Introduction
Lung cancer is the leading cause of cancer-related
death worldwide (1). To improve the
outcomes of patients with lung cancer, various novel therapeutic
agents have been developed, including epidermal growth factor
receptor (EGFR)-tyrosine kinase inhibitors (TKIs). EGFR-TKIs show
significant efficacy against non-small cell lung cancers (NSCLCs)
harboring EGFR mutations, by inhibiting EGFR-AKT signaling
(2–4). However, most of these tumors
eventually acquire resistance to EGFR-TKIs (5,6).
Several mechanisms of acquired resistance to EGFR-TKIs have been
identified, such as secondary EGFR T790M (7) and minor mutations (8), and MET amplification (9). In addition, we also previously
demonstrated an association between resistance to EGFR-TKIs and
stem cell-like properties of the cells (10).
The 90-kDa heat shock protein (Hsp90) is a chaperone
protein that modulates degradation, folding, and/or transport of a
diverse set of critical cellular regulatory proteins (11). Critical oncogenic proteins,
including receptor tyrosine kinases (RTKs) (e.g. EGFR) and their
downstream proteins (e.g. AKT) are client proteins of Hsp90
(12,13), and mutated oncogenic proteins are
more dependent on the functions of Hsp90 (14). Therefore, it was considered that
Hsp90 may be a therapeutic target to overcome the resistance to
EGFR-TKIs. Actually, Hsp90 inhibitors are effective against
EGFR-mutated cell lines, even in those that are resistant to
EGFR-TKIs (15–17). Furthermore, Hsp90 inhibitors are
known to exert a radiosensitizing effect through hypoxia-inducible
factor-1α (HIF-1α), ataxia-telangiectasia mutated (ATM), checkpoint
kinase 1 (CHK1), WEE1 G2 checkpoint kinase (WEE1)
(18–21) and other radioresistance-related
client proteins. The radiosensitizing potential of Hsp90 inhibitors
has been evaluated previously in NSCLC cell lines such as A549 and
NCI-H460 (22,23). However, there are no reports
focusing on the radiosensitizing effect of Hsp90 inhibitors on
EGFR-mutated NSCLCs with acquired resistance to
EGFR-TKIs.
In the present study, we evaluated the effect of the
novel Hsp90 inhibitor NVP-AUY922 (AUY) in overcoming the major
mechanisms of acquired resistance to EGFR-TKIs, such as EGFR
T790M mutation and MET amplification, and the
radiosensitizing effect of this compound. We also studied the
radiosensitizing effect of AUY in overcoming acquired resistance
induced by the acquisition of stem cell-like properties of the
cells.
Materials and methods
Cell lines and reagents
EGFR-mutant cell lines HCC827 (exon19 del.
E746-A750), and PC-9 (exon 19 del. E746-A750) were used. HCC827 was
kindly gifted by Dr Adi F. Gazdar (The University of Texas
Southwestern Medical Center, Dallas, TX, USA), who established this
line with Dr John D. Minna (24,25).
PC-9 was obtained from Immuno-Biological Laboratories (Takasaki,
Gunma, Japan). Their gefitinib-resistant sublines, HCC827-GRmet
with MET amplification, HCC827-GRstem with stem-cell like
properties, and PC-9-GRt790m harboring the EGFR T790M
mutation, were previously established by our group (10). All the cell lines were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS),
and grown in a humidified incubator with 5% CO2 at 37°C.
AUY was obtained from Novartis Pharmaceuticals (Basel, Switzerland)
and dissolved in dimethyl sulfoxide (DMSO) at the concentration of
10 mM as a stock solution and stored at −20°C until they were used
for the in vitro experiments.
Cell proliferation assays
The proliferative ability of the cells was
determined by a modified MTS assay using CellTiter 96®
AQueous One solution reagent (Promega, Madison, WI, USA), as
previously reported (26). The
antiproliferative effects of AUY were determined based on the 10%
and 50% inhibitory concentration (IC10 and
IC50), which denote the concentrations of AUY required
to inhibit cell proliferation by 10% and 50%, respectively.
Clonogenic cell survival assays
Specified numbers of cells were seeded into each
well in 6-well tissue culture plates, and after the cells became
adherent (12 h), they were exposed to various concentrations of
AUY, according to the obtained IC10 values, which were
determined by cell proliferation assays. After a 24-h drug
exposure, the plates were irradiated at 2, 4 or 6 Gy (ionizing
radiation; IR), followed immediately by replacement of the culture
medium with a drug-free conditioned medium. At 14 days after the
IR, the colonies were fixed and stained using 0.4% crystal violet.
The number of colonies containing at least 50 cells was counted.
The survival data were fitted to a linear quadratic model as
previously reported (23): SF = exp
(-αX - βX2), where SF is the survival fraction, X is the
radiation dose, and α and β are the fitted parameters. The results
were evaluated using the surviving cell fractions at 2 Gy (SF2) and
the radiation doses required for 10% survival (D10), and
the radiosensitizing effects of AUY were evaluated using the ratio
of the D10 of the control cells to the D10
for each AUY concentration.
Cell cycle analysis
The cell cycle distribution was evaluated by
propidium iodide staining-based assay using the CycleTest™ Plus DNA
reagent kit and FACSCalibur™ (both from Becton Dickinson, Franklin
Lakes, NJ, USA). The cells were irradiated at 0 or 6 Gy (IR) after
exposure or no exposure to 100 nM AUY for 24 h. At 48 h after IR,
the cells were harvested and analyzed. Doublets, cell debris and
fixation artifacts were gated out, and cell cycle analysis was
performed using the software, CellQuest™, ver. 3.1.
Immunofluorescence staining for
phosphorylated histone H2AX (γH2AX)
DNA double-strand breaks (DNA DSBs) were evaluated
by immunofluorescence staining for γH2AX (27). Each cell line was plated into
chamber slides and after allowing the cells to become adherent (12
h), the medium was changed to that containing or not containing 100
nM of AUY. After a 24-h drug exposure, the plates were irradiated
at 6 Gy, followed immediately by a change of the medium to a
drug-free conditioned medium. The cells were fixed in 4% formalin
for 15 min at 6, 24 and 48 h after the IR. Permeabilization and
blocking were performed for 1 h using 10X PBS with 5% goat serum
and 0.3% Triton X-100. Anti-γH2AX antibody at a 1:200 dilution was
added as the primary antibody (Millipore, Billerica, MA, USA),
followed by incubation overnight at 4°C. Goat anti-mouse IgG
conjugated Alexa Fluor® 555 (Life Technologies,
Carlsbad, CA, USA) at a 1:1,000 dilution was added as the secondary
antibody for 1 h and DAPI staining was performed using
ProLong® Gold antifade reagent with DAPI (Life
Technologies). The number of γH2AX foci in each nucleus was counted
in at least 30 cells in each sample.
Statistical analysis
The Mann-Whitney U test was used to compare the data
between the 2 groups. Data are expressed as the means ± standard
deviations. Probability values (P) <0.05 were considered to
indicate statistical significance. All the data were analyzed using
GraphPad Prism, ver. 6.0.3, J (GraphPad Software, San Diego, CA,
USA).
Results
AUY is effective for overcoming EGFR-TKI
resistance in all of the cell lines examined, except for the cell
line with stem cell-like properties
The IC10 and IC50 values of
AUY in the parental cell lines and EGFR-TKI-resistant sublines are
shown in Table I. HCC827-GRmet and
PC-9-GRt790m cells were more sensitive to AUY than the parental
cell lines. The IC50 value for HCC827-GRstem was over
20-fold as high as that for the other cell lines.
 | Table IInhibitory concentration values of
NVP-AUY922. |
Table I
Inhibitory concentration values of
NVP-AUY922.
| Cell lines | Resistant
mechanism | IC10
(nM) | IC50
(nM) |
|---|
| HCC827 | - | 2.5 | 21.0 |
| HCC827-GRmet | MET
amplification | 2.7 | 7.0 |
| HCC827-GRstem | Stem cell-like
features | 14.0 | 402.0 |
| PC-9 | - | 2.8 | 9.1 |
| PC-9-GRt790m | T790M mutation | 2.9 | 6.7 |
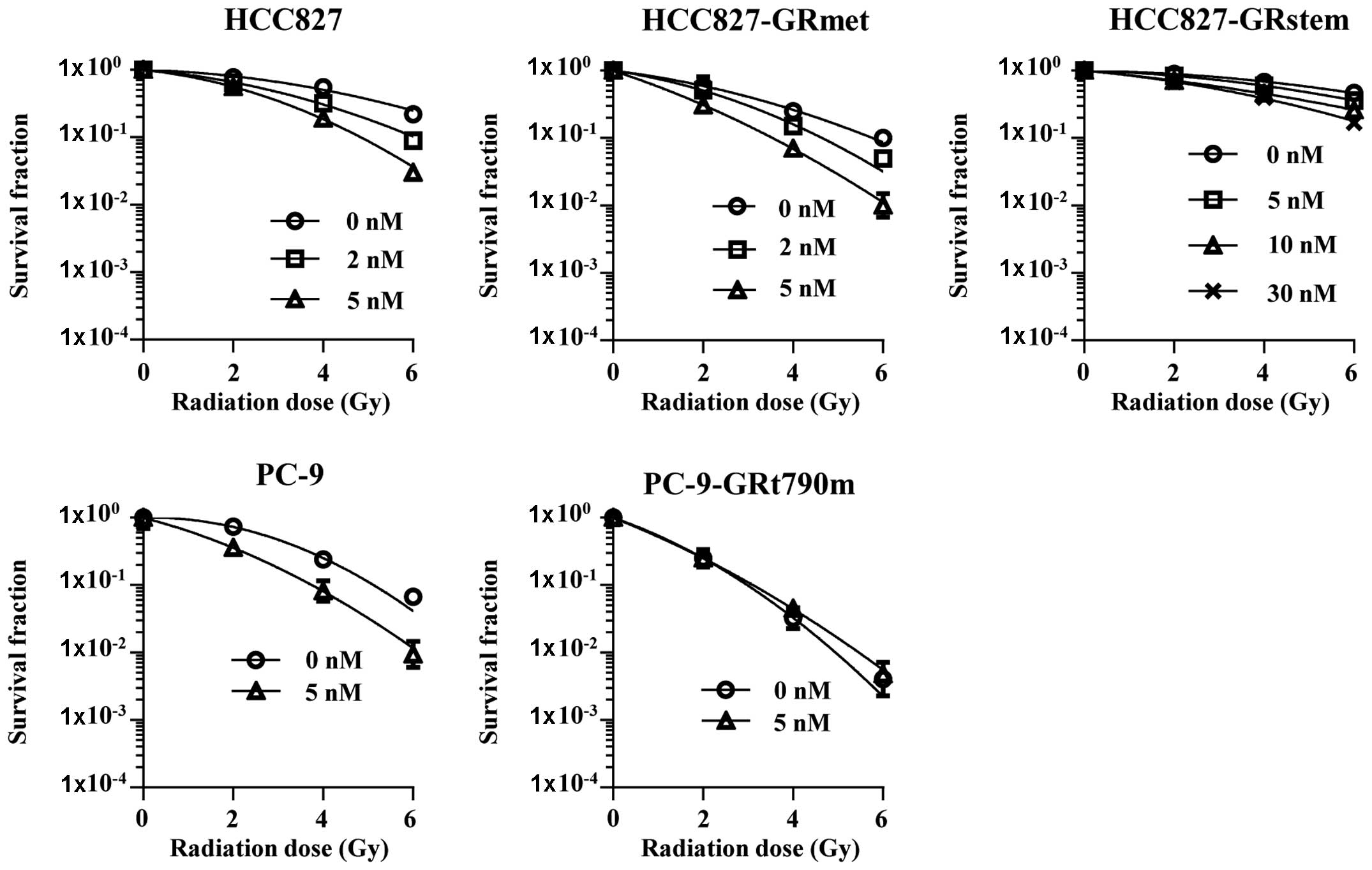
Radiosensitizing effect of AUY
The survival curves and parameters of the clonogenic
cell survival assays are shown in Fig.
1 and Table II, respectively.
The D10 values for HCC827-GRmet or PC-9-GRt790m cells
were lower than those for the parental cell lines, while the value
for HCC827-GRstem was higher than that for the parental cell line.
Therefore, these sublines were more sensitive to IR than the
parental cell lines, except for HCC827-GRstem. Radiosensitization
was defined as a ratio of the D10 value for the control
cells to that for the AUY-treated cells of >1.5. Our study
revealed that both the parental cell line and HCC827-GRmet were
radiosensitized, while HCC827-GRstem and PC-9-GRt790m were not
radio-sensitized by 5 nM of AUY.
| Table IICloning efficiencies and
radiosensitivity parameters. |
Table II
Cloning efficiencies and
radiosensitivity parameters.
| Cell lines | Plating
efficiency | SF2 | α
(Gy−1) | β
(Gy−1) | D10
(Gy)b | D10
control/D10 + AUY922 |
|---|
| HCC827 | 0.3±0.1 | 0.78±0.12 | 0.05±0.04 | 0.03±0.01 | 5 | - |
| +2 nM AUY | 0.3±0.1 | 0.65±0.04 | 0.13±0.01 | 0.04±0.004 | 3.6 | 1.4 |
| +5 nM AUY | 0.3±0.1 | 0.56±0.02 | 0.16±0.01 | 0.01±0.003 | 2.9 | 1.7 |
| HCC827-GRmet | 0.4±0.1 | 0.60±0.22 | 0.19±0.01 | 0.04±0.003 | 3.3 | - |
| +2 nM AUY | 0.4±0.02 | 0.51±0.04 | 0.22±0.03 | 0.06±0.01 | 2.6 | 1.2 |
| +5 nM AUY | 0.2±0.01 | 0.31±0.01 | 0.5±0.01 | 0.04±0.004 | 1.7 | 1.9 |
| HCC827-GRstem | 0.5±0.1 | 0.90±0.25 | 0.02±0.05 | 0.02±0.01 | 6.9 | - |
| +5 nM AUY | 0.5±0.1 | 0.84±0.17 | 0.04±0.02 | 0.02±0.01 | 5.9 | 1.2 |
| +10 nM AUY | 0.6±0.04 | 0.71±0.08 | 0.14±0.01 | 0.01±0.005 | 4.8 | 1.4 |
| +30 nM AUY | 0.5±0.04 | 0.68±0.02 | 0.14±0.01 | 0.02±0.003 | 4.2 | 1.7 |
| PC-9 | 0.85±0.27 | 0.74±0.08 | 0.03±0.1 | 0.09±0.04 | 3.4 | - |
| +5 nM AUY | 0.06±0.01 | 0.36±0.09 | 0.4±0.1 | 0.06±0.05 | 2 | 1.8 |
| PC-9-GRt790m | 0.4±0.1 | 0.25±0.09 | 0.5±0.4 | 0.08±0.17 | 1.5 | - |
| +5 nM AUY | 0.2±0.02 | 0.25±0.02 | 0.6±0.2 | 0.04±0.07 | 1.5 | 1.0 |
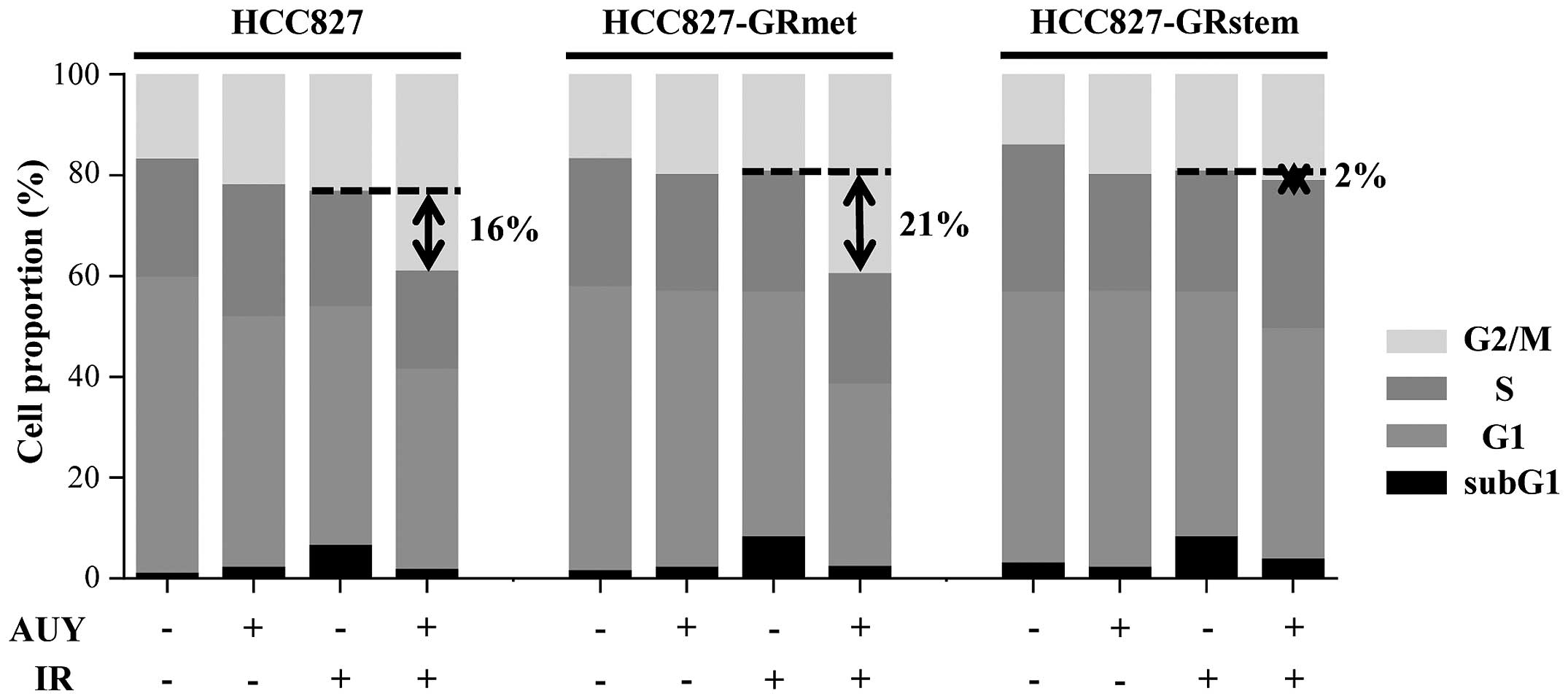
G2/M arrest is caused by IR
with AUY
The proportions of HCC827 cells in the
G2/M phase following exposure to only IR and following
exposure to both IR and AUY were 24 and 40%, respectively. The
proportions of HCC827-GRmet cells in G2/M phase
following exposure to only IR and following exposure to both IR and
AUY were 20 and 41%, respectively. In brief, exposure to both IR
and AUY caused G2/M arrest. However, in the case of the
HCC827-GRstem cells, the proportion of cells in the G2/M
phase following exposure to only IR and following exposure to both
IR and AUY were 20 and 22%, respectively. Therefore, HCC827-GRstem
was resistant to G2/M arrest even after combined IR plus
AUY treatment (Fig. 2).
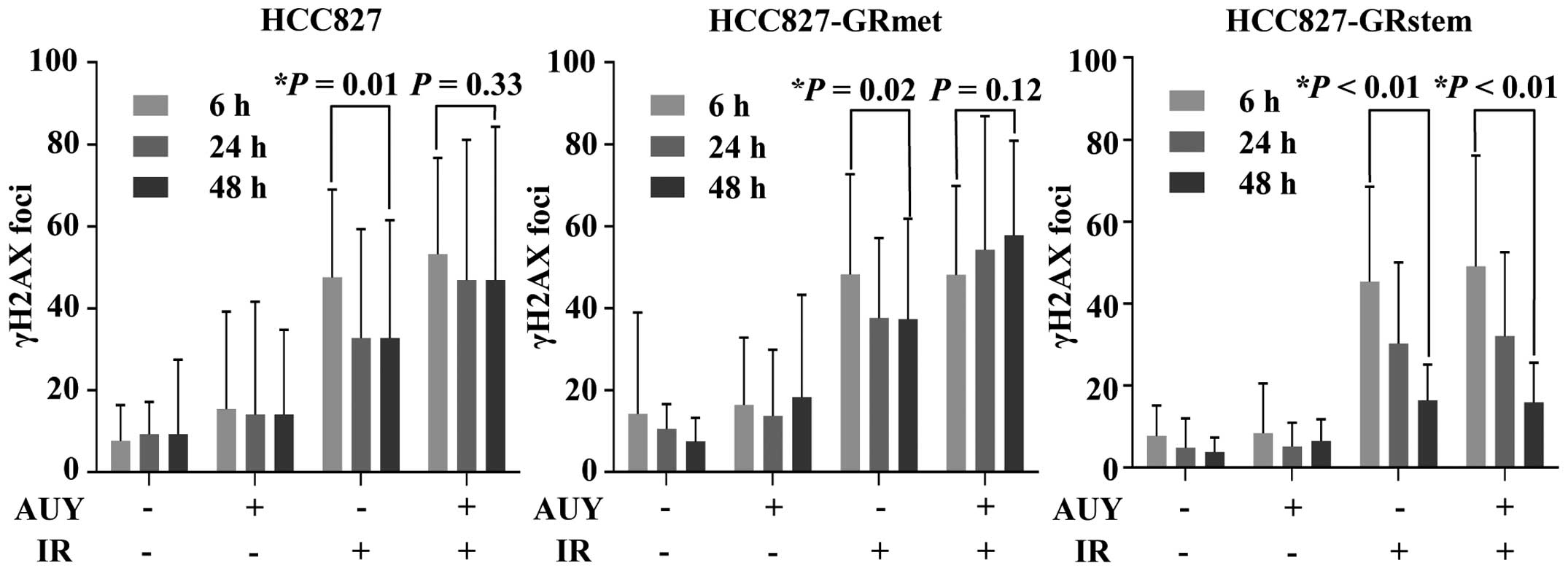
DNA repair ability of the
EGFR-TKI-resistant cell lines
Repair of DNA DSBs was evaluated by determining the
decrease in the number of γH2AX foci (Fig. 3). The numbers of γH2AX foci in the
HCC827 cells after only IR were 47.6±21.4 and 32.7±28.8 at 6 and 48
h, respectively (P=0.01), while the corresponding values after
exposure to both IR and AUY were 53.2±23.5 and 46.9±37.4 (P=0.33).
The numbers of γH2AX foci in the HCC827-GRmet cell line after only
IR were 48.2±24.5 and 37.3±24.5 at 6 and 48 h, respectively
(P=0.02), and the corresponding values in the cells exposed to both
IR and AUY were 48.2±22.7 and 57.8±23.1 (P=0.12). Furthermore, the
numbers of γH2AX foci in the HCC827-GRstem cells exposed to IR
alone were 45.4±23.2 and 16.4±8.7 at 6 and 48 h, respectively
(P<0.01), and the corresponding values in the cells exposed to
both IR and AUY were 49.1±27.1 and 15.9±9.7 (P<0.01). In the IR
treatment group, HCC827 and HCC827-GRmet cells at 48 h showed a
significant decrease in the numbers of γH2AX foci compared to those
at 6 h. However, in the IR plus AUY group, the significant
difference was not achieved between the numbers of γH2AX at 6 h and
those at 48 h. In contrast HCC827-GRstem cells showed a significant
decrease in the number of γH2AX foci after both IR treatment alone
and after combined IR plus AUY treatment.
Discussion
In the present study, AUY was effective against
EGFR-TKI-resistant cells with secondary mutation of EGFR or
other RTK dependence. These results are concordant with previous
reports (15–17). We demonstrated that combined
exposure to IR and AUY caused G2/M arrest and inhibition
of DNA DSB repair, and radiosensitized EGFR-TKI-resistant cell
lines with major resistance mechanisms such as T790M mutation and
MET amplification. However, the DNA repair ability of the
EGFR-TKI-resistant cell line with stem cell-like properties was
maintained even after combined treatment with IR and AUY, and the
radiosensitizing effect of AUY on this cell line was limited.
Notably, the cell lines with acquired resistance to
EGFR-TKIs associated with T790M mutation or MET
amplification were more sensitive to IR than the parental cell
lines in our study. Das and colleagues showed that NSCLC with
activating EGFR mutations were sensitive to IR (28). They proposed two possible mechanisms
to explain this finding. i) Elevated or aberrant signaling from the
mutant EGFR may override the IR-induced checkpoint. ii)
Translocated EGFR binds to the promoter region of DNA-dependent
protein kinase (DNA-PK) (29),
while mutated EGFR may not be able to bind to it. Although the
precise reasons for the greater radiosensitivity of the
EGFR-TKI-resistant-sublines than that of the parental cell lines in
our cohort could not be clearly elucidated, the acquired resistance
mechanisms may have an influence. The signaling from
amplified-MET may also override the IR-induced checkpoint,
or secondary EGFR mutations such as the T790M mutation may
also affect the binding of EGFR to the DNA-PK promoter.
Previously, several mechanisms to explain the
radiosensitizing effect of Hsp90 inhibitors have been reported
(18–21). IR causes G2/M arrest
through the ATM-CHK pathway (30).
As ATM and CHK are client proteins of Hsp90, Hsp90 inhibitors
enhance the G2/M arrest caused by IR (23,31–33).
Alternatively, Hsp90 inhibitors impair non-homologous end joining
(NHEJ) through DNA-PK/ATM (23,31,33–35).
These phenomena were also shown in our cohort, except in the
HCC827-GRstem cell line.
Cancer stem cells show activation of DNA DSB repair
by NHEJ through the DNA-PK/ATM-CHK pathway, and several cancers,
including NSCLCs, show radioresistance (35–38).
HCC827-GRstem, an EGFR-TKI-resistant cell line with stem cell-like
properties, also showed radioresistance and activation of DNA DSB
repair. Therefore, it was expected that the Hsp90 inhibitor might
allow the radioresistance of this cell line to be overcome, since
DNA-PK, ATM and CHK are client proteins of Hsp90. However, combined
treatment with IR and AUY of the HCC827-GRstem cell line produced
neither G2/M arrest nor inhibition of DNA DSB
repair.
As secondary mutations of EGFR or other RTK
dependence accounts for EGFR-TKI resistance in over 60% of cases
(39,40), these can be defined as the major
resistance mechanisms. Combined therapy with IR and AUY is a
promising option to overcome these major EGFR-TKI resistances; on
the other hand, other minor resistance mechanisms, such as those in
cells with stem cell-like properties, require other approaches.
In conclusion, combined therapy with IR and AUY is
effective to overcome major acquired resistance to EGFR-TKIs such
as that associated with the T790M mutation or MET
amplification, while the effect on resistance associated with stem
cell-like properties of the cells was limited. Further
investigation is warranted to elucidate the mechanism of acquired
resistance to EGFR-TKIs associated with stem cell-like properties
of cells.
Acknowledgements
The authors thank Mr. Seiji Tabara and Mr. Hirofumi
Uno (Department of Radiology, Okayama University Hospital) for
irradiating the cell lines and Ms. Fumiko Isobe (Department of
Thoracic, Breast and Endocrinological Surgery, Okayama University
Graduate School of Medicine, Dentistry and Pharmaceutical Sciences,
Okayama, Japan) for her technical support. This study was supported
by a Grant-in-Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology of Japan (grant
no. 24791462 to H.Y.).
Abbreviations:
|
Hsp90
|
90-kDa heat shock protein
|
|
AUY
|
NVP-AUY922
|
|
EGFR
|
epidermal growth factor receptor
|
|
TKI
|
tyrosine kinase inhibitor
|
|
NSCLC
|
non-small cell lung cancer
|
|
IR
|
ionizing radiation
|
|
IC10 and IC50
|
10% and 50% inhibitory
concentrations
|
|
DNA DSB
|
DNA double-strand break
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. New Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitsudomi T, Morita S, Yatabe Y, et al:
Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): an open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar
|
|
4
|
Sequist LV, Martins RG, Spigel D, et al:
First-line gefitinib in patients with advanced non-small-cell lung
cancer harboring somatic EGFR mutations. J Clin Oncol.
26:2442–2449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosell R, Moran T, Queralt C, et al:
Screening for epidermal growth factor receptor mutations in lung
cancer. N Engl J Med. 361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shien K, Toyooka S, Yamamoto H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Young JC, Moarefi I and Hartl FU: Hsp90: a
specialized but essential protein-folding tool. J Cell Biol.
154:267–273. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wright L, Barril X, Dymock B, et al:
Structure-activity relationships in purine-based inhibitor binding
to HSP90 isoforms. Chem Biol. 11:775–785. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Solit DB, Basso AD, Olshen AB, Scher HI
and Rosen N: Inhibition of heat shock protein 90 function
down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer
Res. 63:2139–2144. 2003.PubMed/NCBI
|
|
14
|
Trepel J, Mollapour M, Giaccone G and
Neckers L: Targeting the dynamic HSP90 complex in cancer. Nat Rev
Cancer. 10:537–549. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ueno T, Tsukuda K, Toyooka S, et al:
Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on
non-small cell lung cancer. Lung Cancer. 76:26–31. 2012. View Article : Google Scholar
|
|
16
|
Shimamura T, Li D, Ji H, et al: Hsp90
inhibition suppresses mutant EGFR-T790M signaling and overcomes
kinase inhibitor resistance. Cancer Res. 68:5827–5838. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koizumi H, Yamada T, Takeuchi S, et al:
Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs
in EGFR-mutant lung cancer by decreasing client protein expression
and angiogenesis. J Thorac Oncol. 7:1078–1085. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ha K, Fiskus W, Rao R, et al: Hsp90
inhibitor-mediated disruption of chaperone association of ATR with
hsp90 sensitizes cancer cells to DNA damage. Mol Cancer Ther.
10:1194–1206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim WY, Oh SH, Woo JK, Hong WK and Lee HY:
Targeting heat shock protein 90 overrides the resistance of lung
cancer cells by blocking radiation-induced stabilization of
hypoxia-inducible factor-1alpha. Cancer Res. 69:1624–1632. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tse AN, Sheikh TN, Alan H, Chou TC and
Schwartz GK: 90-kDa heat shock protein inhibition abrogates the
topoisomerase I poison-induced G2/M checkpoint in p53-null tumor
cells by depleting Chk1 and Wee1. Mol Pharmacol. 75:124–133. 2009.
View Article : Google Scholar :
|
|
21
|
Arlander SJ, Felts SJ, Wagner JM,
Stensgard B, Toft DO and Karnitz LM: Chaperoning checkpoint kinase
1 (Chk1), an Hsp90 client, with purified chaperones. J Biol Chem.
281:2989–2998. 2006. View Article : Google Scholar
|
|
22
|
Lee JH, Choi KJ, Seo WD, et al:
Enhancement of radiation sensitivity in lung cancer cells by
celastrol is mediated by inhibition of Hsp90. Int J Mol Med.
27:441–446. 2011.PubMed/NCBI
|
|
23
|
Stingl L, Stuhmer T, Chatterjee M, Jensen
MR, Flentje M and Djuzenova CS: Novel HSP90 inhibitors, NVP-AUY922
and NVP-BEP800, radiosensitise tumour cells through cell-cycle
impairment, increased DNA damage and repair protraction. Br J
Cancer. 102:1578–1591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gandhi J, Zhang J, Xie Y, et al:
Alterations in genes of the EGFR signaling pathway and their
relationship to EGFR tyrosine kinase inhibitor sensitivity in lung
cancer cell lines. PLoS One. 4:e45762009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Girard L, Zochbauer-Muller S, Virmani AK,
Gazdar AF and Minna JD: Genome-wide allelotyping of lung cancer
identifies new regions of allelic loss, differences between small
cell lung cancer and non-small cell lung cancer, and loci
clustering. Cancer Res. 60:4894–4906. 2000.PubMed/NCBI
|
|
26
|
Kubo T, Toyooka S, Tsukuda K, et al:
Epigenetic silencing of microRNA-34b/c plays an important role in
the pathogenesis of malignant pleural mesothelioma. Clin Cancer
Res. 17:4965–4974. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Das AK, Sato M, Story MD, et al:
Non-small-cell lung cancers with kinase domain mutations in the
epidermal growth factor receptor are sensitive to ionizing
radiation. Cancer Res. 66:9601–9608. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dittmann K, Mayer C, Fehrenbacher B, et
al: Radiation-induced epidermal growth factor receptor nuclear
import is linked to activation of DNA-dependent protein kinase. J
Biol Chem. 280:31182–31189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pawlik TM and Keyomarsi K: Role of cell
cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol
Biol Phys. 59:928–942. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koll TT, Feis SS, Wright MH, et al: HSP90
inhibitor, DMAG, synergizes with radiation of lung cancer cells by
interfering with base excision and ATM-mediated DNA repair. Mol
Cancer Ther. 7:1985–1992. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eccles SA, Massey A, Raynaud FI, et al:
NVP-AUY922: a novel heat shock protein 90 inhibitor active against
xenograft tumor growth, angiogenesis, and metastasis. Cancer Res.
68:2850–2860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stecklein SR, Kumaraswamy E, Behbod F, et
al: BRCA1 and HSP90 cooperate in homologous and non-homologous DNA
double-strand-break repair and G2/M checkpoint activation. Proc
Natl Acad Sci USA. 109:13650–13655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bakkenist CJ and Kastan MB: DNA damage
activates ATM through intermolecular autophosphorylation and dimer
dissociation. Nature. 421:499–506. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kastan MB and Lim DS: The many substrates
and functions of ATM. Nat Rev Mol Cell Biol. 1:179–186. 2000.
View Article : Google Scholar
|
|
36
|
Lundholm L, Haag P, Zong D, et al:
Resistance to DNA-damaging treatment in non-small cell lung cancer
tumor-initiating cells involves reduced DNA-PK/ATM activation and
diminished cell cycle arrest. Cell Death Dis. 4:e4782013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Phillips TM, McBride WH and Pajonk F: The
response of CD24(−/low)/CD44(+) breast cancer-initiating cells to
radiation. J Natl Cancer Inst. 98:1777–1785. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bao S, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oxnard GR, Arcila ME, Chmielecki J,
Ladanyi M, Miller VA and Pao W: New strategies in overcoming
acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in lung cancer. Clin Cancer Res. 17:5530–5537.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sequist LV, Waltman BA, Dias-Santagata D,
et al: Genotypic and histological evolution of lung cancers
acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|