Introduction
Colorectal cancer (CRC) is the fourth most common
cause of cancer-related mortality (1). The main treatments for patients with
advanced metastatic colorectal cancer (mCRC) include systemic
combination chemotherapies (2–7).
Although the standard therapies are initially effective, many
patients relapse due to the onset of drug resistance and are
subsequently placed on salvage chemotherapy.
TAS-102 is a novel oral nucleoside antitumor agent
that consists of trifluridine (FTD) and tipiracil hydrochloride
(TPI) at a molar ratio of 1:0.5. FTD is the antitumor component of
TAS-102, whereas TPI prevents degradation of FTD through a
first-pass effect as a thymidine phosphorylase inhibitor. FTD is a
well-known antiproliferative agent with two mechanisms of action;
it inhibits thymidylate synthase (TS) and is also incorporated into
DNA (8,9). Recently, TAS-102 was found to
significantly improve overall survival of mCRC patients in whom
systemic chemotherapy is either ineffective or not tolerated
(10).
Since the serum half-life of FTD is relatively short
after intravenous (i.v.) injection, split dose regimens, as well as
single rapid bolus injections, have been used in the clinic
(11). However, i.v. injection of
FTD has been discontinued due to disease recurrence and prolonged
toxicity (12), and the drug is now
currently used as an ophthalmic solution for treatment of
epithelial keratitis caused by the herpes simplex virus (13).
FTD instability in serum is a result of degradation
by thymidine phosphorylase (TP), and therefore co-administration of
TPI with FTD is employed to increase the effective in vivo
FTD concentration (14). We
previously developed a TAS-102 anticancer treatment regimen in
which FTD was orally administered and combined with TPI; this
approach significantly prolonged survival.
These clinical results suggest that the mechanism of
action of orally administered FTD and TPI co-treatment may differ
from that of i.v. administration of FTD alone. We hypothesized that
the balance between TS inhibition and DNA incorporation may be
altered according to the FTD administration mode. Therefore, we
examined both delivery routes in comparative studies to determine
the mechanism by which TAS-102 exerts its antitumor activity.
Materials and methods
Chemicals and reagents
FTD was obtained from Yuki Gosei Kogyo Co., Ltd.
(Tokyo, Japan). 5-fluoro-2′-deoxyuridine (FdUrd) was obtained from
Wako Pure Chemical Industries (Osaka, Japan). TPI was synthesized
at Junsei Chemical (Tokyo, Japan). Tegafur (FT), gimeracil (CDHP)
and oteracil potassium (Oxo) were synthesized in-house at Good
Manufacturing Practice grade. Cytarabine (Ara-C) was obtained from
Sigma-Aldrich K.K. (Tokyo, Japan). Gemcitabine (dFdC) was
synthesized in-house. [6-3H] FTD (10.0 Ci/mmol),
[6-3H] FdUrd (13.5 Ci/mmol), [5−3H] Ara-C
(26.7 Ci/mmol) and [5-3H] dFdC (26.0 Ci/mmol) were
obtained from Moravek (Brea, CA, USA).
Animals
Five-week-old male nude mice (BALB/cA
Jcl-nu/nu) were purchased from CLEA Japan Inc. (Tokyo,
Japan) and maintained in accordance with the animal experimental
regulations of Taiho Pharmaceutical Co., Ltd. Mice were allowed
free access to a commercial diet and water (filtered and
chlorinated) and exposed to a 12-h light/12-h dark cycle in a
barrier facility.
Cell culture and human cancer xenografts
in nude mice
Eight human cancer xenografts were used (CRC cell
lines DLD-1, HT-29, HCT116, KM20C and SW480; small cell lung cancer
cells Lu-134; breast cancer cells MC-2 and MX-1). Lu-134, MC-2 and
MX-1 were maintained by serial transplantation in the dorsum of
nude mice. Other xenografts were established by implantation of
cultured cells before each study. Lu-134 and MC-2 were obtained
from the Central Institute for Experimental Animals (Tokyo, Japan).
NUGC-3, HT-29 and MX-1 cells were provided by the Japanese
Foundation for Cancer Research (Tokyo, Japan). DLD-1 and HCT116
cells were obtained from Dainippon Pharmaceutical (Tokyo, Japan).
KM20C cells were kindly provided by the National Cancer Center
(Tokyo, Japan). SW480 cells were purchased from American Type
Culture Collection (Manassas, VA, USA). HeLa cells were obtained
from the Health Science Research Resources Bank (Tokyo, Japan).
These cultured cells were maintained at 37°C with 5%
CO2, in the culture medium recommended by each
provider.
Evaluation of growth inhibitory effect in
vitro
HeLa cells were seeded in 96-well plates at 500
cells/180 μl/well in triplicate, pre-cultured for 24 h, and then 20
μl of each drug solution was added for 24 or 72 h. For the 24-h
treatment, cells were washed with phosphate-buffered saline (PBS)
after treatment, drug-free medium was added to each well, and the
culture was further incubated for 48 h. Cell growth inhibition was
evaluated using a Cell Counting Kit-8 (Dojindo Laboratories)
according to the manufacturer’s instructions. The 50% inhibitory
concentration (IC50) values were calculated from the
absorbance data using SAS (SAS Institute, Cary, NC, USA).
Measurement of dTTP in vitro
Cells were seeded in 175-cm2 culture
flasks at 1×106 cells and pre-cultured for 24 h, prior
to treatment with test compounds. After each treatment, cells were
collected and resuspended in 100 μl PBS, and 50 μl of 1.26 N
perchloric acid (PCA) was added to the cell suspension. The mixture
was left on ice for 10 min and centrifuged for 5 min at 15,000 rpm.
Three hundred microliters of 0.5 N tri-n-octylamine dissolved in
CH2Cl2 was added to the supernatant and
centrifuged for 5 min at 15,000 rpm. The supernatant was applied to
an HPLC system (Prominence, Shimadzu Corporation, Kyoto, Japan)
equipped with an ODS reverse phase column (250×4.6 mm, 5 μm;
Phenomenex), under an analytical method utilizing eluent A [10 mM
KH2PO4, 10 mM tetrabutylammonium hydroxide
(TBA-OH), 0.25% methanol, pH 8.0], and eluent B (50 mM
KH2PO4, 5.6 mM TBA-OH, 50% methanol, pH 6.5)
at a solvent flow rate of 0.80 ml/min in a 120-min gradient elution
as follows: B 60% from 0 to 10 min, B 60–70% from 10 to 20 min, B
70% from 20 to 45 min, B 70–80% from 45 to 55 min, B 80% from 55 to
80 min, B 80–100% from 80 to 90 min, B 100% from 90 to 100 min, B
100-60% from 100 to 101 min, and B 60% from 101 to 120 min.
Evaluation of the nucleoside analogue
levels incorporated into DNA in vitro
After each drug treatment, 1×106 cells
were harvested and DNA was extracted with a DNeasy Tissue kit
(Qiagen GmbH, Hilden, Germany). The DNA solution was dissolved in
10 ml of liquid scintillation fluid (ACS-II; GE Healthcare) to
measure the radioactivity, and the quantity of each tritium-labeled
form in the DNA was calculated. The DNA concentration was
calculated based on the absorbance at 260 nm.
In vivo evaluation of antitumor activity
in mice xenograft models
For experiments, ~8 mm3 cubic fragments
of tumor were implanted subcutaneously into the axilla of mice. For
preparation of the orally administered FTD solution, FTD was
dissolved in a 0.5% aqueous solution of hydroxypropyl
methyl-cellulose (HPMC; Shin-Etsu Chemical Co., Ltd., Tokyo,
Japan). The FTD solution for continuous infusion was prepared by
dissolving FTD in physiological saline. The TAS-102 dosing solution
was composed of FTD and TPI dissolved in a 0.5% aqueous solution of
HPMC at a molar ratio of 1:0.5. The dose of TAS-102 was expressed
on the basis of the amount of FTD. For preparation of the S-1
solution, FT, CDHP and Oxo were dissolved in a 0.5% aqueous
solution of HPMC at a molar ratio of 1:0.4:1. The dose of S-1 was
expressed on the basis of the amount of FT. When the tumor volume
reached ~100 mm3, the mice were randomly assigned into
different treatment groups. Nude mice in the treatment group
received the test compounds. The control group received no
treatment. For continuous infusion, the compound was administered
with an osmotic pump (ALZET osmotic pump model 1002; DURECT
Corporation, Cupertino, CA, USA). Tumor volume was measured twice a
week throughout the experiments. Tumor volume and relative tumor
volume were calculated according to the following formula: Tumor
volume = (width)2 × (length)/2; and relative tumor
volume = (mean tumor volume during treatment)/(mean tumor volume at
the start of treatment). The following formula was used to
calculate the antitumor effect: Tumor growth inhibition rate (%) =
1 - (mean relative tumor volume of the treatment groups/the mean
relative tumor volume of the control group). Body weight changes
(BWCs) were used as a proxy measure of side-effects, and calculated
according to the following formula: BWC (%) = [(BW on evaluation
date) - (BW on grouping date)]/(BW on day grouping date) × 100. The
tumor growth inhibition rate (IR) % on day 15 was used to quantify
the antitumor effect. In parallel, some tumors were isolated,
immersed in liquid nitrogen, and stored at −80°C for additional
experiments described below.
Analysis of FTD incorporation into the
DNA of tumor tissues in mouse xenografts
DNA was extracted from tumors using the DNA
Isolation kit for Cells and Tissues (Roche Diagnostics GmbH,
Penzberg, Germany). The extracted DNA was diluted to 500 μg/ml with
ddH2O and degraded to nucleosides in a 200-μl reaction
mixture consisting of 100 mM Tris-HCl (pH 7.0), 50 mM NaCl, 2.5 mM
CaCl2, 10 mM MgCl2, 1 U DNaseI (Takara Bio
Inc., Tokyo, Japan), 40 μg phosphodiesterase I (Sigma-Aldrich, St.
Louis, MO, USA), 2 U alkaline phosphatase (Nippon Gene Co., Ltd.,
Tokyo, Japan) and 10 μg extracted DNA at 37°C for 2 h. After the
reaction, 20 μl of 4.2 N PCA was added to each reaction mixture and
centrifuged at 15,000 rpm for 3 min. Sixty microliters of 1 M
K2HPO4 was added to each sample for
neutralization and centrifugation was performed to remove debris.
The supernatant was analyzed by HPLC system with ODS reverse phase
column (250×4.6 mm, 5 μm; Phenomenex, Shimadzu GLC Ltd., Tokyo,
Japan), under the analytical method with eluent A [10 mM sodium
phosphate (pH 6.8), 5% CH3CN], eluent B [10 mM sodium
phosphate (pH 6.8), 60% CH3CN], at a solvent flow rate
of 1.0 ml/min in a gradient elution as follows: B 0% from 0 to 5
min, B 0–20% from 5 to 10 min, B 20% from 10 to 15 min, B 20–70%
from 15 to 20 min, B 70% from 20 to 25 min, B 70-0% from 25 to 25.1
min, and B 70-0% from 25.1 to 35 min.
Measurement of deoxyuridine monophosphate
(dUMP) in tumor tissues of mouse xenografts
Collected tumors were homogenized with 0.48 N PCA
and centrifuged for 5 min at 2,000 rpm. Supernatants were collected
and centrifuged for 10 min at 15,000 rpm. Two volumes of 0.5 N
tri-n-octylamine in CH2Cl2 were added and
again centrifuged for 3 min at 15,000 rpm; the supernatant was then
analyzed using HPLC system with ODS reverse phase column (250×4.6
mm, 5 μm; Phenomenex), under the analytical method with eluent A
(10 mM KH2PO4, 10 mM TBA-OH, 0.25% methanol:
50 mM KH2PO4, 10 mM TBA-OH, 30% methanol =
6:4, pH 6.7), and eluent B (50 mM KH2PO4, 10
mM TBA-OH, 30% methanol, pH 6.7) at a solvent flow rate 0.80 ml/min
in a 175-min gradient elution as follows: B 1–23% from 0 to 20 min,
B 23–45% from 20 to 130 min, B 45–99% from 130 to 150 min, B 99-99%
from 150 to 160 min, B 99-1% from 160 to 160.1 min, and B 1-1% from
160.1 to 175 min.
In vivo survival following introduction
of xenografts
KM20C cell suspensions were prepared from in
vitro cell culture and injected into the peritoneal cavity at a
volume of 1×107 cells on day 0. Drug treatment was
started the next day (day 1). TAS-102 (150 mg/kg/day) was orally
administered twice daily for 28 days, and S-1 (8.3 mg/kg/day) was
also orally administered once daily for 28 days. The median
increase of life span (ILS) was calculated as a survival index
according to the following formula: ILS (%) = [(median survival
time of treated group)/(median survival time of control group) - 1]
× 100. The difference in the survival period distribution among
groups was analyzed using the log-rank test.
Ki-67 staining and morphological
analysis
All reagents and antibodies were obtained from Dako,
Tokyo, Japan. Heat-induced antigen retrieval was performed in
Target Retrieval Solution with an autoclave at 120°C for 15 min.
Endogenous peroxidase activity was inhibited with
Peroxidase-Blocking Solution at room temperature (RT) for 5 min.
The sections were then incubated at RT with the anti-Ki-67 mouse
mono-clonal antibody (clone MIB-1; 1:150 dilution). The sections
were washed in wash buffer and incubated with EnVision at RT for 30
min. The reaction products were visualized with a liquid
diaminobenzidine substrate. The nuclei were lightly counterstained
with hematoxylin.
Statistical analysis
All statistical analyses were performed using SAS
(SAS Institute, Cary, NC, USA). A P-value <0.05 was considered
to indicate a statistically significant result.
Results
Comparison of FTD and FdUrd effects on
cell growth and TS activity
We first compared the degree of growth inhibition
and TS inhibition elicited by FTD and FdUrd following 24 and 72 h
drug exposures. Table I shows the
IC50 values derived from cell growth assays under these
conditions. The cytotoxicity of FdUrd was 40.7-fold lower after the
24-h treatment when compared with 72-h treatment. Notably, the
magnitude of this difference with respect to duration of drug
exposure was much lower (2.7-fold) in the case of FTD
treatment.
 | Table IGrowth inhibitory effect of the FTD
and FdUrd on HeLa cells following continuous or intermittent
treatment. |
Table I
Growth inhibitory effect of the FTD
and FdUrd on HeLa cells following continuous or intermittent
treatment.
| Compounds | Treatment duration
(h) |
IC50a (μM) | Ratiob |
|---|
| FTD | 24 | 13.4 | 2.7 |
| 72 | 4.9 | |
| FdUrd | 24 | 37.1 | 41.2 |
| 72 | 0.9 | |
Both FTD and FdUrd can inhibit TS (15). We therefore compared the amount of
dTTP accumulation in HeLa cells after treatment with cytotoxic
concentrations of FTD and FdUrd as an index of the TS inhibitory
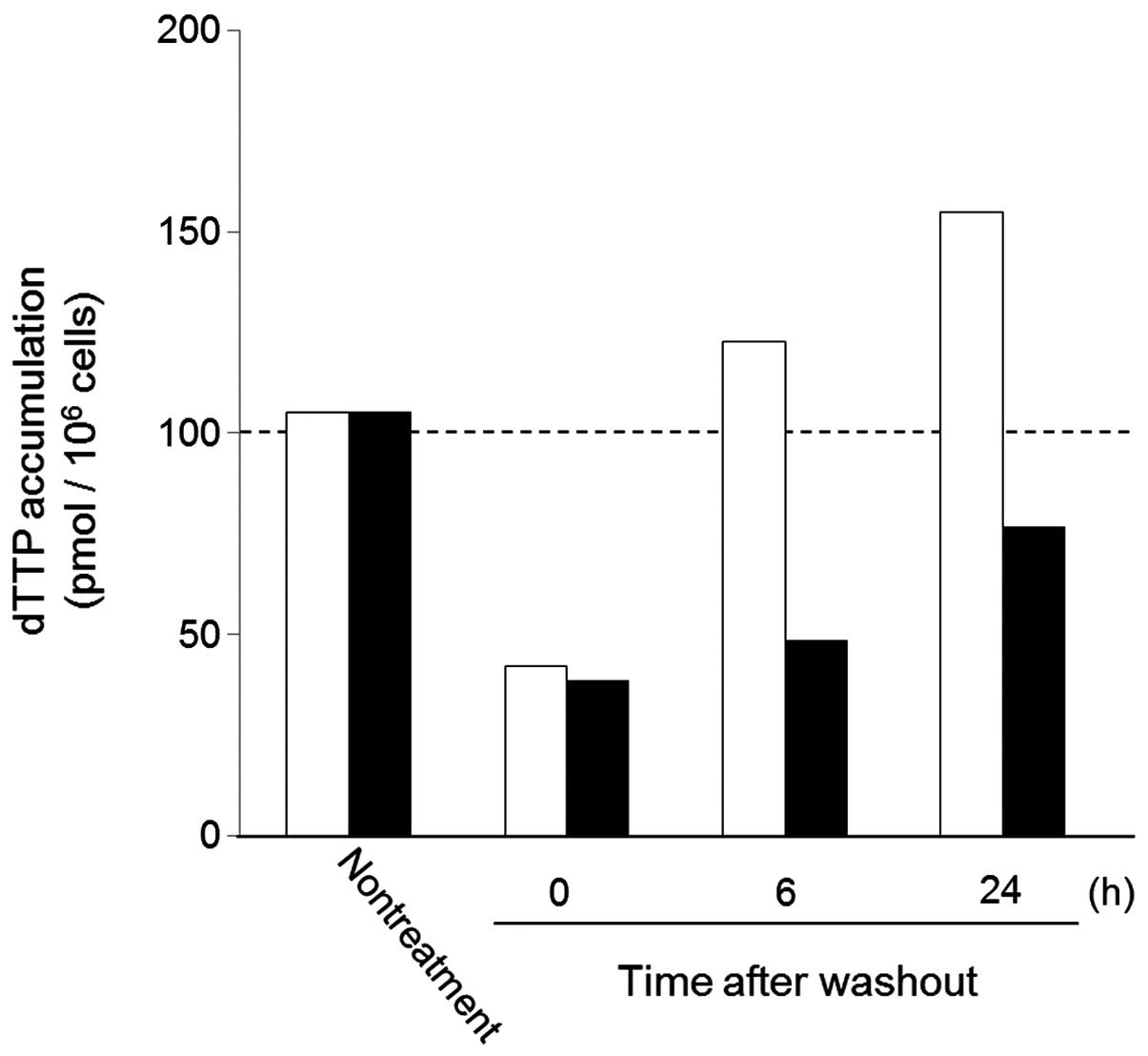
effect (16) (Fig. 1).
The dTTP levels in HeLa cells were markedly
decreased after a 24-h treatment with either FTD or FdUrd. The dTTP
pools did not reach pre-treatment levels following washout of
FdUrd. In contrast, the dTTP pool in HeLa cells was restored to
untreated levels between 6 and 24 h following washout of FTD. These
results indicate that FTD-dependent inhibition of TS is reversible,
and we therefore infer that inhibition of TS is not the major
mechanism by which FTD exerts its cytotoxic effect.
Comparison of incorporation of FTD and
other nucleoside analogs into DNA
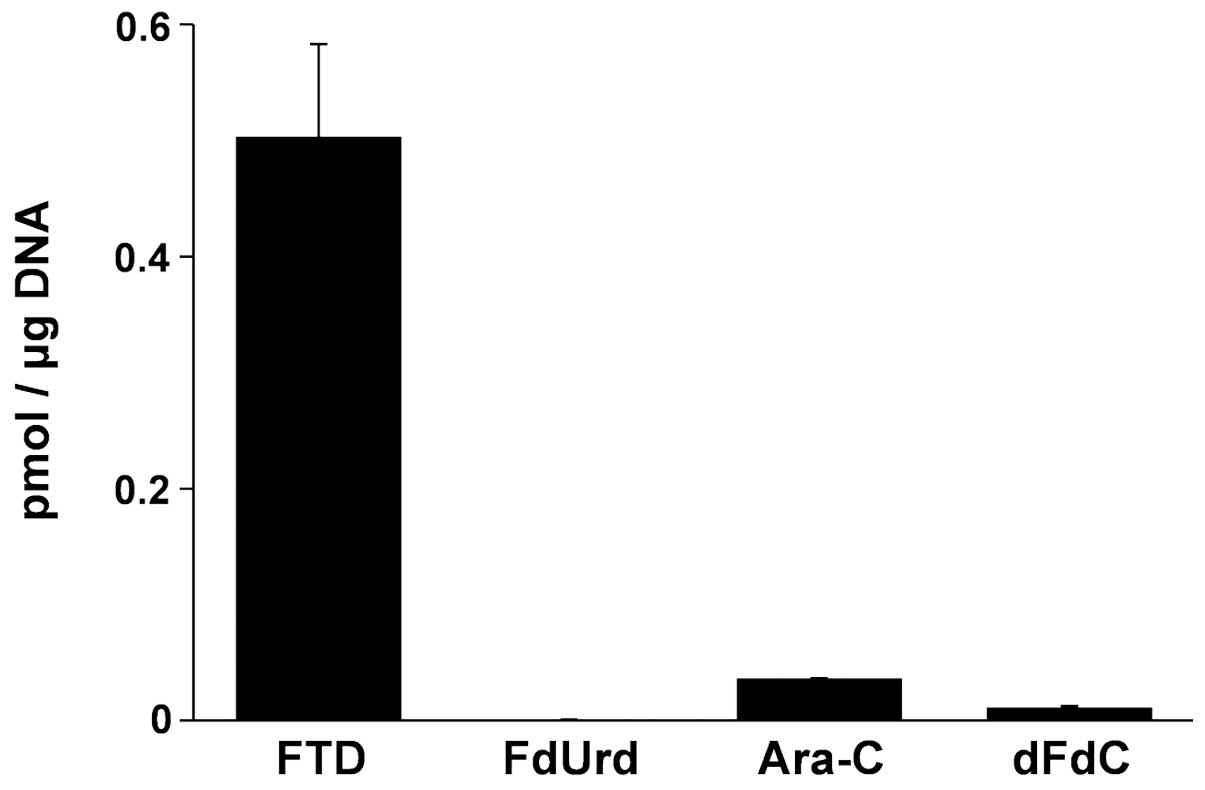
We next compared the DNA incorporation level of FTD
to that of other nucleoside analogs, namely, FdUrd, Ara-C and dFdC,
as they are known to be incorporated into DNA (17). Incorporation of tritium-labeled
nucleosides at concentrations that induced the same level of
cytotoxicity was allowed to proceed for 4 h (Fig. 2). The amount of FTD incorporation
(0.50±0.05 pmol/μg DNA) was significantly higher than that of Ara-C
0.04±0.0001 pmol/μg DNA, dFdC 0.01±0.01 pmol/μg DNA and FdUrd
0.001±0.00004 pmol/μg DNA.
Comparison of the in vivo antitumor
activity, DNA incorporation level and dUMP accumulation following
delivery of FTD via oral or continuous infusion routes
To translate our in vitro findings to a more
relevant model, FTD was administered by oral and continuous
infusion routes at either the maximal tolerated dose (MTD) or 2/3X
MTD for 14 days of treatment.
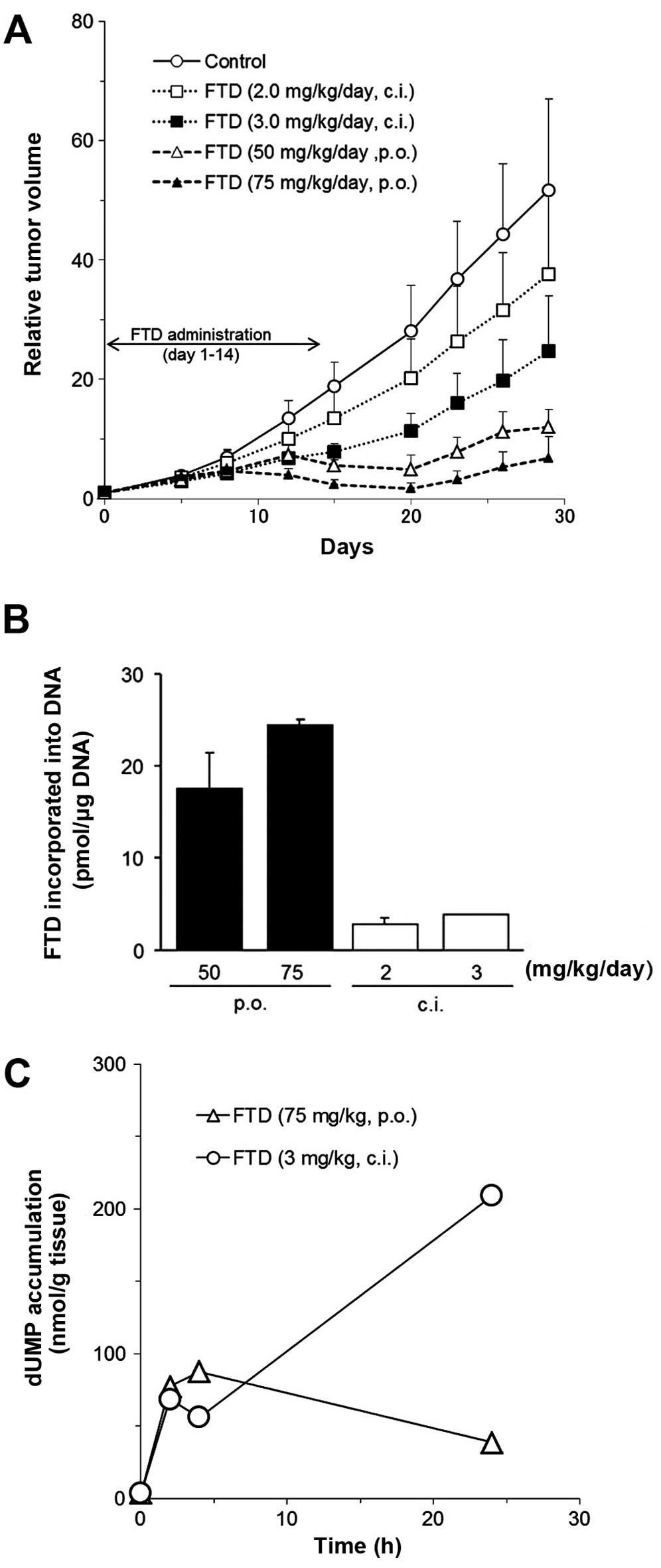
As shown in Table
II, considerably higher doses of FTD were tolerated when the
drug was administered orally rather than as a continuous infusion.
For example, the change in body weight (−8.8±11.1%) with 50
mg/kg/day of oral FTD was similar to the change (7.9±12.0%) induced
by 2 mg/kg/day of infusion FTD. Additionally, oral administration
of FTD had a stronger antitumor effect than continuous infusion of
FTD at the MTD (Table II). As an
example, 50 mg/kg/day FTD oral administration gave an IR of 70.8%
whereas 2 mg/kg/day FTD continuous infusion produced an IR of only
28.7%. Slight tumor shrinkage and tumor growth suppression was
observed after FTD oral administration (Fig. 3A). Consistent with the above
results, the amount of FTD incorporated into the DNA of xenograft
tumor cells was higher after oral administration (p.o., 50 and 75
mg/kg/day FTD, 17.34±3.90 and 24.23±0.50 pmol/μg DNA respectively;
c.i. 2.0 and 3.0 mg/kg/day FTD, 2.56±0.66 and 3.68±0.02 pmol/μg
DNA, respectively). As an index of TS activity, we also evaluated
dUMP accumulation in tumor tissues, as its levels increase when TS
is inhibited (18). As shown in
Fig. 3C, dUMP levels increased
during continuous infusion treatment. The increase reached high
level following continuous infusion treatment (208.9 nmol/g). On
the other hand, the dUMP level after oral administration increased
transiently, and reverted to almost basal level (39 nmol/g) at 24
h.
| Table IIAntitumor efficacy of FTD in human
breast cancer xenografts following oral administration or
continuous infusion. |
Table II
Antitumor efficacy of FTD in human
breast cancer xenografts following oral administration or
continuous infusion.
| Treatment | Administration
route | RTVa (mean ± SD) | IRb (%) | BWCc (%, mean ± SD) |
|---|
| Control | - | 18.9±3.9 | - | 17.6±8.1 |
| 50 mg/kg/day
FTD | p.o. (b.i.d.) | 5.5±1.0e | 70.8 | −8.8±11.1 |
| 75 mg/kg/day
FTD | p.o. (b.i.d.) | 2.4±0.8e | 87.5 | −23.9±4.4 |
| 2.0 mg/kg/day
FTD | c.i. | 13.5±4.7d | 28.7 | 7.9±12.0 |
| 3.0 mg/kg/day
FTD | c.i. | 7.9±1.3e | 58.2 | −13.6±9.1 |
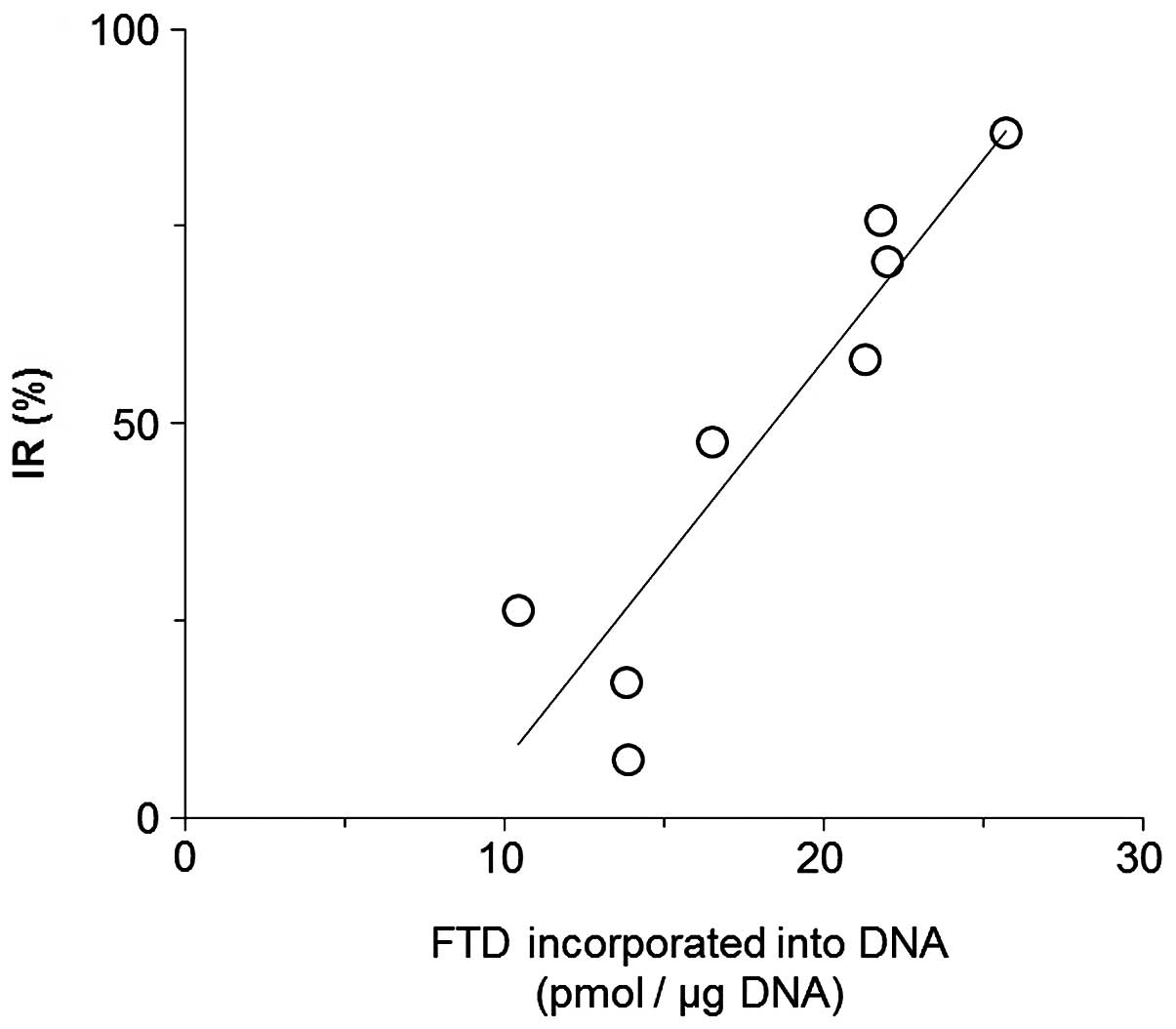
Relationship between TAS-102 antitumor
activity and the amount of FTD incorporation into tumor DNA
Since inhibition of TS did not appear to correlate
with the antitumor activity of FTD, we hypothesized that direct
interaction of the drug with DNA may contribute to its mechanism of
action. TAS-102 (150 mg/kg/day) was orally administered to mice
harboring xenografts in two daily doses for 14 days (Table III). This regimen was effective
against xenografts derived from a broad range of tumor cell types.
Moreover, inclusion of HPLC analysis at day 7 revealed a positive
correlation between the antitumor activity of TAS-102 and the
amount of FTD incorporated into DNA (Pearson correlation
coefficient r=0.92, R2=0.84, P=0.0013 by Pearson
correlation coefficient test, Fig.
4).
| Table IIIAntitumor efficacy of TAS-102 and DNA
incorporation of FTD in xenografts derived from various cell
types. |
Table III
Antitumor efficacy of TAS-102 and DNA
incorporation of FTD in xenografts derived from various cell
types.
| Tumor | Origin | IRa (%) | FTD incorporated
into DNA |
|---|
| DLD-1 | CRC | 7.3 | 13.9 |
| HT-29 | CRC | 17.2 | 13.8 |
| SW480 | CRC | 26.4 | 10.4 |
| HCT116 | CRC | 47.6 | 16.5 |
| KM20C | CRC | 70.5 | 22.0 |
| Lu-134 | SCLC | 75.8 | 21.8 |
| MC-2 | BC | 58.0 | 21.3 |
| MX-1 | BC | 86.9 | 25.7 |
FTD incorporation into DNA of a colon
cancer-derived xenograft following TAS-102 treatment
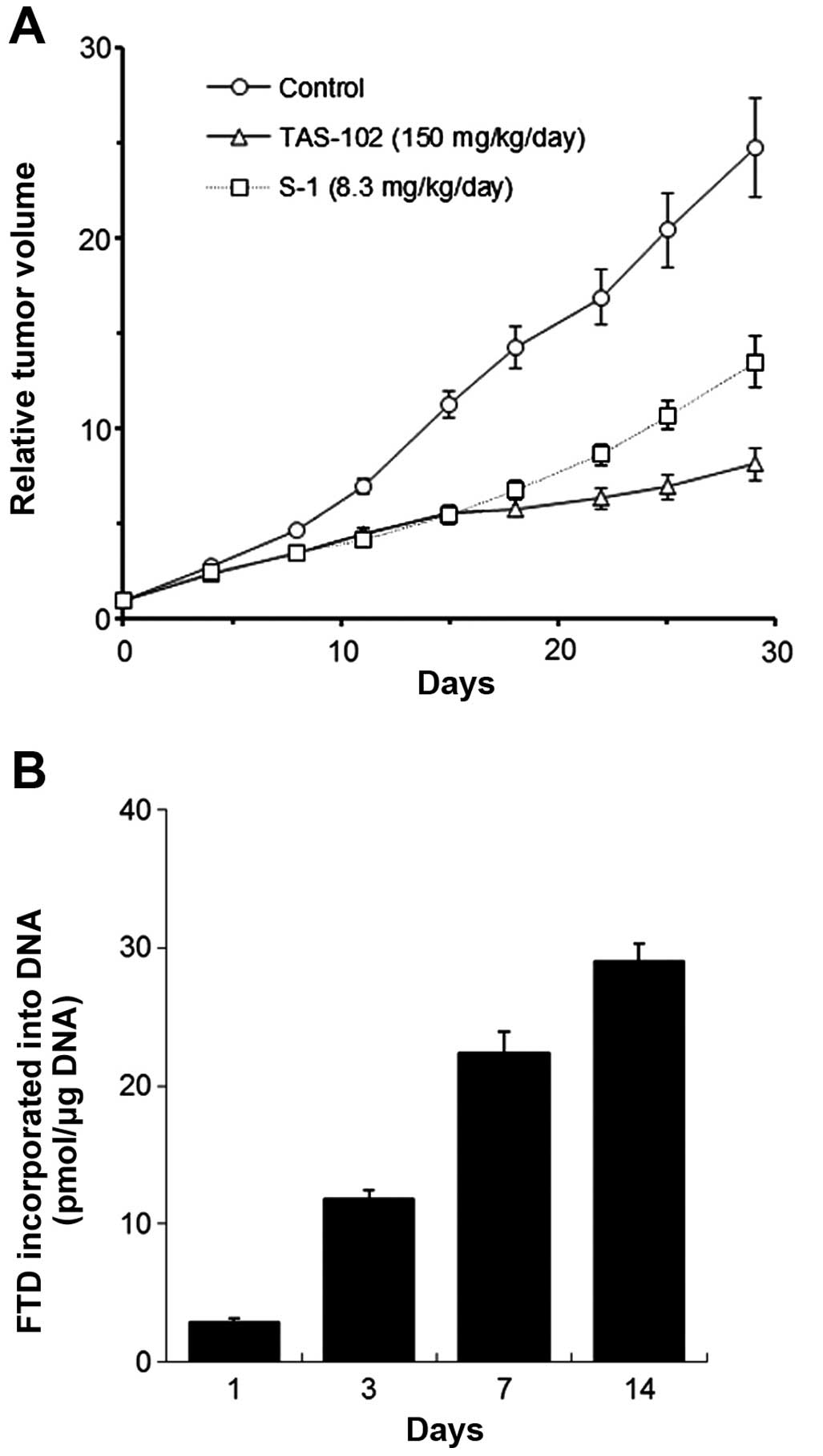
To investigate the kinetics of FTD incorporation
into DNA, we followed the process during TAS-102 treatment of mice
harboring KM20C human colon cancer xenografts (Fig. 5A and B). TAS-102 treatment (150
mg/kg/day, twice daily for 14 days) was compared to oral
administration of S-1 once daily for 14 days. TAS-102 and S-1 had
similar antitumor effects (IR of 50.2 and 51.4%, respectively). FTD
accumulated in the DNA of KM20C xenografts in a time-dependent
manner throughout the administration of TAS-102. Compared to S-1
treatment, the growth suppressive effect of TAS-102 appeared more
sustained (Fig. 5A).
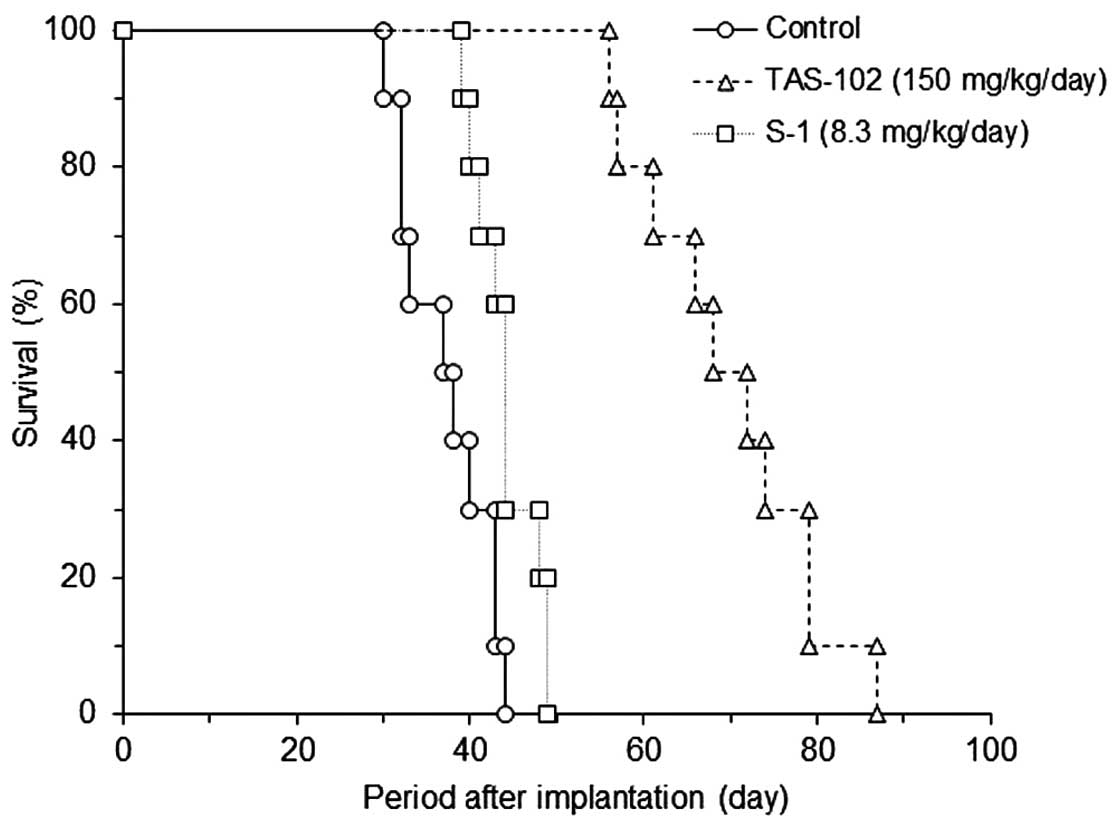
TAS-102 prolongs survival in mice bearing
human colon cancer xenografts
To determine whether TAS-102 could prolong survival,
we used mice implanted with KM20C xenografts (Fig. 6). The median survival time of
control, TAS-102 and S-1 administered group was 38, 70 and 44 day
respectively, and the ILS of TAS-102 and S-1 were 86.7 and 17.3%.
TAS-102 significantly prolonged the survival of mice when compared
to control (P<0.01), S-1 (P<0.01) as determined by the
log-rank test.
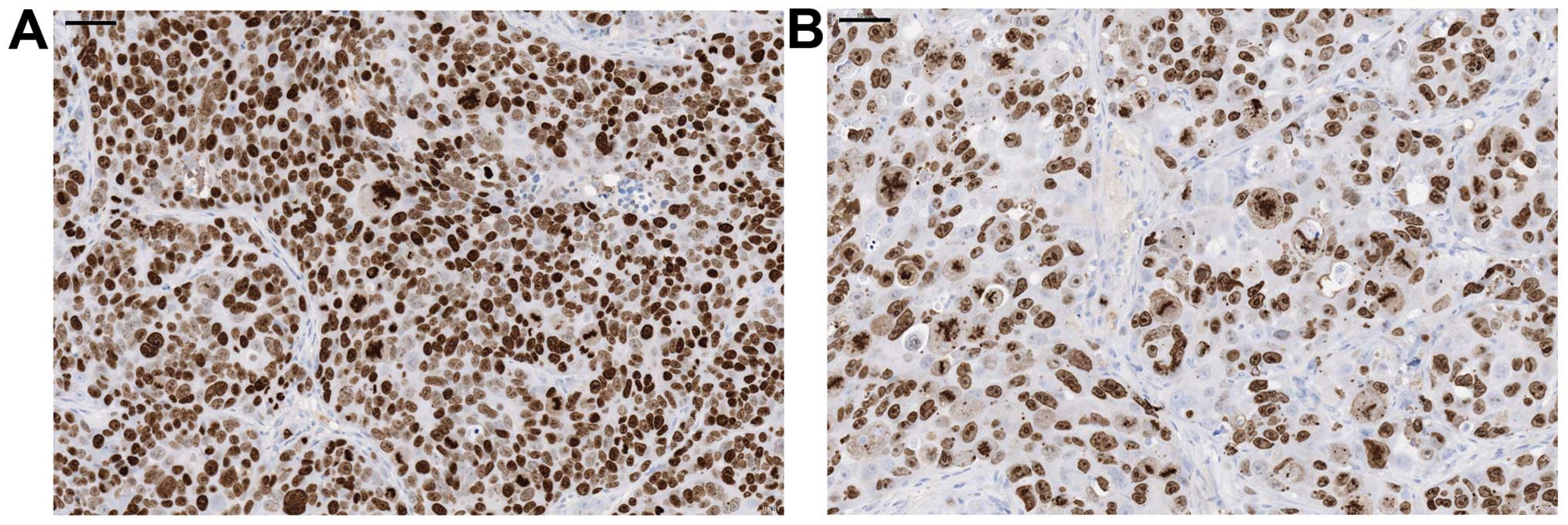
Ki-67 staining and morphological
analysis
To further characterize the growth suppressive
activity of TAS-102 in vivo, we performed
immunohistochemical staining for Ki-67 (a marker of proliferating
cells), as well as morphological analysis (Fig. 7A and B). Tumor samples were
collected from xenografts following a 14-day TAS-102 treatment.
Ki-67 staining intensity was markedly decreased in most cells.
Notably, we also observed a swelling of nuclei in the tumors
treated with TAS-102.
Discussion
FTD exhibits two major cellular activities, namely,
inhibition of TS and incorporation into DNA. In the present study,
we focused on determining which of these two activities plays the
major role in the antitumor activity of TAS-102.
We first noted that both short- and long-term FTD
treatments are cytotoxic; however, TS activity was only sustained
by long-term treatment. In contrast, even short-term FdUrd
treatment was sufficient to block TS activity, whereas the
cytotoxicity of short-term FdUrd was diminished. This is likely
since fluorodeoxyuridine monophosphate (FdUMP) inhibits TS by
forming a covalent bond, whereas FTD monophosphate
(F3dTMP) inhibits TS in a reversible manner (15), and short-term TS inhibition is not
sufficient to exert cytotoxicity. We suggest, therefore, that the
growth inhibition after FTD washout is likely due to DNA
incorporation rather than TS inhibition. There are reports that
FdUrd can be incorporated into DNA (19). However, fluorodeoxyuridine
triphosphate (FdUTP) is efficiently degraded by deoxyuridine
5′-triphosphate diphosphohydrolase (dUTPase) (20). In the present study, we confirmed
that the amount of FTD incorporation into DNA was much higher than
FdUrd and other antitumor nucleosides. We thus infer that FTD and
its cellular derivatives may be less prone to pathways that would
otherwise reduce their ability to incorporate to DNA.
Secondly, both the amount of FTD incorporation into
DNA and antitumor activity was higher, yet the TS inhibitory
activity was lower, when the drug was orally delivered. We infer
that persisting low concentrations of FTD following continuous
infusion block DNA synthesis via inhibition of TS, and this leads
to a small amount of FTD incorporation into DNA, and unwanted
side-effects. Coincidentally, little amount of FTD incorporates
into DNA, since DNA synthesis is inhibited by TS persisting
inhibition. By contrast, oral administration does not appear to
inhibit TS chronically, and, therefore, DNA synthesis is less
affected and FTD incorporates into DNA during DNA synthesis. It is
reported that intracellular F3TTP concentrations reach a
plateau after 8 h, and decline after washout (21). From these considerations, we suggest
that daily oral administration of FTD could be a suitable regimen
for antitumor treatments.
A significant caveat is that FTD is degraded when
administered orally. Indeed, the activity of TP in the liver of
monkeys and humans is high, which can negatively affect systemic
drug concentrations. In the present study, a positive correlation
was observed between the level of FTD incorporated into tumor cell
DNA and the antitumor activity of TAS-102 toward various
xenografts. This relationship suggests that FTD incorporation into
DNA is the major mechanism underlying the antitumor activity of
TAS-102. Notably, the antitumor effect of TAS-102 is sustained
after administration (Fig. 5A), and
a similar result was observed following FTD oral administration
(Fig. 3A). This persistent
antitumor activity likely accounts for the ability of TAS-102 to
prolong the survival of xenograft-bearing mice. FTD is incorporated
in place of thymidine bases in DNA (19), and this only slightly destabilizes
the DNA duplex (22); it may alter
DNA-protein interactions and cause nuclei swelling due to
nucleosomal abnormality. Thus, the ability of FTD to accumulate in
the DNA of tumor cells without causing immediate catastrophic
consequences may allow it to affect tumor cells in a chronic and
persistent manner. Bijnsdorp et al (23) stated that mechanisms by which FTD
induces cell death are distinct from those elicited by 5-FU. It was
also reported that TAS-102 monotherapy significantly improved
overall survival for patients with mCRC and who were insensitive to
fluoropyrimidine-based standard therapies (10). Further analyses are needed to
elucidate the mechanisms responsible for the cytotoxicity of FTD
following its incorporation into DNA.
In conclusion, we suggest that, due to its unique
mechanism of action, which is distinct from that of existing 5-FU
derivatives, TAS-102 has potential as a potent therapeutic option
for malignancies that are insensitive to fluoropyrimidine-based
therapy.
Acknowledgements
The authors thank Professor J. Patrick Barron of the
Department of International Medical Communications of Tokyo Medical
University for his review of this manuscript. We gratefully
acknowledge Professor Godefridus J. Peters of the Department of
Oncology of VU University Medical Center for his scientific advice.
We thank Dr Kiyoshi Morikawa for providing the KM20C cells. All
authors are employees of Taiho Pharmaceutical Co., Ltd., the
company that developed TAS-102.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Hurwitz H, Fehrenbacher L, Novotny W, et
al: Bevacizumab plus irinotecan, fluorouracil, and leucovorin for
metastatic colorectal cancer. N Engl J Med. 350:2335–2342. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Giantonio BJ, Catalano PJ, Meropol NJ, et
al: Bevacizumab in combination with oxaliplatin, fluorouracil, and
leucovorin (FOLFOX4) for previously treated metastatic colorectal
cancer: results from the Eastern Cooperative Oncology Group Study
E3200. J Clin Oncol. 25:1539–1544. 2007. View Article : Google Scholar
|
|
4
|
Jonker DJ, O’Callaghan CJ, Karapetis CS,
et al: Cetuximab for the treatment of colorectal cancer. N Engl J
Med. 357:2040–2048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Cutsem E, Peeters M, Siena S, et al:
Open-label phase III trial of panitumumab plus best supportive care
compared with best supportive care alone in patients with
chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol.
25:1658–1664. 2007.PubMed/NCBI
|
|
6
|
Sobrero AF, Maurel J, Fehrenbacher L, et
al: EPIC: phase III trial of cetuximab plus irinotecan after
fluoropyrimidine and oxaliplatin failure in patients with
metastatic colorectal cancer. J Clin Oncol. 26:2311–2319. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peeters M, Price TJ, Cervantes A, et al:
Randomized phase III study of panitumumab with fluorouracil,
leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as
second-line treatment in patients with metastatic colorectal
cancer. J Clin Oncol. 28:4706–4713. 2010. View Article : Google Scholar
|
|
8
|
Heidelberger C, Parsons DG and Remy DC:
Syntheses of 5-trifluoromethyluracil and
5-trifluoromethyl-2′-deoxyuridine. J Med Chem. 7:1–5. 1964.
|
|
9
|
Fujiwara Y, Oki T and Heidelberger C:
Fluorinated pyrimidines. XXXVII. Effects of
5-trifluoromethyl-2′-deoxyuridine on the synthesis of
deoxyribonucleic acid of mammalian cells in culture. Mol Pharmacol.
6:273–280. 1970.PubMed/NCBI
|
|
10
|
Yoshino T, Mizunuma N, Yamazaki K, et al:
TAS-102 monotherapy for pretreated metastatic colorectal cancer: a
double-blind, randomised, placebo-controlled phase 2 trial. Lancet
Oncol. 13:993–1001. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dexter DL, Wolberg WH, Ansfield FJ, Helson
L and Heidelberger C: The clinical pharmacology of
5-trifluoromethyl-2′-deoxyuridine. Cancer Res. 32:247–253.
1972.
|
|
12
|
Ansfield FJ and Ramirez G: Phase I and II
studies of 2′-deoxy-5-(trifluoromethyl)-uridine (NSC-75520). Cancer
Chemother Rep. 55:205–208. 1971.
|
|
13
|
Skevaki CL, Galani IE, Pararas MV,
Giannopoulou KP and Tsakris A: Treatment of viral conjunctivitis
with antiviral drugs. Drugs. 71:331–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fukushima M, Suzuki N, Emura T, et al:
Structure and activity of specific inhibitors of thymidine
phosphorylase to potentiate the function of antitumor
2′-deoxyribonucleosides. Biochem Pharmacol. 59:1227–1236.
2000.PubMed/NCBI
|
|
15
|
Langenbach RJ, Danenberg PV and
Heidelberger C: Thymidylate synthetase: mechanism of inhibition by
5-fluoro-2′-deoxyuridylate. Biochem Biophys Res Commun.
48:1565–1571. 1972.PubMed/NCBI
|
|
16
|
Miller EM and Kinsella TJ:
Radiosensitization by fluorodeoxyuridine: effects of thymidylate
synthase inhibition and cell synchronization. Cancer Res.
52:1687–1694. 1992.PubMed/NCBI
|
|
17
|
Sampath D, Rao VA and Plunkett W:
Mechanisms of apoptosis induction by nucleoside analogs. Oncogene.
22:9063–9074. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pettersen HS, Visnes T, Vågbø CB, et al:
UNG-initiated base excision repair is the major repair route for
5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends
mainly on RNA incorporation. Nucleic Acids Res. 39:8430–8444. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suzuki N, Emura T and Fukushima M: Mode of
action of trifluorothymidine (TFT) against DNA replication and
repair enzymes. Int J Oncol. 39:263–270. 2011.PubMed/NCBI
|
|
20
|
Quesada-Soriano I, Casas-Solvas JM, Recio
E, et al: Kinetic properties and specificity of trimeric
Plasmodium falciparum and human dUTPases. Biochimie.
92:178–186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Emura T, Nakagawa F, Fujioka A, et al: An
optimal dosing schedule for a novel combination antimetabolite,
TAS-102, based on its intracellular metabolism and its
incorporation into DNA. Int J Mol Med. 13:249–255. 2004.PubMed/NCBI
|
|
22
|
Markley JC, Chirakul P, Sologub D and
Sigurdsson ST: Incorporation of 2′-deoxy-5-(trifluoromethyl)uridine
and 5-cyano-2′-deoxyuridine into DNA. Bioorg Med Chem Lett.
11:2453–2455. 2001.
|
|
23
|
Bijnsdorp IV, Peters GJ, Temmink OH,
Fukushima M and Kruyt FA: Differential activation of cell death and
autophagy results in an increased cytotoxic potential for
trifluorothymidine compared to 5-fluorouracil in colon cancer
cells. Int J Cancer. 126:2457–2468. 2010.
|