Introduction
The epithelial-mesenchymal transition (EMT) program
is the differentiation switch epithelial polarized cells into
motile mesenchymal cells, which plays a key role during various
biological events such as embryonic development, fibrotic diseases
and tumor metastasis. EMT enables cells to acquire fibroblast-like
properties and shows reduced intercellular adhesion and increased
motility (1–5). Furthermore, EMT is dysregulated in
cancer cells and is characterized by the acquisition of mesenchymal
phenotype, leading to enhanced motility and allow tumor cells to
metastasize and establish secondary tumors at distant sites
(3,6). Additionally, EMT contributes to
enhanced chemoresistance and the acquisition of
stem/progenitor-like characteristics by regulating signaling
pathways (7). Findings of those
studies suggest that EMT is involved in the modulation of drug
resistance and enhances the ability of tumor metastasis.
Transforming growth factor-β (TGF-β), a secreted
cytokine, was found to act as a suppressor in tumor cell growth
control, however, it may also promote tumor invasion and metastasis
(8). Accumulating evidence reveals
that TGF-β is often used as the inducer of EMT (9,10) to
increase tumor metastasis. Although several molecules involved in
TGF-β-mediated EMT have been identified, the mechanisms involved in
the induction of EMT remain to be determined.
Cathepsin L is a cysteine protease that belongs to
the papain-like family (peptidase C1A), which is reported to be
associated with cancer tumorigenesis, proliferation and migration
(11–14). It plays an important role in the
degradation and renewal of intracellular proteins, and is involved
in several important physiological processes including the
activation of prohormone, presenting antigens and development of
organs (15–17). Cathepsin L has been proven to be
upregulated in a variety of malignancies: breast, lung, gastric,
colon, melanomas and gliomas. Moreover, the level of cathepsin L
expression is associated with the degree of malignancy (18). Results of previous studies showed
that cathepsin L inhibition increases apoptosis via the
responsiveness of IGF-1 receptor (19), or the activity of arsenite in U87
cells (20). Simultaneously, in
cathepsin L antisense clones of U87 cells, the apoptotic rate is
increased when induced by intrinsic or extrinsic stimulation
(21). Furthermore, accumulating
evidence suggests cathepsin L has been associated with tumor
invasion and migration (22,23).
When the extracellular activity of cathepsin L is increased, the
cell-cell adhesion is reduced and degradation of the ECM is
increased (24,25–27).
In addition, overexpression of cathepsin L was contributed to the
increased malignant properties following the Ras-transformation of
NIH/3T3 cells (28). Frade et
al, Yang et al and Rousselet et al proved that
the upregulation of cathepsin L also may switch the melanoma cell
phenotype from non-metastatic to highly metastatic and increased
tumor invasion and migration (29–31).
The aforementioned studies showed that cathepsin L increased the
invasion and migration of ovarian cancer cells and may therefore be
a molecular target for cancer metastasis (32).
Cathepsin L is known to regulate tumor progression,
invasion and migration. However, the mechanisms involved in the
regulation of tumor invasion and migration remain to be determined.
Findings of previous studies showed that cathepsin L may contribute
its proteolytic action to the migration and invasion of tumor cells
(24–27). Increased activity of cathepsin L
decreased the cell-cell adhesion and enhanced degradation of the
ECM, suggesting that downregulation of cathepsin L decreased
metastatic tumor development (33).
However, Goulet et al suggested that the role of cathepsin L
in cancer metastasis may be associated with its extracellular
activities and may involve its processing function of the
transcription factor CUTL1 (34).
Other authors reported that cathepsin L is also involved in
proteolytic activation cascades, which include u-PA and cathepsin
B. Of note, this proteolytic network plays an important role in
tumor invasive processes (35). On
the other hand, it has been reported that inhibition of cathepsin L
did not affect invasion of 8863 and LM melanoma cell lines
(36). Nevertheless, the exact role
of cathepsin L in tumor cell invasion and migration remains
unidentified, and many studies suggest that it is
context-dependent.
On the basis of the crucial role of cathepsin L and
its involvement in tumor invasion and migration, we investigated
the effect of cathepsin L knockdown on tumor invasion and migration
and whether cathepsin L enhanced tumor via invasion and migration
TGF-β-mediated EMT. We demonstrated that cathepsin L is a regulator
of tumor invasion and migration in cancer cells, and the regulation
of tumor invasion and migration by cathepsin L is mediated through
EMT. Thus, cathepsin L is a novel target for reducing tumor
progression.
Materials and methods
Materials
Cell culture reagents and the Lipofectamine reagent
were purchased from Invitrogen Life Technologies (Carlsbad, CA,
USA). Human recombinant TGF-β was purchased from R&D Systems
(Minneapolis, MN, USA). Phalloidin was obtained from Sigma-Aldrich
(St. Louis, MO, USA). The antibodies used in this study were:
N-cadherin (Abcam, Cambridge, MA, USA); E-cadherin (BD Biosciences,
Franklin Lakes, NJ, USA); cathepsin L (Abcam); Akt and p-Akt (both
from Cell Signaling Technology, Danvers, MA, USA); Snail and Wnt-5a
(both from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA);
β-actin (MultiSciences Biotech Hangzhou, China); and Cxcr-7 (Anbo
Biotechnology, Sunnyvale, CA, USA).
Cell culture
A549 and MCF-7 cells and their derivative lines were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS), penicillin (100 U/ml)/streptomycin (100 U/ml), and 300 or
500 ng/ml of puromycin at 37°C in a humidified atmosphere of 5%
CO2. All the knockdown and control sublines from these
cell lines were generated using cathepsin L shRNA plasmid and
control vectors shRNA plasmid (both from Santa Cruz Biotechnology,
Inc.) under puromycin selection.
Wound-healing and invasion assays
For the wound-healing assay the cells were grown in
6-well plates. When confluency was achieved, the cells were
scratched by a pipette tip, rinsed to remove debris, then further
incubated with fresh RPMI-1640 containing 1% FCS in the presence or
absence of TGF-β for 24 h. Cell migration images were captured at 0
and 24 h, quantitative analysis of the wound-healing index
determined as a percentage was calculated using 20 randomly
selected distances across the wound at 0 and 24 h, divided by the
distance measured at 0 h.
A cell invasion assay was performed using 24-well
Matrigel invasion chambers (BD Biosciences). The cells were
trypsinized and reseeded in the upper chamber at a concentration of
1×105/ml in 200 μl of RPMI-1640 supplemented with 1%
FCS. The lower chamber contained 700 μl of RPMI-1640 supplemented
with 10% FCS. After 24 h, the cells on the upper surface of the
filters were removed, and the cells on the lower surface were fixed
with methanol and stained with crystal violet.
Determination of Snail mRNA levels by
reverse transcription-PCR
Cells were grown in 6-well plates, and treated or
not with TGF-β for 24 h. Total RNA was isolated with TRIzol reagent
according to the manufacturer’s instructions. RNA was reverse
transcribed and amplified by PCR using the primers: Cathepsin L
upstream: 5′-AAACACAGCTTCACA ATGGCC-3′ and downstream:
5′-TTTGAAAGCCATTCA TCACCTG-3′; Snail upstream: 5′-CCTCCCTGTCAGATG
AGGAC-3′ and downstream: 5′-CCAGGCTGAGGTATTCC TTG-3′. The
amplification products were analyzed in a 1.0% agarose gel.
Immunofluorescence
Twenty-four hours after seeding on coverslips with
TGF-β the cells were fixed with methanol for 10 min at 4°C and
permeabilized for 10 min with 0.1% Triton X-100. The cells were
then incubated for 1 h in blocking buffer (1% BSA and 0.1% Triton
X-100) at 4°C. For immunofluorescence (IF), the cells were
incubated with antibodies against E- and N-cadherin at 4°C
overnight. The cells were then rinsed three times with PBS, and
incubated with appropriate biotinylated secondary antibodies for 1
h. Alex Fluor 488 (green) (1:500; Molecular Probes, Eugene, OR,
USA) was used as a third antibody for 1 h. Nuclei were
counterstained with 0.5 ng/ml DAPI for 15 min at room temperature.
Coverslips were mounted on slides with vectashield mounting medium
for fluorescence and analyzed by confocal microscopy.
Actin cytoskeleton staining
Cells were fixed for 10 min in 4% paraformaldehyde,
permeabilized with 0.2% Triton X-100 in PBS for 10 min, blocked in
2% BSA for 30 min, and incubated for 1 h with phalloidin. The
nuclei were counterstained with DAPI.
Western blot analysis
The samples were harvested and lysed with ice-cold
lysis buffer. The protein concentration of the lysates was
determined by Protein Assay Kit (Thermo Fisher Scientific,
Rockford, IL, USA). The lysates were loaded and separated on 10%
SDS-PAGE gels and the proteins were transferred onto a
polyvinylidene difluoride membrane. The membrane was blocked in 5%
BSA for 1 h, and incubated with primary antibodies overnight. After
washing three times, the blots were incubated with secondary
antibodies for 1 h. Immunoblots were detected by the Odyssey
Infrared Imaging System (Li-COR Biosciences, Lincoln, NE, USA).
In vivo assay and immunohistochemical
staining
Five-week-old male nude (BALB/c) mice (Animal
experiment Center of Suzhou University, Suzhou) wsere used in the
present study A549-shVector and A549-shCathepsin L cells
(5×107 cells/0.1 ml of medium/mouse) were injected into
mice subcutaneously to generate the mouse model. On day 42 after
tumor injection, the tumors were removed from mice and analyzed for
western blotting and immunohistochemical staining. The samples were
stained using VECTASTAIN ABC kit and ImmPACT DAB (Vector
Laboratories, Burlingame, CA, USA) according to the supplier’s
instructions. Tumor size was measured with a caliper and was
calculated using the formula: π/6 × larger diameter × (smaller
diameter)2.
Results
TGF-β induces morphological changes and
enhances the expression of cathepsin L in cancer cells
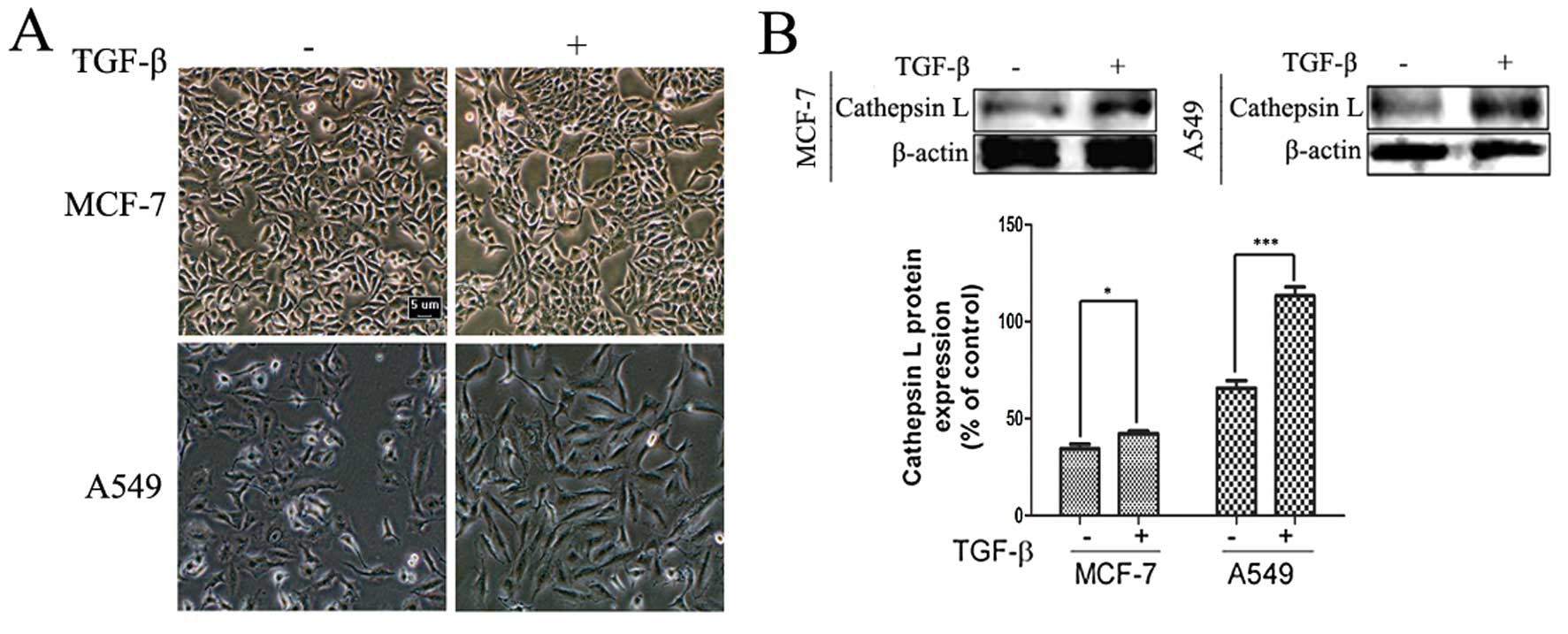
To determine the effect of TGF-β on cell morphology
and the expression level of cathepsin L, we observed the
morphological changes and examined the activity of invasion and
migration in A549 human lung and in human MCF-7 breast cancer cell
lines treated with TGF-β for 24 h. As shown in Fig. 1A, treatment of the human A549 and
MCF-7 cancer cell lines with TGF-β revealed morphological changes,
with a more scattered and spindle-shaped appearance, especially in
A549 cells (Fig. 1A). TGF-β also
significantly increased the expression of cathepsin L, which has
been reported to play an important role in cancer invasion and
migration (Fig. 1B). Furthermore,
TGF-β induces EMT characteristics such as loss of cell-cell
adhesion, reorganization of actin cytoskeleton and acquisition of
increased migratory characteristics and promotion of cell migration
(3,37). On the basis of previous exploration,
an association was identified between cathepsin L and the EMT
induced by TGF-β. Thus, we determined whether cathepsin L is a
target of TGF-β in regulating EMT, which regulates tumor invasion
and migration.
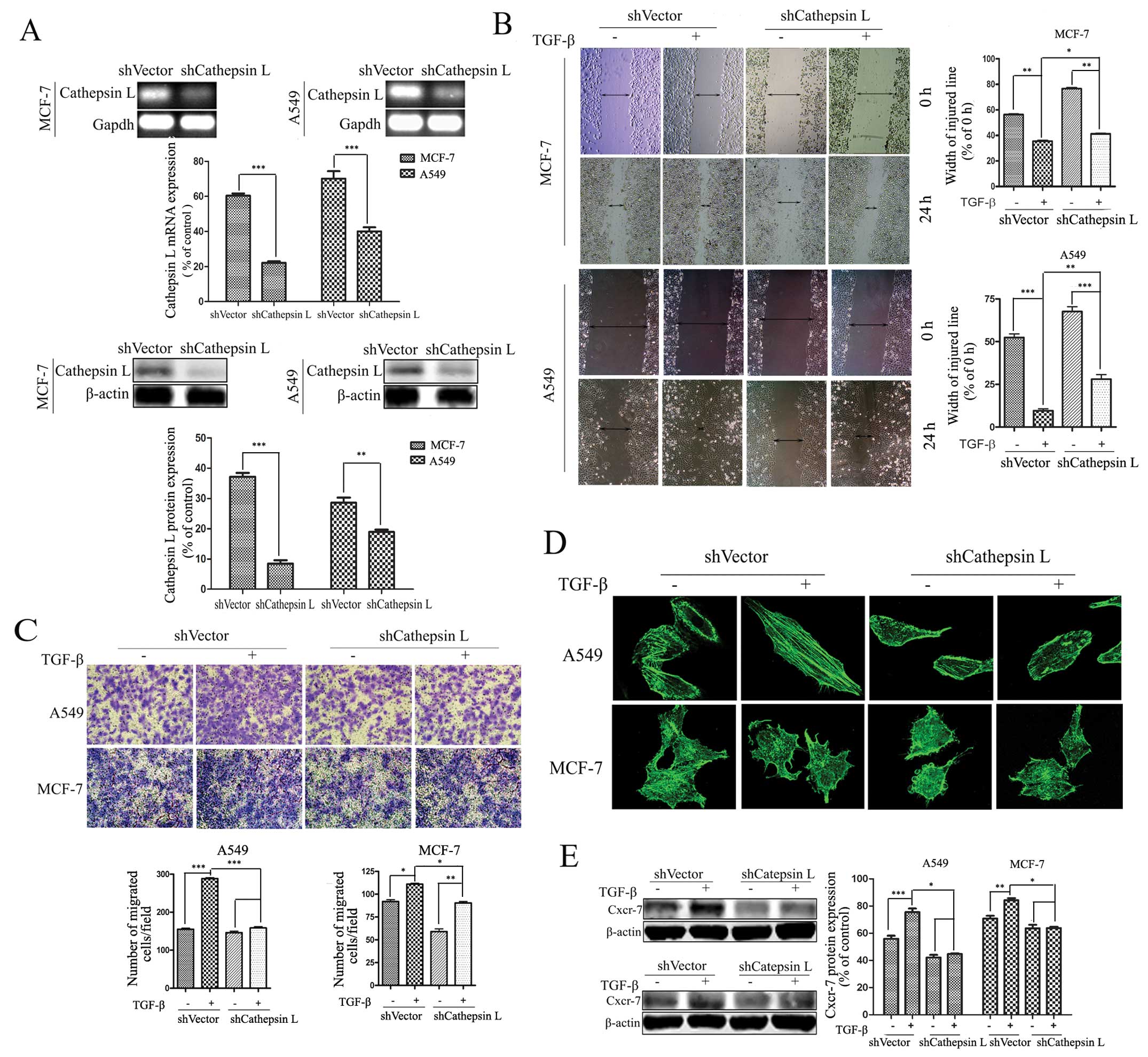
Cathepsin L knockdown attenuates
TGF-β-induced cell migration and invasion
The regulation of cathepsin L by TGF-β suggests that
cathepsin L is involved in TGF-β-induced cell migration and
invasion. To verify this role of cathepsin L, shRNA expression
vectors were introduced into A549 and MCF-7 cells, and stably
transfected cell clones were obtained by limiting dilution culture
under the pressure of puromycin. RT-PCR and western blot analysis
were conducted to reveal loss of cathepsin L expression in RNA and
protein levels in the cathepsin L knockdown (shcathepsin L) cells
(Fig. 2A). In tumor cell lines
treated with TGF-β, respectively, the effect of cathepsin L
knockdown on migration and invasion was determined by a
wound-healing assay. The results showed an increase in the width of
the injured line. Results of the colony formation assay exhibited a
decrease in the number of colonies, suggesting that suppression of
cathepsin L inhibits TGF-β-induced cell migration and invasion in
A549 and MCF-7 cells (Fig. 2B and
C). The effect of cathepsin L was more significant in A549 than
in MCF-7 cells. Furthermore, we demonstrated that suppression of
cathepsin L markedly suppressed TGF-β-induced actin remodeling
associated with cell motility in A549 and MCF-7 cells (Fig. 2D). In addition, emerging evidence
suggests that multiple pairs of chemokines and their receptors are
likely to play important roles in mediating tumor growth and
metastasis (38). TGF-β treatment
enhanced the expression of Cxcr-7, one of the chemokine receptors,
and this effect was inhibited by cathepsin L knockdown in A549 and
MCF-7 cells (Fig. 2E). The enhanced
ability of tumor invasion and migration by inhibiting cathepsin L
in the TGF-β-treated cells suggest an important role of cathepsin L
in the progression of tumor invasion and migration.
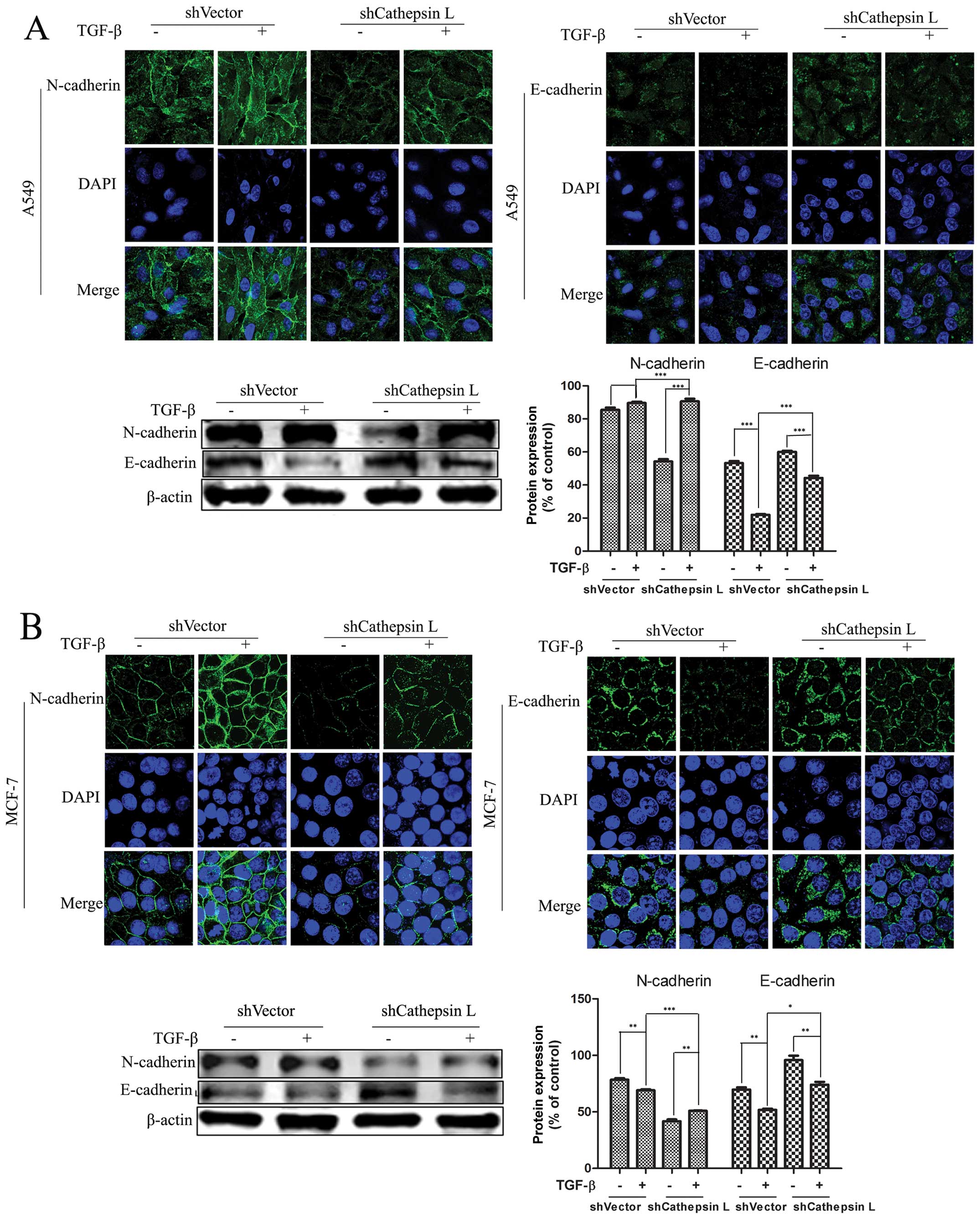
Cathepsin L knockdown blocks
TGF-β-induced EMT
Suppression of cathepsin L in breast cancer and
several other types of carcinomas has been reported to correlate
with tumor invasion and migration. Nevertheless, the mechanisms by
which cathepsin L weakens the ability of cell invasion and
migration remain to be clarified. To examine whether changes of the
cathepsin L protein level are associated with TGF-β-mediated EMT,
we knocked down the expression of cathepsin L in the two cancer
lines treated with TGF-β. At the end of 24 h, compared with the
untreated cells, we found that EMT was induced in TGF-β-treated
cells. Furthermore, when cathepsin L was switched from a high
expression to a low expression state, we assessed the modulation of
E- and N-cadherin protein, two hallmarks of EMT, using IF and
western blot analysis. Notably, in A549 cells, the knockdown of
cathepsin L result in blunting of TGF-β-mediated EMT, as
demonstrated by a decrease in the amount of N-cadherin and an
increase of E-cadherin. By contrast, these alterations were not
observed in the untransfected cells with shCathepsin L (Fig. 3). The same experiments were
performed in the MCF-7 cell line and the results were consistent
with results for the A549 cells. These results suggested that
blocking of EMT by inhibiting cathepsin L indicates a regulatory
role for cathepsin L in mediating the TGF-β-induced EMT and cell
migration.
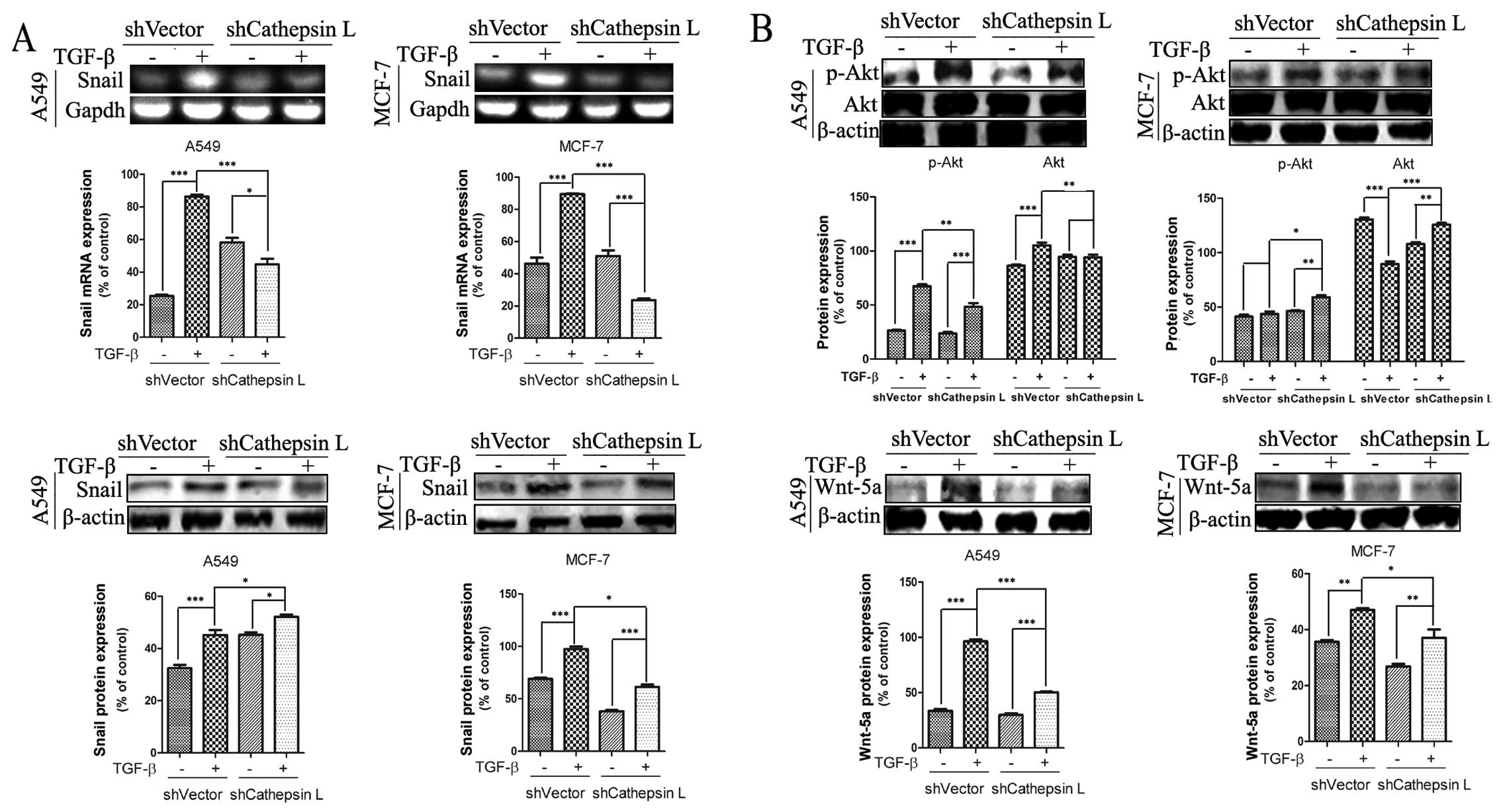
Cathepsin L regulates the expression of
molecules involved in EMT
Overexpression of cathepsin L in lung cancer and
other types of carcinoma has been reported to be correlated with
tumorigenesis and development. Nevertheless, the mechanism by which
cathepsin L promotes the invasive and migratory ability of tumor
cells remains to be clarified. Findings of recent studies showed
that Snail is an essential regulator of EMT and mediates
invasiveness as well as metastasis in various malignant tumors
(2,39,40).
To verify whether there was an association of the function of Snail
and activity of cathepsin LA549 and MCF-7 cells lines were used in
which shCathepsin L was or was not introduced. We observed that
TGF-β treatment induced an increase in the mRNA and protein levels
of Snail in A549 and MCF-7 cells. By contrast, stable transfection
of shCathepsin L suppressed TGF-β-induced Snail gene expression
(Fig. 4A). These results support
the hypothesis that cathepsin L regulates the expression of Snail
and is involved in the regulation of TGF-β-induced EMT in A549 and
MCF-7 cells. In addition, Snail was induced by other signaling
pathways such as Wnt and phosphatidylinositol 3-kinase (PI3K)-AKT
pathways to induce EMT (41–43).
As shown in Fig. 4B, transfection
of cathepsin L abrogated the TGF-β-induced increase of p-Akt and
Wnt-5a protein levels in A549 and MCF-7 cells. These data indicate
that cathepsin L, an EMT regulator functions via a mechanism
involving the effect of Snail expression, Wnt and PI3K-AKT
pathways.
Cathepsin L contributes to regulating EMT
in vivo
Based on the in vitro experiment results, the
effect of cathepsin L on TGF-β-mediated EMT was more significant in
A549 than MCF-7 cells. Thus, to further validate the role of
cathepsin L in EMT, two clones of A549 derivative cells with
cathepsin L knockdown (shCathepsin L) and a non-target control
(shVector) were injected into athymic nude mice subcutaneously to
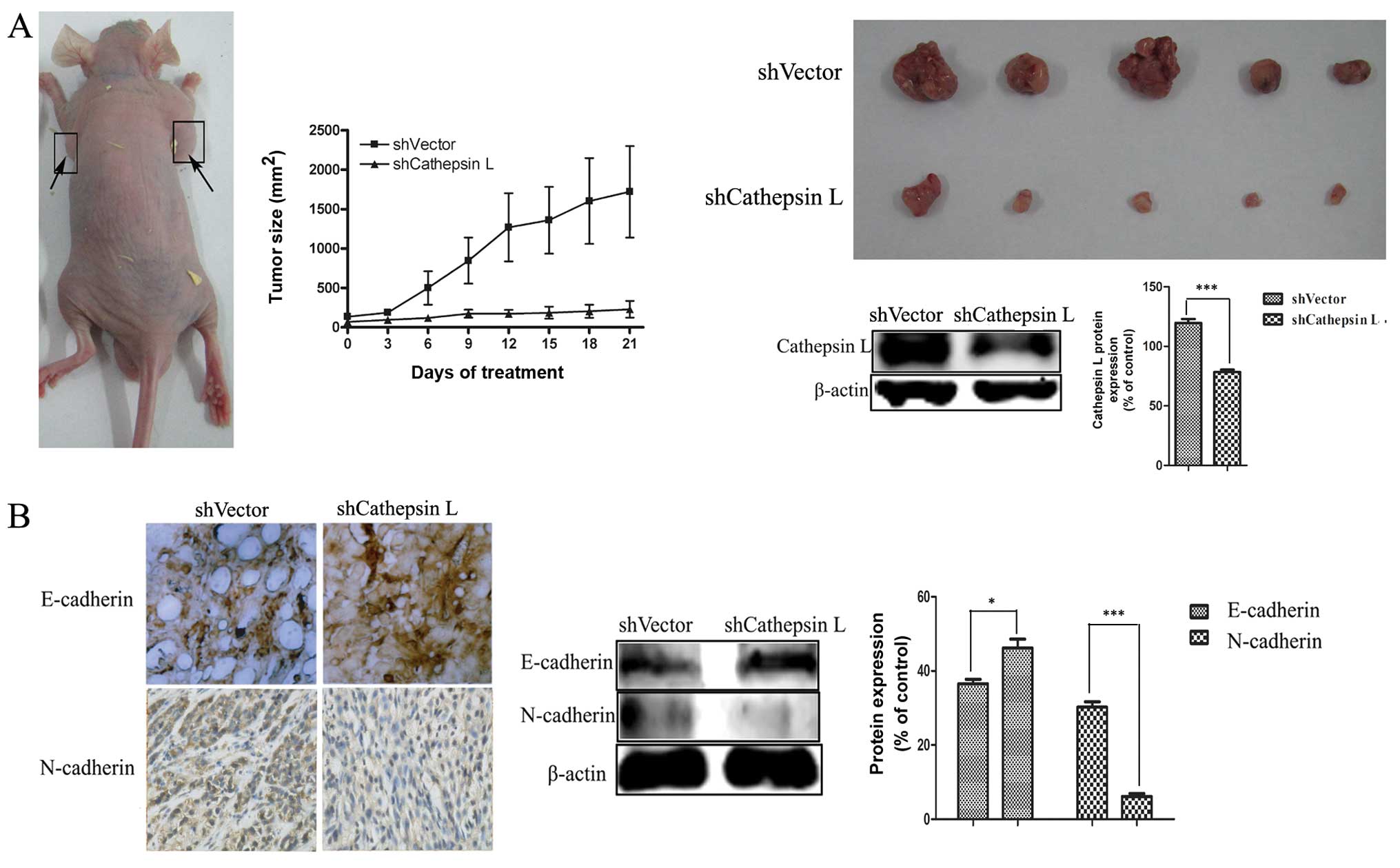
generate the mouse model.
After 42 days, the tumor size was measured and the
tumor volume in A549 cells with cathepsin L knockdown was found to
have significant shrinkage compared with the non-target control
(Fig. 5A), suggesting the ability
of cell proliferation was blunted. Downregulation of E-cadherin,
with upregulation of N-cadherin is known to be a key indicator of
the EMT process (44).
Immunohistochemical staining and western blot analysis were used to
analyze the protein expression of the epithelial markers E-cadherin
and the mesenchymal markers N-cadherin in tumor tissue. As shown in
Fig. 5B, an increased level of
E-cadherin and a decreased level of N-cadherin were observed in
cathepsin L knockdown (shCathepsin L) tumor tissue, compared with
the non-target control tumor tissue.
These data, along with the results of the in
vitro analysis, proved that cathepsin L knockdown suppresses
tumor invasion and migration by inhibiting EMT, confirming the role
of cathepsin L in further regulating EMT.
Discussion
In the present study, we report a previously
unrecognized role of cathepsin L in regulating EMT and provide a
possible mechanistic role for how cathepsin L inhibition
contributes to the decreased cell invasion and migration. In the
present study, two types of epithelial tumor cells, human MCF-7
breast and human A549 lung carcinoma cell lines were selected as
the model system. Our results show that the effect of cathepsin L
knockdown on the suppression of TGF-β-induced cell migration and
invasion was associated with its regulation of EMT. Its mechanism
may be explained by suppression of the EMT-inducing molecules, such
as Snail, which is associated with the PI3K-AKT and Wnt signaling
pathways. The findings concerning the function of cathepsin L in
TGF-β-induced EMT provide new evidence on the regulation of cancer
invasion and migration.
Accumulating evidence reveals that TGF-β is often
used as the inducer of EMT to increase tumor invasion and
migration. A549 is K-Ras mutation, whereas MCF-7 is not and it has
been shown that active Ras is required for TGF-β-induced EMT
(45). Thus, we treated A549 and
MCF-7 cells with different concentrations of TGF-β. The results
showed that the cell morphology changes of EMT occurred ~24 h after
TGF-β treatment (Fig. 1A), while
the expression of cathepsin L was increased (Fig. 1B). Therefore, the expression of
cathepsin L is associated with EMT.
Cathepsin L, which is less well studied than
cathepsin B, has been found to be associated with tumor invasion
and migration (23). In various
tumor types, such as melanoma, lung and breast cancer (21,31,46),
the functional significance of cathepsin L in invasion is
appreciated. Our results show that cathepsin L may induce the
morphological changes of A549 and MCF-7 cells and enhance the
migration and invasion in the two cell lines. Of note, the
migration and invasion of cells was positively correlated with the
expression of cathepsin L. Thus, when the invasion ability peaked,
the expression of cathepsin L also peaked. To determine whether the
migration and invasion change of A549 and MCF-7 cells corresponded
with the expression of cathepsin L, we suppressed the expression of
cathepsin L via transfection with shCathepsin L. The results show
that cathepsin L knockdown strongly suppressed TGF-β-mediated cell
migration, invasion and actin remodeling associated with cell
motility. Moreover, cathepsin L knockdown inhibited the expression
of Cxcr-7, which is important in tumor invasion and migration.
The expression of cathepsin L is known to regulate
invasion and migration in various types of cells. However, the
underlying mechanisms remain to be clarified. Krueger et al
demonstrated that cathepsin L activates cathepsin B, triggering a
chain reaction and inducing the activation of uPA, which
synergistically promotes tumor cell invasion and migration
(47). Cathepsin L stimulated the
release of some growth factors by degrading the extracellular
matrix in order to further promote tumor invasion and migration
(33). Furthermore, Gocheva et
al suggested that cathepsin L decreases the cell-cell adhesion
to enhance tumor invasion and migration by its direct cleavage of
E-cadherin (48), a marker protein
of EMT. Considering the existing results obtained in our
laboratory, it is likely that cathepsin L suppresses cancer
invasion and migration by inhibiting the EMT process. To detect
whether the effect of cathepsin L on cell migration and invasion
was associated with EMT, we performed IF and western blot analysis
to assess expression of E- and N-cadherin, which are EMT-associated
proteins. Inactivation of E-cadherin is one of the key events in
EMT. On the other hand, activation of an inappropriate cadherin,
such as N-cadherin, may be a subsequent event and promote tumor
invasion and migration (37). Our
results show that high N-cadherin but low E-cadherin levels were
observed after TGF-β treatment, suggesting that TGF-β induced EMT
in A549 and MCF-7 cells. However, cathepsin L knockdown inhibited
EMT in response to TGF-β and restored epithelial phenotypes in
tumor cells (Fig. 3), suggesting
that cathepsin L may function as an EMT regulator in various types
of epithelial tumor cells.
Regulation of EMT is a complex process involving
multiple genes and the pathways in embryonic development and in
normal and transformed cell lines, such as extracellular
signal-regulated protein kinases (ERKs), PI3K/Akt, Smads and Wnts,
and is significantly associated with the aggressiveness and
metastatic potential of cancer (49,50).
In addition, it is more likely that the majority of signaling
pathways known to trigger EMT converge at the induction of the
E-cadherin repressors (49). Loss
of E-cadherin expression is considered an important step in the
progression of tumor metastasis, and a fundamental event in EMT
(49).
Snail, a member of the Snail family of zinc finger
transcription factors, is a suppressor of the transcription of
shotgun (an E-cadherin homologue). It is a widely used inducer of
EMT in various types of human cancer of epithelial origin, is known
as a repressor of E-cadherin gene and is important in
TGF-β-induced EMT (51,52). Snail was able to regulate the
changes in gene expression patterns that underlie EMT (6,51).
Our results show that cathepsin L knockdown
abrogated TGF-β-induced expression of Snail at the mRNA and protein
levels (Fig. 4A). This result
suggests that Snail may be significantly involved in the regulation
of EMT by cathepsin L. It also shows that PI3K/Akt and Wnt
signaling pathways affect the expression of epithelial markers to
regulate the EMT process (49,50).
PI3K-Akt signaling pathways participate in regulating cell
proliferation, differentiation, survival and migration. In recent
years, we found that the activity of this pathway abnormality may
cause malignant cell transformation and is associated with invasion
and migration of tumor cell. Wnt signaling has an important role in
embryonic development, and its deregulation is closely associated
with the occurrence of a number of malignant tumors, including
breast and colon cancer. Lee et al suggested that it induces
Snail-dependent EMT, which is responsible for tumor invasion and
migration (53). In addition,
WNT-5A is a transcriptional target of TGF-β whose expression was
induced by TGF-β (54).
In the present study, we observed the enhanced
activation of PI3K/Akt and Wnt signaling pathways in TGF-β-treated
cells. However, cathepsin L knockdown suppressed the enhanced
expression of p-Akt and Wnt-5a. Therefore, we considered that the
mechanism of how cathepsin L knockdown regulates EMT may be
explained by the suppression of Snail genes, and the
PI3K-AKT and Wnt signaling pathways (Fig. 4B).
In the process of tumor progression, cells
proliferate and differentiate abnormally, with the proliferative
and invasive ability becoming stronger via the EMT. To confirm the
role of cathepsin L in regulating EMT, we determined whether
cathepsin L knockdown in vivo may prevent EMT. In this
experiment, the tumor growth rate and expression level of
EMT-associated proteins were measured to detect EMT rangeability.
The results demonstrate the in vitro findings that,
cathepsin L regulates EMT in vivo. Tumor volume in cathepsin
L knockdown cells had significant shrinkage compared with the
non-target control. An increase in E-cadherin and a decreased in
the N-cadherin levels were observed in cathepsin L knockdown
(shCathepsin L) tumor tissue (Fig.
5) were observed, providing further experimental evidence
supporting the role of cathepsin L in the regulation of EMT. These
results show that cathepsin L knockdown is efficacious for EMT
inhibition in tumor cells.
In summary, the present study has identified
cathepsin L as a novel regulator of EMT and its involvement in the
modulation of tumor cell invasion and migration. Additonally,
inhibition of cathepsin L suppresses the increased cell invasion
and migration induced by TGF-β. These findings may expand current
knowledge of EMT networks and shed light on how EMT is regulated in
epithelial tumor cells. Thus, cathepsin L may be examined as a
novel therapeutic target for the prevention of tumor metastasis by
regulating EMT.
References
|
1
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan F, Samuel S, Evans KW, Lu J, Xia L,
Zhou Y, Sceusi E, Tozzi F, Ye XC, Mani SA and Ellis LM:
Overexpression of snail induces epithelial-mesenchymal transition
and a cancer stem cell-like phenotype in human colorectal cancer
cells. Cancer Med. 1:5–16. 2012. View
Article : Google Scholar
|
|
3
|
Shi J, Wang DM, Wang CM, Hu Y, Liu AH,
Zhang YL, Sun B and Song JG: Insulin receptor substrate-1
suppresses transforming growth factor-β1-mediated
epithelial-mesenchymal transition. Cancer Res. 69:7180–7187. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.PubMed/NCBI
|
|
5
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial - mesenchymal
and mesenchymal - epithelial transitions in carcinoma progression.
J Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saito RA, Watabe T, Horiguchi K, Kohyama
T, Saitoh M, Nagase T and Miyazono K: Thyroid transcription
factor-1 inhibits transforming growth factor-β-mediated
epithelial-to-mesenchymal transition in lung adenocarcinoma cells.
Cancer Res. 69:2783–2791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim S, Becker A, Zimmer A, Lu J, Buettner
R and Kirfel J: SNAI1-mediated epithelial-mesenchymal transition
confers chemoresistance and cellular plasticity by regulating genes
involved in cell death and stem cell maintenance. PLoS One.
8:e665582013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pardali K and Moustakas A: Actions of
TGF-beta as tumor suppressor and pro-metastatic factor in human
cancer. Biochim Biophys Acta. 1775:21–62. 2007.
|
|
9
|
Gupta M, Korol A and West-Mays JA: Nuclear
translocation of myocardin-related transcription factor-A during
transforming growth factor beta-induced epithelial to mesenchymal
transition of lens epithelial cells. Mol Vis. 19:1017–1028.
2013.PubMed/NCBI
|
|
10
|
Naber HP, Drabsch Y, Snaar-Jagalska BE, et
al: Snail and Slug, key regulators of TGF-β-induced EMT, are
sufficient for the induction of single-cell invasion. Biochem
Biophys Res Commun. 435:58–63. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levicar N, Nuttall RK and Lah TT:
Proteases in brain tumor progression. Acta Neurochir. 145(Wien):
825–838. 2003. View Article : Google Scholar
|
|
12
|
Yong VW: Metalloproteinases: mediators of
pathology and regeneration in the CNS. Nat Rev Neurosci. 6:931–944.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harada T, Arii S, Mise M, et al:
Membrane-type matrix metalloproteinase-1(MT 1-MMP) gene is
overexpressed in highly invasive hepatocellular carcinomas. J
Hepatol. 28:231–239. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang JR, Li XH, Gao XJ, et al: Expression
of MMP-13 is associated with invasion and metastasis of papillary
thyroid carcinoma. Eur Rev Med Pharmacol Sci. 17:427–435.
2013.PubMed/NCBI
|
|
15
|
Brix K, Dunkhorst A, Mayer K and Jordan S:
Cysteine cathepsins: cellular roadmap to different functions.
Biochimie. 90:194–207. 2008. View Article : Google Scholar
|
|
16
|
Bylaite M, Moussali H, Marciukaitiene I,
et al: Expression of cathepsin L and its inhibitor hurpin in
inflammatory and neoplastic skin diseases. Exp Dermatol.
15:110–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stahl S, Reinders Y, Asan E, et al:
Proteomic analysis of cathepsin B- and L-deficient mouse brain
lysosomes. Biochimica Biophysica Acta. 1774:1237–1246. 2007.
View Article : Google Scholar
|
|
18
|
Skrzydlewska E, Sulkowska M, Koda M and
Sulkowski S: Proteolytic-antiproteolytic balance and its regulation
in carcinogenesis. World J Gastroenterol. 11:1251–1266. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Navab R, Pedraza C, Fallavollita L, et al:
Loss of responsiveness to IGF-I in cells with reduced cathepsin L
expression levels. Oncogene. 27:4973–4985. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pucer A, Castino R, Mirković B, et al:
Differential role of cathepsins B and L in autophagy-associated
cell death induced by arsenic trioxide in U87 human glioblastoma
cells. Biol Chem. 391:519–531. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zajc I, Hreljac I and Lah T: Cathepsin L
affects apoptosis of glioblastoma cells: a potential implication in
the design of cancer therapeutics. Anticancer Res. 26:3357–3364.
2006.PubMed/NCBI
|
|
22
|
Strojnik T, Kos J, Zidanik B, et al:
Cathepsin B immunohistochemical staining in tumor and endothelial
cells is a new prognostic factor for survival in patients with
brain tumors. Clin Cancer Res. 5:559–567. 1999.PubMed/NCBI
|
|
23
|
Kirschke H, Eerola R, Hopsu-Havu VK, et
al: Antisense RNA inhibition of cathepsin L expression reduces
tumorigenicity of malignant cells. Eur J Cancer. 36:787–795. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gocheva V and Joyce JA: Cysteine
cathepsins and the cutting edge of cancer invasion. Cell Cycle.
6:60–64. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jedeszko C and Sloane BF: Cysteine
cathepsins in human cancer. Biol Chem. 385:1017–1027. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mohamed MM and Sloane BF: Cysteine
cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer.
6:764–775. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Strojnik T, Kavalar R, Trinkaus M and Lah
TT: Cathepsin L in glioma progression: comparison with cathepsin B.
Cancer Detect Prev. 29:448–455. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chambers AF, Colella R, Denhardt DT and
Wilson SM: Increased expression of cathepsins L and B and decreased
activity of their inhibitors in metastatic, ras-transformed NIH 3T3
cells. Mol Carcinog. 5:238–245. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Frade R, Rodrigues-Lima F, Huang S, et al:
Procathepsin-L, a proteinase that cleaves human C3 (the third
component of complement), confers high tumorigenic and metastatic
properties to human melanoma cells. Cancer Res. 58:2733–2736.
1998.PubMed/NCBI
|
|
30
|
Yang Z and Cox JL: Cathepsin L increases
invasion and migration of B16 melanoma. Cancer Cell Int. 7:82007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rousselet N, Mills L, Jean D, et al:
Inhibition of tumorigenicity and metastasis of human melanoma cells
by anti-cathepsin L single chain variable fragment. Cancer Res.
64:146–151. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang SM, Li L, Zhang W, et al:
Relationship between cathepsin L and invasion and metastasis of
ovarian carcinoma cells. Zhonghua Fu Chan Ke Za Zhi. 45:598–602.
2010.(In Chinese). PubMed/NCBI
|
|
33
|
Lah TT, Durán Alonso MB and Van Noorden
CJ: Antiprotease therapy in cancer: hot or not? Expert Opin Biol
Ther. 6:257–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Goulet B, Sansregret L, Leduy L, et al:
Increased expression and activity of nuclear cathepsin L in cancer
cells suggests a novel mechanism of cell transformation. Mol Cancer
Res. 5:899–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Krueger S, Kellner U, Buehling F, et al:
Cathepsin L antisense oligonucleotides in a human osteosarcoma cell
line: effects on the invasive phenotype. Cancer Gene Ther.
8:522–528. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Matarrese P, Ascione B, Ciarlo L, et al:
Cathepsin B inhibition interferes with metastatic potential of
human melanoma: an in vitro and in vivo study. Mol Cancer.
9:2072010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shintani Y, Okimura A, Sato K, et al:
Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hao M, Zheng J, Hou K, et al: Role of
chemokine receptor CXCR7 in bladder cancer progression. Biochem
Pharmacol. 84:204–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bi WR, Jin CX, Xu GT and Yang CQ: Bone
morphogenetic protein-7 regulates Snail signaling in carbon
tetrachloride-induced fibrosis in the rat liver. Exp Ther Med.
4:1022–1026. 2012.PubMed/NCBI
|
|
40
|
Nieto MA: The Snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shin SY, Rath O, Zebisch A, et al:
Functional roles of multiple feedback loops in extracellular
signal-regulated kinase and Wnt signaling pathways that regulate
epithelial-mesenchymal transition. Cancer Res. 70:6715–6724. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eger A, Stockinger A, Park J, et al:
beta-Catenin and TGFbeta signalling cooperate to maintain a
mesenchymal phenotype after FosER-induced epithelial to mesenchymal
transition. Oncogene. 23:2672–2680. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xiao D and He J: Epithelial mesenchymal
transition and lung cancer. J Thorac Dis. 2:154–159.
2010.PubMed/NCBI
|
|
44
|
Chunhacha P, Sriuranpong V and
Chanvorachote P: Epithelial-mesenchymal transition mediates anoikis
resistance and enhances invasion in pleural effusion-derived human
lung cancer cells. Oncol Lett. 5:1043–1047. 2013.PubMed/NCBI
|
|
45
|
Davies M, Robinson M, Smith E, et al:
Induction of an epithelial to mesenchymal transition in human
immortal and malignant keratinocytes by TGF-beta1 involves MAPK,
Smad and AP-1 signalling pathways. J Cell Biochem. 95:918–931.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Amuthan G, Biswas G, Ananadatheerthavarada
HK, et al: Mitochondrial stress-induced calcium signaling,
phenotypic changes and invasive behavior in human lung carcinoma
A549 cells. Oncogene. 21:7839–7849. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Krueger S, Kellner U, Buehling F and
Roessner A: Cathepsin L antisense oligonucleotides in a humall
osteosarcoma cell line: efects on the invasive phenotype. Cancer
Gene Ther. 8:522–528. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gocheva V, Zeng W, Ke D, et al: Distinct
roles for cysteine cathepsin genes in multistage tumorigenesis.
Genes Dev. 20:543–556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang Q, Bai X, Chen W, et al:
Wnt/β-catenin signaling enhances hypoxia-induced
epithelial-mesenchymal transition in hepatocellular carcinoma via
crosstalk with hif-1α signaling. Carcinogenesis. 34:962–973. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: an alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hanahan D: Heritable formation of
pancreatic beta-cell tumours in transgenic mice expressing
recombinant insulin/simian virus 40 oncogenes. Nature. 315:115–122.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee SY, Jeon HM, Ju MK, et al: Wnt/Snail
signaling regulates cytochrome c oxidase and glucose metabolism.
Cancer Res. 72:3607–3617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kumawat K, Menzen MH, Slegtenhorst RM,
Halayko AJ, Schmidt M and Gosens R: TGF-β-activated kinase 1 (TAK1)
signaling regulates TGF-β-induced WNT-5A expression in airway
smooth muscle cells via Sp1 and β-catenin. PLoS One. 9:e948012014.
View Article : Google Scholar
|