Introduction
Cervical carcinoma is the second most common type of
cancer in women and is the most prevalent female malignancy in many
developing countries (1,2). Although the efficacy of chemotherapy
for the majority of cancer types has improved over the last three
decades, the severe reduction in quality of life caused by highly
toxic effects of chemotherapeutic drugs is still a formidable
problem in clinical medicine (3,4).
Therefore, the development and mechanistic investigation of
chemotherapeutic or chemopreventive agents against cervical
carcinoma are important steps towards reducing the incidence and
mortality of this disease (5).
Apoptotic and cell cycle control have recently attracted the
attention of investigators for the development of novel anticancer
agents (6,7).
Apoptosis is a highly regulated cellular process
that maintains the physiological balance of an organism by
eliminating unwanted and defective cells through an orderly process
of cell disintegration (8).
Apoptosis is characterized by morphological changes such as
membrane blebbing, cytoplasmic and chromatin condensation, and
apoptotic body formation (9).
Biochemical changes including cysteine-dependent aspartate-directed
protease (caspase) activation, DNA fragmentation, and
phosphatidylserine translocation are also associated with apoptosis
(10). The death receptor and
mitochondrial pathway are known to lead to apoptosis. The death
receptor pathway is triggered by members of the death receptor
super family. The formation of a death-inducing signaling complex,
clustered by CD95, leads to the recruitment of multiple
procaspase-8 molecules via the adapter molecule FADD
(Fas-associated death domain protein), resulting in caspase-8
activation. The mitochondrial pathway, which is regulated primarily
by the Bcl-2 family, may be caused by the opening of the
mitochondrial permeability transition pore, resulting in a decline
of mitochondrial membrane potential (MMP), failure of
Ca2+ control (11),
generation of reactive oxygen species (ROS) (12), and the release of pro-apoptotic
proteins (13). In the early stage
of apoptosis, the pro-apoptotic protein Bax translocates from the
cytosol to the mitochondria, leading to an efflux of cytochrome
c (14), which then binds to
Apaf-1 and forms the apoptosome. The apoptosome recruits and
activates procaspase-9, which in turn activates downstream
executioner caspases, such as caspase-3, −6, and −7 (15,16).
The cell cycle is defined as a set of events
responsible for cell duplication (17). The progression of cell cycle events
is monitored at checkpoints that occur at the G1/S boundary, during
the S phase, and during the G2/M phase (18). In a normal cell cycle, the M phase
always follows the S phase and does not occur until the S phase is
complete. The cell cycle checkpoints are activated by DNA damage
and by the misalignment of chromosomes at the mitotic spindle
(19). In this case, the growth
arrest caused by checkpoint mechanisms allows the cell to repair
the damage, after which progression through the cell cycle resumes.
If the damage cannot be repaired, the cell is eliminated through
apoptosis (20,21).
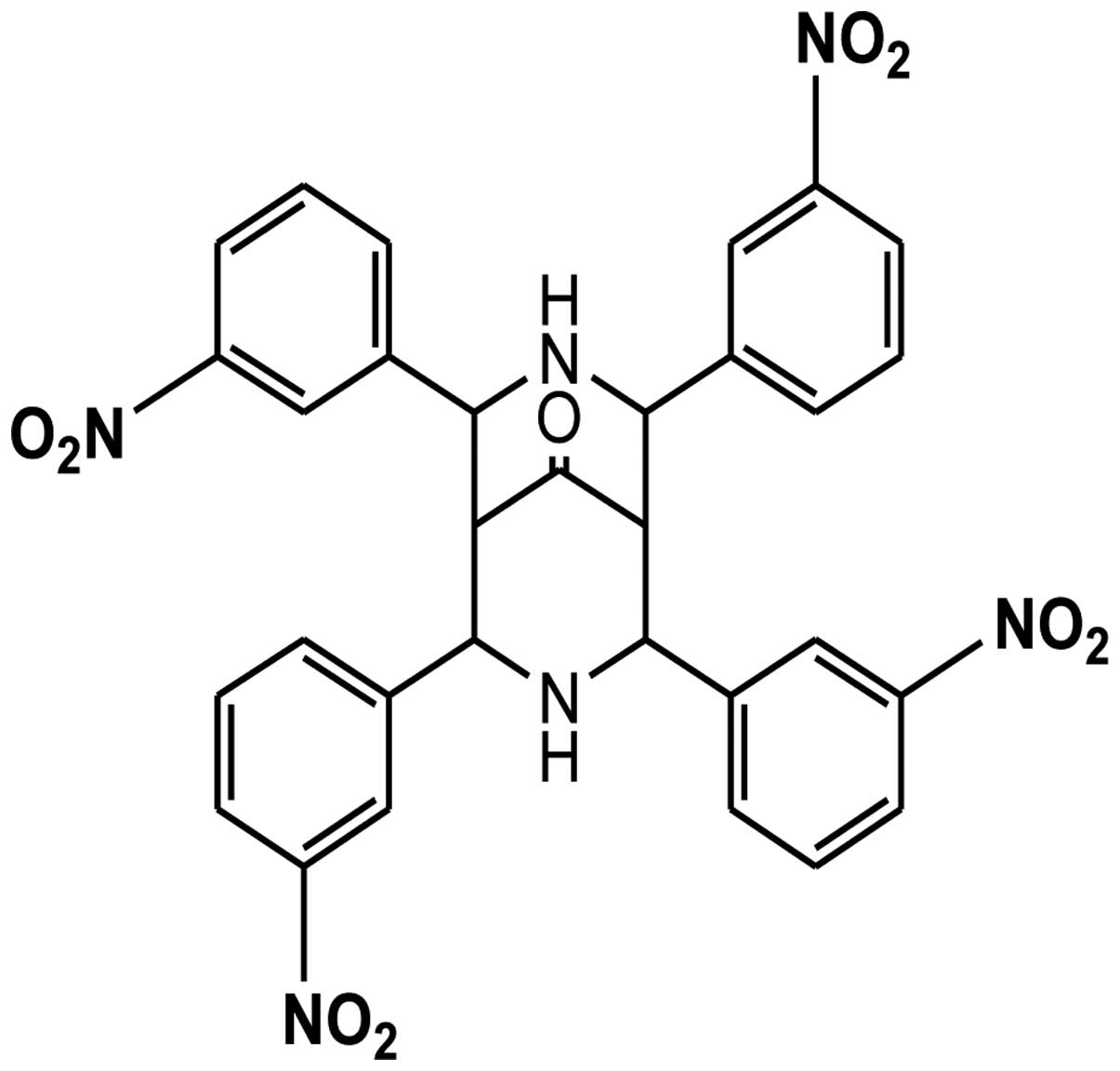
3,7-Diazabicyclo[3.3.1]nonane, known as bispidinone,
is of biological interest owing to its presence in the molecular
structure of various alkaloids, such as diterpene, norditerpene,
and lupin (22). The antibacterial
and antifungal efficiency of bispidinone analogs have led to their
use as templates for the improvement of antimicrobial agents
(23). Furthermore, it has been
confirmed that bispidinone analogs exert anticancer effects in HeLa
cells via mitochondrial-mediated apoptosis. In this study, we
determined the anticancer effects of a novel bispidinone analog,
2,4,6,8-(3)-tetranitrophenyl-3,7-diazabicyclo[3.3.1]nonan-9-one
(B16) (Fig. 1), and investigated
the underlying molecular mechanism of its activities.
Materials and methods
Chemicals
Bispidinone and bispidinone analogs were obtained
from the laboratory of Dr D.H. Park. Stock solution of the samples
was dissolved in dimethyl sulfoxide (DMSO) as 20 mM and kept at
4°C. Further dilutions were made immediately prior to each
experiment. Eagle’s minimal essential medium (EMEM),
penicillin-streptomycin and trypsin-EDTA were purchased from
HyClone (Logan, UT, USA). Fetal bovine serum (FBS) was obtained
from Gibco-BRL (Carlsbad, CA, USA). The Cell Counting Kit-8 (CCK-8)
was obtained from Dojindo (Osaka, Japan). The propidium iodide
(PI)/RNase staining buffer and Annexin-FITC kit for apoptosis were
from BD Pharmingen (San Diego, CA, USA).
Cell culture
HeLa cells were obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA) and were cultured in
EMEM supplemented with 10% FBS and 1% penicillin-streptomycin at
37°C in a humidified atmosphere with 5% CO2.
Cell viability and proliferation
assay
HeLa cells were plated at 5×103
cells/well in a 96-well microplate. After 24 h, the media were
substituted with fresh media containing B16 at concentrations of
7.5, 15 and 30 μM. The plate was incubated for 48 h and cell
viability was assessed using a WST-8 assay according to the
manufacturer’s recommendations (24). CCK-8 solution (10 μl) was added to
each well of the plate followed by 2-h incubation. The optical
density for viable cells was read at 450 nm in a multimicroplate
reader (Synergy HT; BioTek Instruments, Inc. Winooski, VT, USA).
For the effect of B16 on HeLa cell proliferation, the cells were
seeded at 5×103 cells/ml media in 96-well plates and
treated with or without B16 (25 μM) at various time points. Each
experiment was repeated at least three times.
Measurement of apoptotic cell
morphology
HeLa cells were distributed (2×105
cells/well) into a 6-well plate and allowed to adhere overnight.
The cells were treated with B16 (25 μM) for 24 and 48 h.
Non-treated wells received an equivalent volume of DMSO (<0.1%)
as a control. Optic phase-contrast images were captured with a
Phase Contrast-2, ELWD 0.3 inverted microscope (Nikon, Japan).
Annexin V-FITC/PI apoptotic analysis
Cells (3×105) in a 60-mm Petri dish,
treated with or without B16, were collected by trypsinization and
washed with ice-cold phosphate buffered saline (PBS) via
centrifugation at 2,500 g for 3 min. Cells (1×105) were
resuspended in 100 μl of binding buffer and stained with 5 μl of
Annexin V-FITC and 10 μl of PI (50 μg/ml) for 15 min at room
temperature, in the dark. Analysis was performed by FACSCalibur
flow cytometer (Becton-Dickinson, San Jose, CA, USA) with 10,000
events/analysis. The data were analyzed using Cell Quest Pro
software (Becton-Dickinson Instruments, Franklin Lakes, NJ,
USA).
Cell cycle arrest assay
Cells (3×105) in a 60-mm Petri dish,
treated with or without B16, were collected by trypsinization and
washed with ice-cold PBS via centrifugation as above. The cells
were suspended in PBS and fixed with 70% ethanol (v/v). Samples
were washed with ice-cold PBS and stained with PI/RNase staining
buffer for 15 min at room temperature. The number of cells in
different phases of the cell cycle was analyzed using a FACScan
flow cytometer analysis system and 20,000 events were analyzed each
time. The percentage of cells in different phases of the cell cycle
was determined using Modifit software (Becton-Dickinson
Instruments).
[3H]-thymidine incorporation
assay
Briefly, the HeLa cells were applied to 12-well
plates in growth medium (EMEM + 10% FBS + 1%
penicillin-streptomycin). After the cells had grown to 70–80%
confluence, they were rendered quiescent by incubation for 24 h in
EMEM containing 2% FBS. The cells were treated with or without B16
in EMEM supplemented with 10% FBS and the cultures were incubated
for 21 or 45 h. [3H]-dTTP was added at 1 μCi/ml (1
μCi=37 kBq) and incubated for an additional 3 h. Incorporated
[3H]-dTTP was extracted in cell lysis buffer and
measured in a liquid scintillation analyzer (Tris-Crab 2910TR;
Perkin-Elmer Inc., Waltham, MA, USA) (25).
Measurement of intracellular ROS
Generation of ROS was assessed using the fluorescent
indicator 2,7-dichlorodihy-drofluorescein (H2DCF-DA), a
cell-permeable indicator for ROS shown to react with
H2O2 (26).
As previously described, H2DCF-DA was oxidized to highly
green fluorescent 2,7-dichlorofluorescein (DCF) by the generation
of ROS. Cells (3×105) in a 60-mm dish, treated with or
without B16, were collected by trypsinization and centrifugation as
describe above. The samples were washed with ice-cold PBS and
stained with 2 μl H2DCF-DA for 30 min at 37°C in the
dark. Relative fluorescence intensities were monitored using the
FACSCalibur flow cytometer and analyzed using CellQuest software
with the FL-1 channel (green) set to 530 nm.
Measurement of mitochondrial-membrane
potential
Rhodamine 123 was used as a fluorescent probe to
detect the depolarization of mitochondrial-membrane potential.
During apoptosis, the integrity of the mitochondrial membrane is
disrupted, leading to depolarization of the membrane, opening of
mitochondrial permeability transition pores and release of
sequestered rhodamine 123. Cells (3×105) in a 60-mm
Petri dish), treated with or without B16, were collected by
trypsinization and centrifugation as described above. The samples
were washed with ice-cold PBS and stained with 5 μl rhodamine 123
for 30 min at 37°C in the dark. Relative fluorescence intensities
were monitored using a FACSCalibur flow cytometer and analyzed
using Cell Quest software with the FL-1 channel.
Western blot analysis
Following treatment of cells with or without B16,
total cell lysates was prepared as previously described. Protein
contents of the lysates were determined by the Bradford protein
assay (Bio-Rad, Hercules, CA, USA). Proteins (8 μg) were resolved
by sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto nitrocellulose membranes
(Schleicher & Schuell, BioScience, Inc., Keene, NH, USA) by
western blotting. The results were quantified using Image J v. 1.43
software (National Institutes of Health, Bethesda, MD, USA). The
following primary polyclonal antibodies were used: β-actin, Bcl-2,
procaspase-9, procaspase-8 (1:1,000 dilution; Cell Signaling
Technology Inc., Danvers, MA, USA), procaspase-3, p53 (1:300; Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and Bax (1:1,000; BD
Pharmingen, San Diego, CA, USA).
Statistical analysis
Results reported were obtained from at least three
independent experiments with similar results. The results are
presented as mean ± standard deviation (SD) in quantitative
experiments. Statistical analysis was performed by one-way analysis
of variance (ANOVA). *P<0.05 was considered to
indicate a statistically significant difference. Microsoft Excel
2007 was used for statistical and graphical evaluations.
Results
Cytotoxic effect of B16 in HeLa
cells
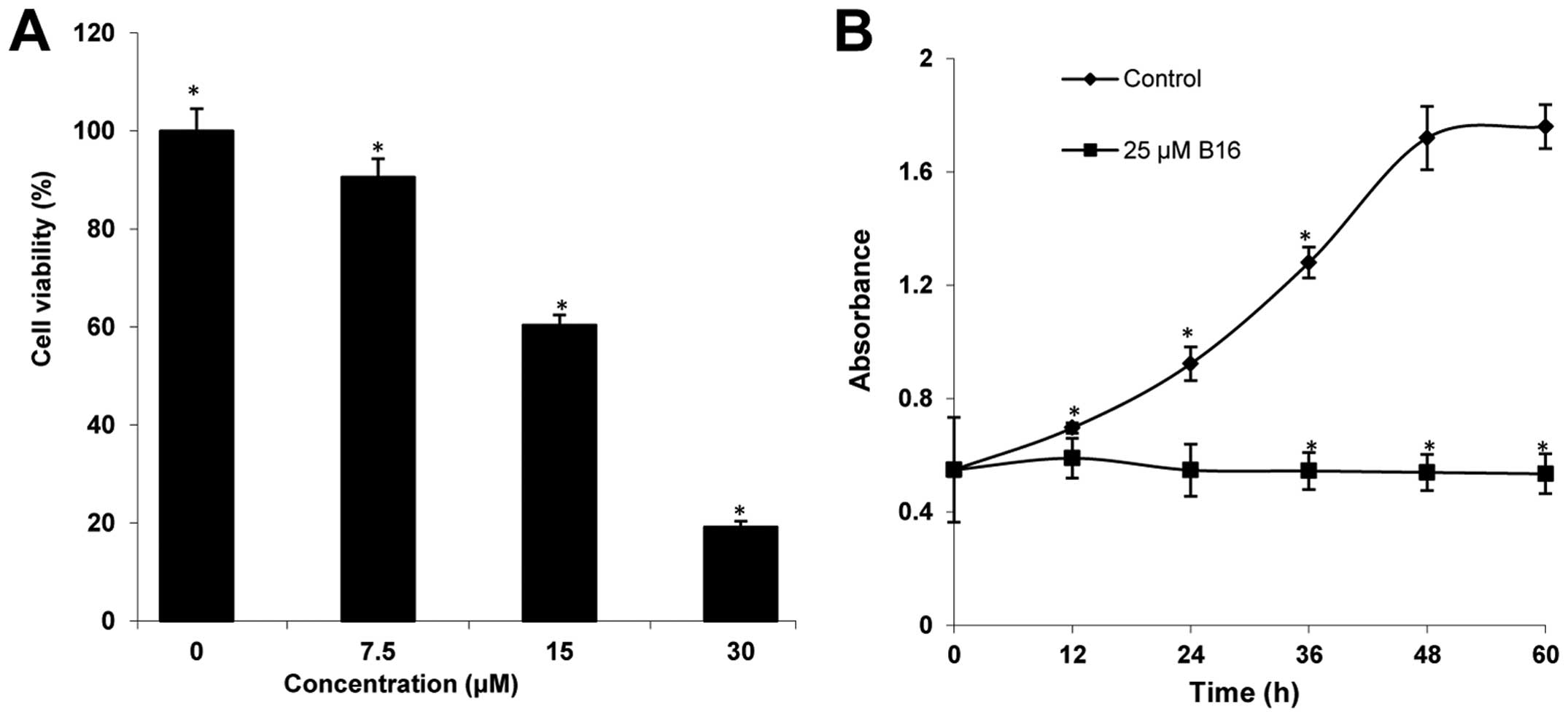
The cytotoxicity of B16 was determined using WST-8
assays. After incubation with increasing concentrations (7.5, 15,
and 30 μM) for 48 h, B16 showed significant cytotoxicity in HeLa
cells in a concentration-dependent manner, compared to the control
(Fig. 2A). The IC50
value of B16 was 22.66 μM. In addition, the proliferation of HeLa
cells treated with 25 μM of B16 was measured at 12-h intervals for
60 h. As shown in Fig. 2B, the
proliferation of HeLa cells treated with B16 continuously increased
during the first 12 h, and then the proliferation rate observed at
12 h was maintained until 60 h. By contrast, the control cells
maintained an exponential proliferation state throughout the 60-h
incubation. These results suggested that B16 reduced the viability
of HeLa cells in a dose- and time-dependent manner.
Morphological changes and early apoptosis
induced by B16 in HeLa cells
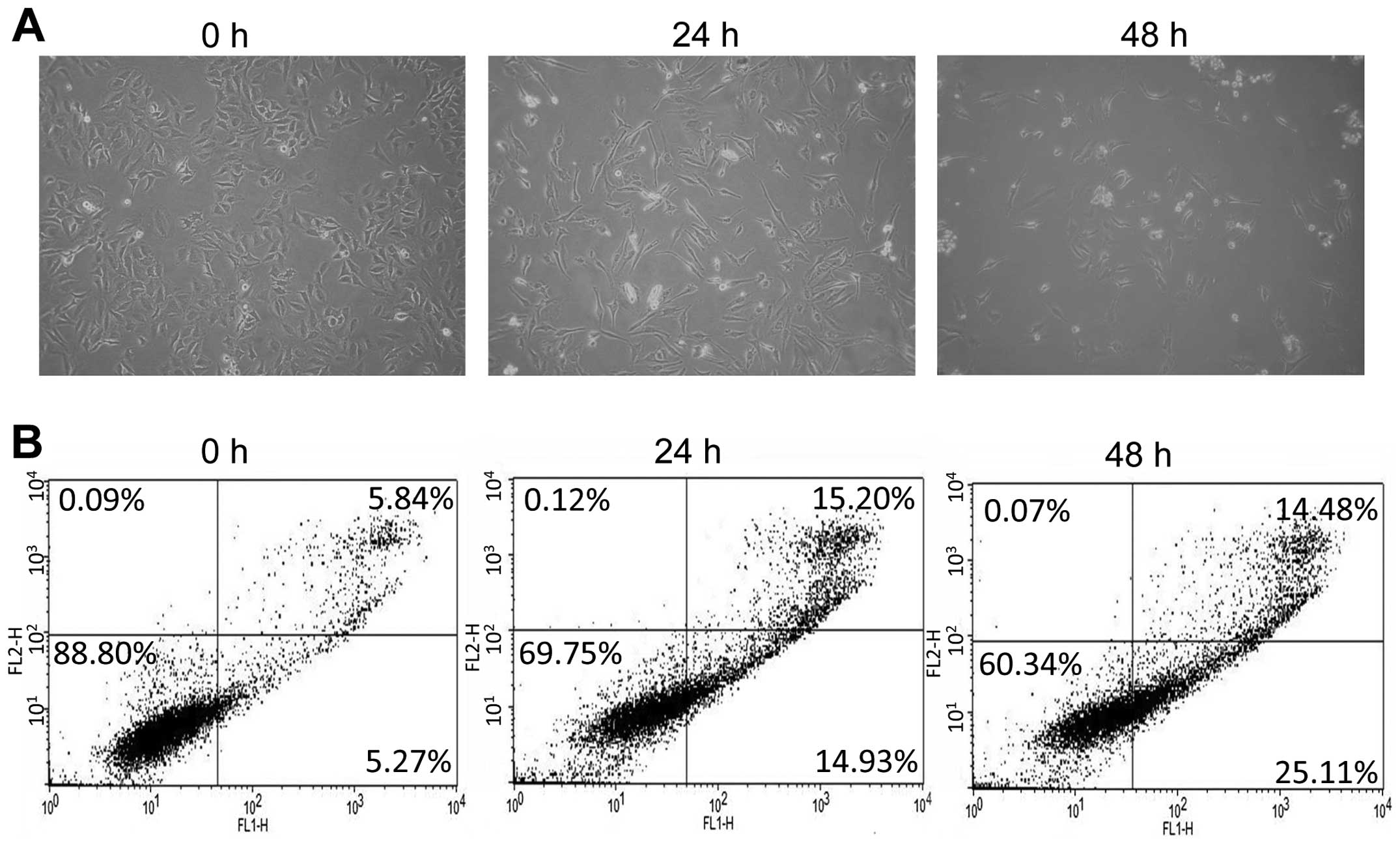
Morphological observation using light microscopy was
performed to study B16-induced apoptosis in HeLa cells. As shown in
Fig. 3A, the untreated cells spread
regularly in culture plates and grew to near confluence. After 24 h
of treatment with B16, some of the cells floated in the culture
plates, while the majority of the attached cells maintained a
shrunken shape. After 48 h of treatment, a significant proportion
of the HeLa cells dislodged from the plates and the remaining
adherent cells showed typical morphological changes, such as
shrinkage, floating, and an increase in intercellular space. To
assess the induction of apoptosis by B16 in HeLa cells, an Annexin
V and PI double staining assay was performed. A 48-h treatment with
B16 significantly increased the percentage of cells undergoing
early apoptosis from 5.27 (0 h) to 25.11% (48 h) (Fig. 3B). In addition, the percentage of
cells in late apoptosis (necrotic cells) was also increased by B16
treatment (Fig. 3B).
Induction of cell cycle arrest by
B16
Cell growth and inhibition are tightly mediated
through cell cycle control mechanisms (27). To examine the effects of B16 on the
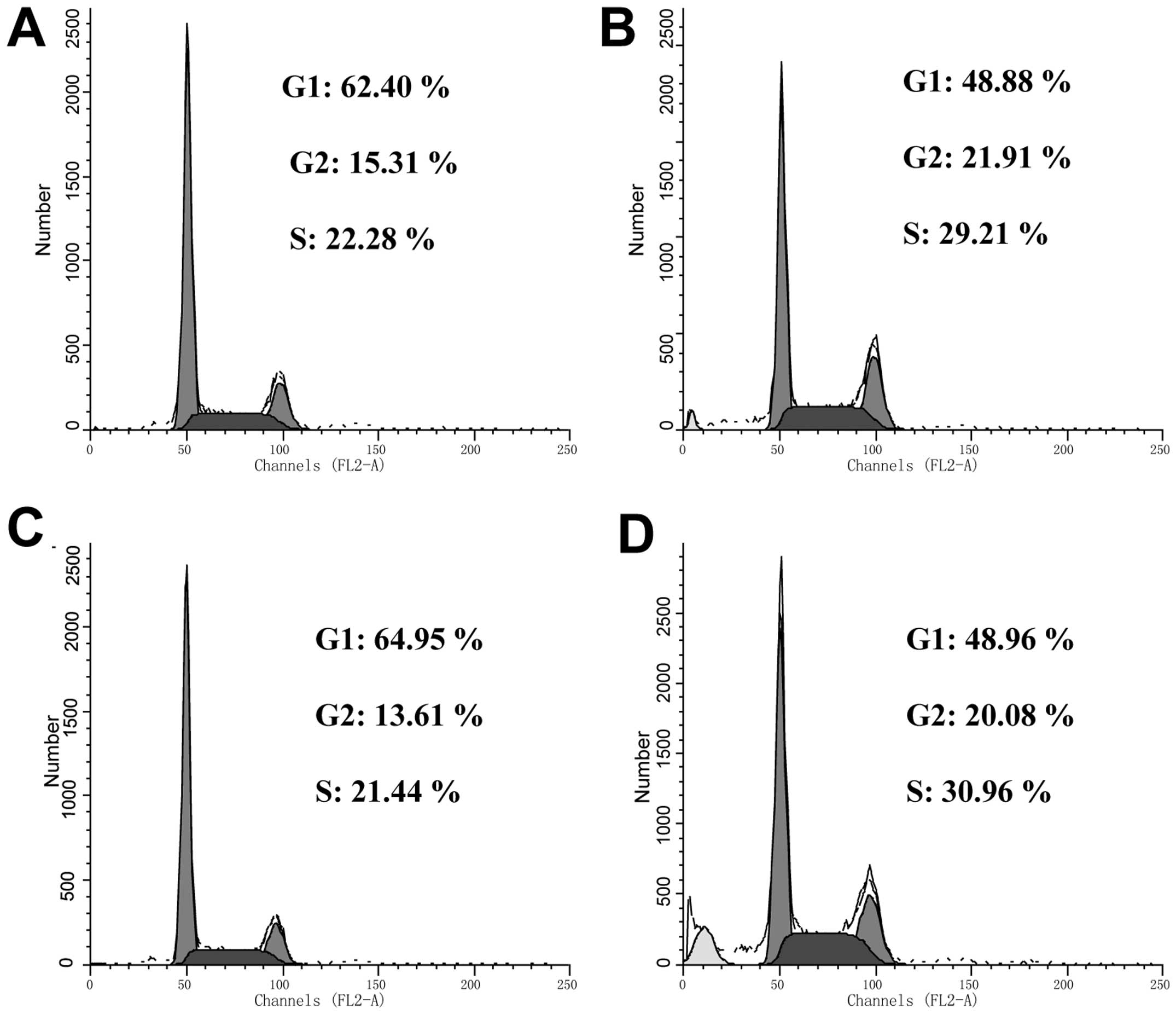
cell cycle, a DNA content assay was performed to analyze the cell
cycle distribution of HeLa cells treated with 25 μM B16. In the
cells undergoing apoptosis, a DNA peak that is characteristic of
apoptosis (usually known as the sub-G0/G1 peak or apoptotic peak)
appeared (Fig. 4). After 24 h of
treatment, B16 induced a significant accumulation of cells in the S
phase (21.44% in control cells vs. 29.21% in the B16-treated
cells). Concomitantly, there was a significant decrease of cells in
the G1 phase from 64.95 in the control cells to 48.88% in the
B16-treated cells. The level of S-phase arrest induced by B16 in
HeLa cells did not appear to be time dependent.
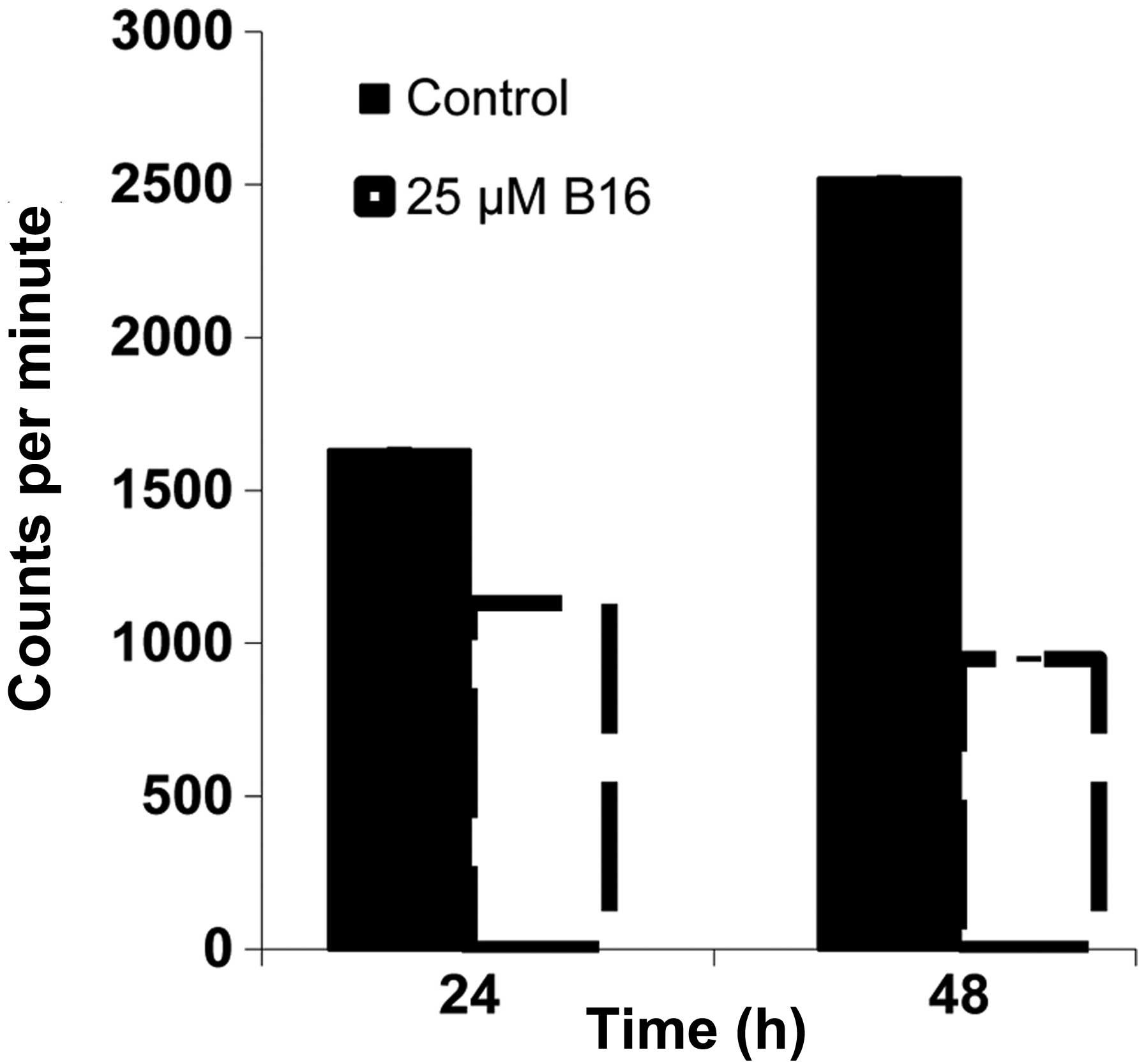
Inhibition of [3H]-thymidine
incorporation by B16 in HeLa cells
The results of the DNA content assay suggested that
HeLa cells treated with B16 accumulated in the S phase. To confirm
this hypothesis, we analyzed DNA replication in HeLa cells treated
with B16 by determining the incorporation of [3H]-dTTP.
In cells treated with B16 for 24 h and 48 h, [3H]-dTTP
incorporation was reduced by 30 and 60%, respectively, compared to
the controls (Fig. 5). This result
suggested that B16 had an inhibitory effect on the process of DNA
replication.
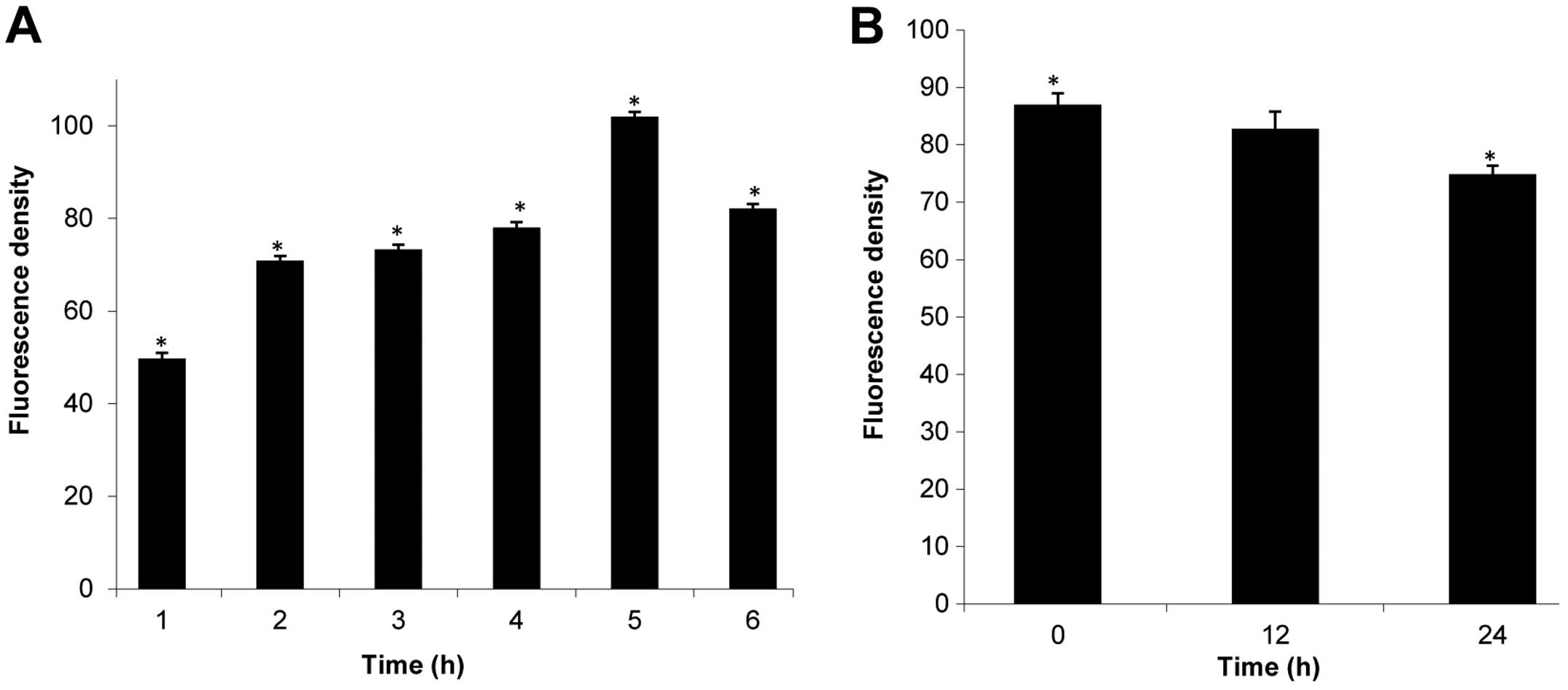
Effect of B16 on ROS generation and MMP
levels in HeLa cells
ROS plays an increasingly important role in
apoptosis-induced cell death (28).
Therefore, we determined the effect of B16 on ROS generation by
measuring the fluorescence of the cell-permeable dye
H2DCF-DA in HeLa cells during a 6-h treatment with 25 μM
B16. The mean H2DCF-DA fluorescence value increased from
49.78 (0 h) to 101.98 (3 h) following treatment with B16, whereas
the value of H2DCF-DA fluorescence decreased after the
3-h time point (Fig. 6A). These
results indicate that treatment with B16 induced changes in ROS
generation.
The generation of ROS is associated with the
disruption of the mitochondrial-membrane potential, and
mitochondrial-membrane depolarization is considered to be a crucial
hallmark of apoptosis. In our study, rhodamine 123 was used as a
fluorescent probe to detect the loss of MMP. Fig. 6B shows that the mean rhodamine 123
fluorescence decreased from 86.97 (0 h) to 82.8 (12 h) and 74.83
(24 h) following treatment with 25 μM B16, suggesting that
mitochondria were involved in the observed B16-induced
apoptosis.
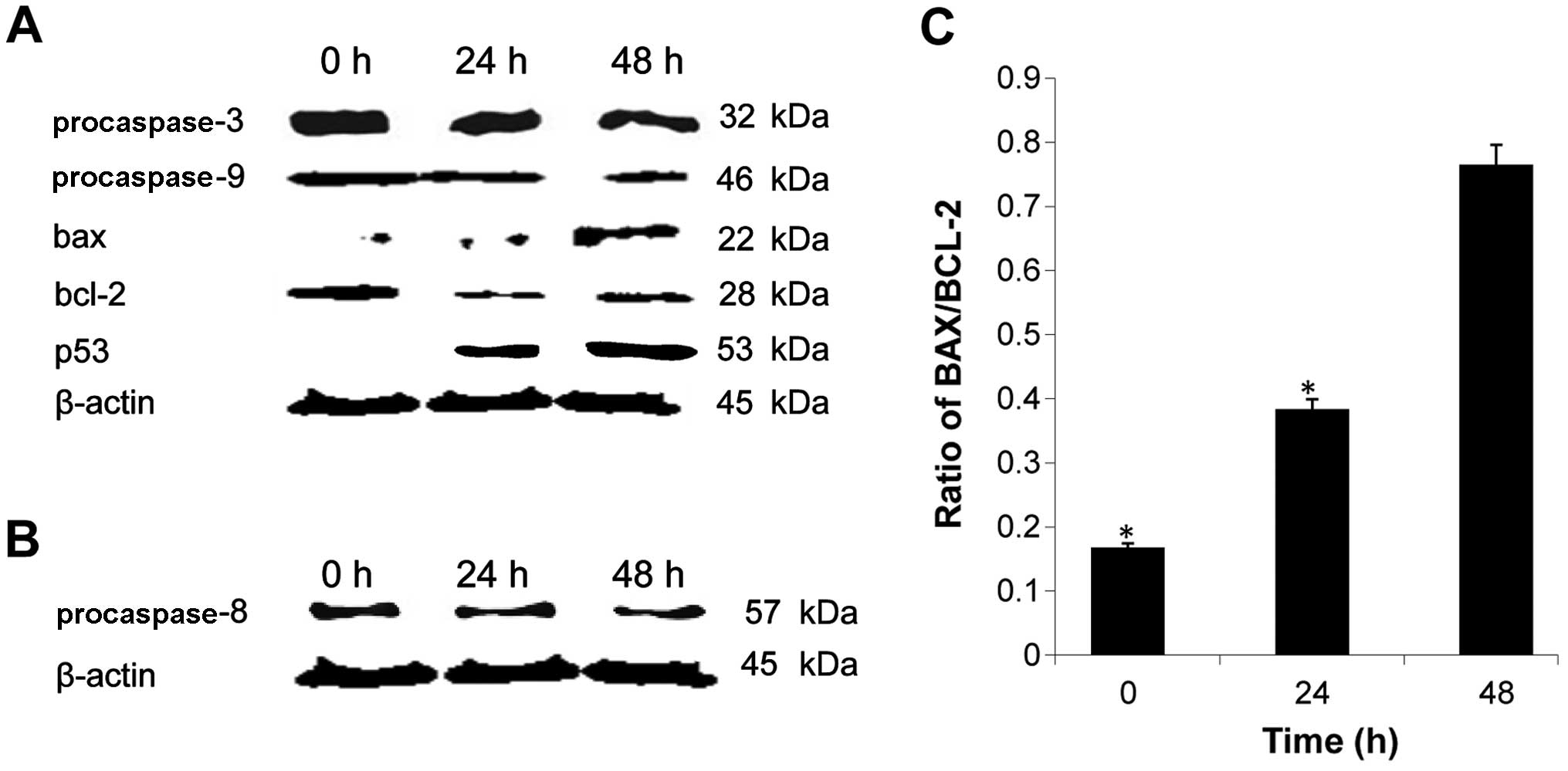
Involvement of protein activation in
B16-induced apoptosis
To investigate the underlying mechanism of apoptosis
induced by B16, the expression levels of several
apoptosis-associated proteins were determined by western blot
analysis. As shown in Fig. 7A, a
decrease in procaspase-3 and −9 expression levels was observed in
cells treated with B16, demonstrating the activation of the caspase
cascade in these cells. We also assessed the expression level of
p53, a known tumor suppressor, and found that it increased
following treatment with B16 (Fig.
7A). This result suggests that B16 suppresses tumor growth. B16
treatment also reduced the expression of procaspase-8 (Fig. 7B). As caspase-8 is known to be
involved in the Fas signaling apoptotic pathway, this result
suggested that B16-induced apoptosis is mediated by the Fas
signaling pathway. The expression level of the pro-apoptotic
protein Bax was significantly upregulated by B16 treatment in a
time-dependent manner, whereas the expression level of the
anti-apoptotic protein Bcl-2 showed a tendency for downregulation
compared to the control. Correspondingly, a marked increase in the
Bax/Bcl-2 ratio was observed (Fig.
7C). The changes in expression of Bcl-2 family members further
confirmed that the mitochondrial pathway is involved in B16-induced
apoptosis. Moreover, the caspase cascade is associated with the
intrinsic and extrinsic apoptotic pathways. Thus, taken together,
these results suggested that B16 induced apoptosis in HeLa cells
via a caspase- and mitochondrial-dependent pathway.
Discussion
Bispidinone (3,7-diazabicyclo[3.3.1]nonane) is
present in the molecular structure of various alkaloids (e.g.,
diterpene, norditerpene and lupin); thus, this compound is of
significant biological interest. To the best of our knowledge,
there is no reported evidence for the use of bispidinone for cancer
treatment, whereas its cytotoxicity has been documented. B16 is a
novel bispidinone analog that can significantly suppress the
proliferation of HeLa cells in a dose- and time-dependent manner.
To the best of our knowledge, this is the first study on B16
induction of apoptosis in human cervical cancer cells and suggests
a potential therapeutic role for B16 in the treatment of cervical
cancer.
To examine the anticancer effects of B16 and ensure
the accuracy of the results, we used several methods in this study
to determine the effects of B16 on apoptosis. Cell cycle arrest and
apoptosis can be induced by inhibitor-mediated blockage of DNA
replication or DNA template damage due to radiation or other
factors (29,30). In this study, a DNA content assay
determined that B16 inhibited HeLa cell proliferation via arrest of
the cell division cycle during the S phase, in which cells
replicate their DNA (31).
Moreover, [3H]-thymidine incorporation was inhibited by
B16 treatment, indicating that the S-phase arrest was caused by
inhibition of DNA replication. B16 also induced clear
apoptosis-associated morphological changes, and the result of
Annexin V-FITC/PI double staining indicated that B16 induced early
apoptosis in HeLa cells.
The B16-induced HeLa cell apoptosis observed in the
present study was associated with a significant increase in the
levels of intracellular ROS and a disruption of MMP. The Bcl-2
family proteins Bax and Bcl-2 play important roles in the
mitochondrial-mediated apoptotic pathway (32,33).
Bcl-2 prevents apoptosis by preserving mitochondrial integrity, and
the ratio of Bax to Bcl-2 is crucial to sustaining drug-induced
apoptosis via the mitochondrial-mediated apoptotic pathway
(34). The present study has
demonstrated that B16 upregulated the expression level of Bax and
downregulated the expression level of Bcl-2, leading to an increase
in the ratio of Bax/Bcl-2 protein levels. These findings add to the
increasing evidence that Bcl-2 family proteins play central roles
in apoptosis.
In the intrinsic pathway of apoptosis, caspase
activation is closely linked to the permeabilization of the outer
mitochondrial membrane. The release of mitochondrial cytochrome
c facilitates the formation of the apoptosome complex,
consisting of Apaf-1 and caspase-9, which subsequently activates
effector caspases such as caspase-3 and leads to apoptosis
(35,36). In the present study, caspase
activation was determined by western blot assays using antibodies
against activated procaspase-3, −8, and −9. The activation of
caspase-3 and −9 was detected, indicating that B16 induced
apoptosis via the intrinsic pathway. However, B16 also reduced the
expression of caspase-8, which is an initiator caspase first
activated through an external signal for cell death. Moreover, the
level of p53, a well-known tumor-suppressor protein, was increased.
Taken together, these results suggest that B16 induced apoptosis
via the intrinsic and extrinsic pathways.
In conclusion, our results have demonstrated that
B16 exerted an inhibitory effect on the growth of HeLa human
cervical cancer cells, and that this effect was associated with the
induction of S-phase arrest and the promotion of apoptosis. B16
induced apoptosis via an increase in ROS generation and
depolarization of the mitochondrial membrane. Moreover, caspase
cascades were activated following B16 treatment, confirming that
the apoptosis induced by B16 was associated with the intrinsic and
extrinsic pathways. The results of our study suggest that B16
suppresses cancer cell proliferation, induces cancer cell
apoptosis, and inhibits cancer cell growth.
Acknowledgements
The authors would like to thank Dr Paramasivam
Parthiban for providing bispidinone analogs. This study was partly
supported by NRF-2011-0010160.
References
|
1
|
Bray F, Jemal A, Grey N, Ferlay J and
Forman D: Global cancer transitions according to the Human
Development Index (2008–2030): a population-based study. Lancet
Oncol. 13:790–801. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith HO, Tiffany MF, Qualls CR and Key
CR: The rising incidence of adenocarcinoma relative to squamous
cell carcinoma of the uterine cervix in the United States - a
24-year population-based study. Gynecol Oncol. 78:97–105. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rein DT and Kurbacher CM: The role of
chemotherapy in invasive cancer of the cervix uteri: current
standards and future prospects. Anticancer Drugs. 12:787–795. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gascoigne KE and Taylor SS: Cancer cells
display profound intra-and interline variation following prolonged
exposure to antimitotic drugs. Cancer Cell. 14:111–122. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Preg. 19:1893–1907. 2010.
View Article : Google Scholar
|
|
6
|
Sherwood SW, Sheridan JP and Schimke RT:
Induction of apoptosis by the anti-tubulin drug colcemid:
relationship of mitotic checkpoint control to the induction of
apoptosis in HeLa S3 cells. Exp Cell Res. 215:373–379. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pucci B, Kasten M and Giordano A: Cell
cycle and apoptosis. Neoplasia. 2:2912000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiao J, Huang G, Zhu C, Ren D and Zhang S:
Morphological study on apoptosis HeLa cells induced by
soyasaponins. Toxicol In Vitro. 21:820–826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dewson G and Kluck RM: Mechanisms by which
Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci.
122:2801–2808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zamzami N and Kroemer G: The mitochondrion
in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol.
2:67–71. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simizu S, Takada M, Umezawa K and Imoto M:
Requirement of caspase-3(-like) protease-mediated hydrogen peroxide
production for apoptosis induced by various anticancer drugs. J
Biol Chem. 273:26900–26907. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu MJ, Wang Z, Li HX, Wu RC, Liu YZ and
Wu QY: Mitochondrial dysfunction as an early event in the process
of apoptosis induced by woodfordin I in human leukemia K562 cells.
Toxicol Appl Pharmacol. 194:141–155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: a
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Putcha GV, Harris CA, Moulder KL, Easton
RM, Thompson CB and Johnson EM: Intrinsic and extrinsic pathway
signaling during neuronal apoptosis lessons from the analysis of
mutant mice. J Cell Biol. 157:441–453. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Day TW, Huang S and Safa AR: c-FLIP
knockdown induces ligand-independent DR5-, FADD-, caspase-8-, and
caspase-9-dependent apoptosis in breast cancer cells. Biochem
Pharmacol. 76:1694–1704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Graña X and Reddy EP: Cell cycle control
in mammalian cells: role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
18
|
Hall M and Peters G: Genetic alterations
of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human
cancer. Adv Cancer Res. 68:67–108. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dulić V, Kaufmann WK, Wilson SJ, Tisty TD,
Lees E, Harper JW, Elledge SJ and Reed SI: p53-dependent inhibition
of cyclin-dependent kinase activities in human fibroblasts during
radiation-induced G1 arrest. Cell. 76:1013–1023. 1994. View Article : Google Scholar
|
|
20
|
MacLachlan TK, Sang N and Giordano A:
Cyclins, cyclin-dependent kinases and cdk inhibitors: implications
in cell cycle control and cancer. Crit Rev Eukaryot Gene.
5:127–156. 1995. View Article : Google Scholar
|
|
21
|
Murray A: Cell cycle checkpoints. Curr
Opin Cell Biol. 6:872–876. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parthiban P, Aridoss G, Rathika P,
Ramkumar V and Kabilan S: Synthesis, stereochemistry and
antimicrobial studies of novel oxime ethers of aza/diazabicycles.
Bioorg Med Chem Lett. 19:6981–6985. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haridas V, Rajgokul KS, Sadanandan S,
Agrawal T, Sharvani V, Gopalakrishna M, Bijesh M, Kumawat KL, Basu
A and Medigeshi GR: Bispidine-amino acid conjugates act as a novel
scaffold for the design of antivirals that block Japanese
encephalitis virus replication. PLoS Negl Trop Dis. 7:20052013.
View Article : Google Scholar
|
|
24
|
Tominaga H, Ishiyama M, Ohseto F, Sasamoto
K, Hamamoto T, Suzuki K and Watanabe M: A water-soluble tetrazolium
salt useful for colorimetric cell viability assay. Anal Commun.
36:47–50. 1999. View
Article : Google Scholar
|
|
25
|
Lin SY, Liu JD, Chang HC, Yeh SD, Lin CH
and Lee WS: Magnolol suppresses proliferation of cultured human
colon and liver cancer cells by inhibiting DNA synthesis and
activating apoptosis. J Cell Biochem. 84:532–544. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bobyleva V, Pazienza TL, Maseroli R,
Tomasi A, Salvioli S, Cossarizza A, Franceschi C and Skulachev VP:
Decrease in mitochondrial energy coupling by thyroid hormones: a
physiological effect rather than a pathological hyperthyroidism
consequence. FEBS Lett. 430:409–413. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Collins KL and Kelly TJ: Effects of T
antigen and replication protein A on the initiation of DNA
synthesis by DNA polymerase alpha-primase. Mol Cell Biol.
11:2108–2115. 1991.PubMed/NCBI
|
|
28
|
Tang XQ, Feng JQ, Chen J, Chen PX, Zhi JL,
Cui Y, Guo RX and Yu HM: Protection of oxidative preconditioning
against apoptosis induced by H2O2 in PC12 cells: mechanisms via
MMP, ROS, and Bcl-2. Brain Res. 1057:57–64. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shiff SJ, Qiao L, Tsai LL and Rigas B:
Sulindac sulfide, an aspirin-like compound, inhibits proliferation,
causes cell cycle quiescence, and induces apoptosis in HT-29 colon
adenocarcinoma cells. J Clin Invest. 96:491–503. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boehme SA and Lenardo MJ: Propriocidal
apoptosis of mature T lymphocytes occurs at S phase of the cell
cycle. Eur J Immunol. 23:1552–1560. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang HS, Gavin M, Dahiya A, Postigo AA,
Ma D, Luo RX, Harbour JW and Dean DC: Exit from G1 and S phase of
the cell cycle is regulated by repressor complexes containing
HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 101:79–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thomas A, El Rouby S, Reed JC, Krajewski
S, Silber R, Potmesil M and Newcomb EW: Drug-induced apoptosis in
B-cell chronic lymphocytic leukemia: relationship between p53 gene
mutation and bcl-2/bax proteins in drug resistance. Oncogene.
12:1055–1062. 1996.PubMed/NCBI
|
|
33
|
Yin X, Oltvai ZN and Korsmeyer SJ: BH1 and
BH2 domains of Bcl-2 are required for inhibition of apoptosis and
heterodimerization with Bax. Nature. 369:321–323. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miyashita T, Krajewski S, Krajewska M,
Wang HG, Lin H, Liebermann DA, Hoffman B and Reed JC: Tumor
suppressor p53 is a regulator of bcl-2 and bax gene expression in
vitro and in vivo. Oncogene. 9:1799–1805. 1994.PubMed/NCBI
|
|
35
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saitoh M, Nagai K, Nakagawa K, Yamamura T,
Yamamoto S and Nishizaki T: Adenosine induces apoptosis in the
human gastric cancer cells via an intrinsic pathway relevant to
activation of AMP-activated protein kinase. Biochem Pharmacol.
67:2005–2011. 2004. View Article : Google Scholar : PubMed/NCBI
|