zhang X, Liu P, Zhang B, Mao H, Shen L and
Ma Y: Inhibitory effects of STAT3 decoy oligodeoxynucleotides on
human epithelial ovarian cancer cell growth in vivo. Int J
Mol Med. 32:623–628. 2013.PubMed/NCBI
|
|
43
|
Kanda N, Seno H, Konda Y, et al: STAT3 is
constitutively activated and supports cell survival in association
with survivin expression in gastric cancer cells. Oncogene.
23:4921–4929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Asanuma K, Tsuji N, Endoh T, Yagihashi A
and Watanabe N: Survivin enhances Fas ligand expression via
up-regulation of specificity protein 1-mediated gene transcription
in colon cancer cells. J Immunol. 172:3922–3929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lin L, Liu A, Peng Z, et al: STAT3 is
necessary for proliferation and survival in colon cancer-initiating
cells. Cancer Res. 71:7226–7237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Forgue-Lafitte ME, Coudray AM, Breant B
and Mester J: Proliferation of the human colon carcinoma cell line
HT29: autocrine growth and deregulated expression of the c-myc
oncogene. Cancer Res. 49:6566–6571. 1989.PubMed/NCBI
|
|
47
|
Abukhdeir AM and Park BH: P21 and p27:
roles in carcinogenesis and drug resistance. Expert Rev Mol Med.
10:e192008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nourazarian AR, Najar AG, Farajnia S,
Khosroushahi AY, Pashaei-Asl R and Omidi Y: Combined EGFR and c-Src
antisense oligodeoxynucleotides encapsulated with PAMAM Denderimers
inhibit HT-29 colon cancer cell proliferation. Asian Pac J Cancer
Prev. 13:4751–4756. 2012. View Article : Google Scholar
|
|
49
|
Boehm AL, Sen M, Seethala R, et al:
Combined targeting of epidermal growth factor receptor, signal
transducer and activator of transcription-3, and Bcl-X(L) enhances
antitumor effects in squamous cell carcinoma of the head and neck.
Mol Pharmacol. 73:1632–1642. 2008. View Article : Google Scholar : PubMed/NCBI
|