Introduction
Several diseases, especially cancer, are caused by
aberrant epigenetic alterations in addition to genetic mutations
(1,2). Histone acetylation, an example of
epigenetic regulation, is a process that is under intensive study
since it regulates gene transcription. Histone deacetylases (HDAC),
which regulate histone acetylation, are often inappropriately
expressed in cancer cells, which lead to the silencing of
tumor-suppressor genes or the activation of oncogenes (3). HDAC inhibition abrogates epigenetic
silencing commonly observed in cancer, and a number of HDAC
inhibitors have been developed for cancer treatment (4). Several HDAC inhibitors have been
identified through synthetic or natural sources and separated into
several structurally distinct classes: short chain fatty, and
hydroxamic acids, benzamides, cyclic tetrapeptides, and
electrophilic ketones. Members of those classes show substantially
different potency and target specificity (3,5).
Apicidin, a cyclic peptide HDAC inhibitor, was the first reported
reversible inhibitor of the in vitro development of
apicomplexan parasites and was later shown to have
antiproliferative and cyto-differentiation activity in mammalian
cells (6,7). Apicidin has been shown to exhibit
antitumor activity in several human cancer cells. Previous studies
reported that apicidin induced apoptosis and cell cycle arrest in
ovarian, endometrial and breast cancer cells (8–10).
Nevertheless, the precise molecular mechanism of cell death effect
by apicidin remains unclear and, to the best of our knowledge, has
not been previously examined in salivary mucoepidermoid carcinoma
(MEC), the most common malignant salivary tumor corresponding to
34% of all salivary malignancies (11).
Cell death occurs through at least three
morphologically distinct subroutines, known as apoptosis, necrosis
and autophagy (12). Apoptosis is a
well-defined, programmed mechanism that allows cells to commit
suicide and is the major type of cell death induced by various
antitumor drugs including HDAC inhibitors (3,5,13).
Autophagy plays an important homeostatic role, mediating the
removal of dysfunctional or damaged organelles, which are digested
and recycled for cellular metabolic needs (14). Autophagy recently attracted
considerable attention in cancer research. However, present
autophagy studies are often viewed as confounding, because of its
association with apparently contradictory roles, such as survival
and cell death, depending on the model used (15,16).
Apoptosis and necrosis are irreversible forms of cell death,
whereas autophagy leads to cell death or paradoxically allows cells
to escape cell death (17). In
recent studies autophagy has been identified in tumor cell lines
treated with HDAC inhibitors. However, whether autophagy
contributes to the anticancer activity of HDAC inhibitors remains
to be clarified (16,18). Previous studies reported that
apicidin induces autophagy in oral squamous cell carcinoma (OSCC)
cells, however, the molecular mechanism of apicidin has yet to be
thoroughly identified (18). The
present study evaluated the antitumor effect of apicidin and
examined whether the molecular mechanisms of apicidin induce cell
death in human salivary MEC cells.
Materials and methods
Chemicals and cell culture
Apicidin [Cyclo
(N-O-methyl-L-tryptophanyl-L-isoleucinyl-D-pipecolinyl-L-2-amino-8-oxo
decanoyl)] was purchased from Sigma-Aldrich (St. Louis, MO,
USA). Apicidin was dissolved in sterile dimethyl sulfoxide (DMSO;
Sigma-Aldrich) to generate a 5 mM stock solution, and stored at
−80°C. Subsequent dilutions were made in RPMI-1640 (Gibco-BRL,
Rockville, MD, USA). Caspase pan-inhibitor z-VAD-fmk was purchased
from MBL Co. (Woburn, MA, USA). Extracellular signal-regulated
kinase (ERK) inhibitor U0126 was purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). c-Jun NH2-terminal kinase
(JNK) inhibitor SP600125 and PI3K/AKT inhibitor LY294002 were
purchased from Cayman Chemical Co. (Ann Arbor, Michigan, USA). All
other chemicals were purchased from Sigma-Aldrich.
The YD-15 human salivary MEC cells were purchased
from the Korea Cell Line Bank (KCLB, Seoul, Korea) (19). The cells were maintained as
monolayers at 37°C in an atmosphere containing 5%
CO2/air in RPMI-1640 containing 10% heat-inactivated
fetal bovine serum (FBS; Gibco-BRL, Gaithersburg, MD, USA) and 1%
penicillin/streptomycin (Gibco-BRL).
MTT assay
The cells were grown in 96-well plates at a density
of 1×104 cells/well. The cells were allowed to attach
for 24 h, and were then exposed to apicidin. At the end of the
treatment period, 20 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
5 mg/ml, Sigma-Aldrich) reagent was added to each well. After 4 h
incubation in the dark at 37°C, the supernatant was aspirated and
formazan crystals were dissolved in 100 μl of DMSO at 37°C for 10
min with gentle agitation. The absorbance per well was measured at
540 nm using a VersaMax microplate reader (Molecular Devices Corp.,
Sunnyvale, CA, USA).
HDAC activity
The HDAC enzyme activity was measured using a
fluorogenic HDAC Assay kit purchased from BD Biosciences (San
Diego, CA, USA) according to the manufacturer’s instructions.
Briefly, the cell lysates were incubated with vehicle or various
concentrations of apicidin and SAHA at 37°C for 30 min in the
presence of an HDAC fluorimetric substrate. The HDAC assay
developer, which produces a fluorophore in the reaction mixture,
was added, and the fluorescence was measured using Victor3
(Perkin-Elmer, Waltham, MA, USA) with excitation at 360 nm and
emission at 460 nm. The measured activity was assessed using
GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Trypan blue exclusion assay
The trypan blue exclusion assay was based on the
ability of viable cells to exclude the dye. Five minutes after 0.4%
trypan blue was added to the cells, they were loaded into a
hematocytometer and counted for the dye uptake. The number of
viable cells was calculated as a percentage of the total cell
population.
Western blot analysis
The cells were seeded in 6-well plates and exposed
to the indicated concentrations (1.0 and 5.0 μM) of apicidin. After
treatment, the cells were harvested using lysis buffer (Pro-Prep,
Daejeon, Intron, Korea). Protein concentrations were measured using
the Protein Assay kit (Bio-Rad, Hercules, CA, USA) according to the
manufacturer’s instructions. Total protein was then separated using
10–15% sodium dodecyl sulfate (SDS)-polyacrylamide gels and
transferred to polyvinylidene difluoride (PVDF) membrane (NEN Life
Science Products, Boston, MA, USA). The membranes were probed
sequentially with antibodies against acetylated histone H3,
acetylated histone H4 (Upstate, Lake Placid, NY, USA), Bax,
cytochrome c, cleaved caspase-9, -7 and -3, pro-caspase 3,
poly ADP ribose polymerase (PARP), light change 3B (LC3B),
insulin-like growth factor 1 receptor (IGF-1R), ERK, p-ERK, JNK,
p-JNK, p38, p-p38, AKT, p-AKT, p-mTOR (Cell Signaling Technology
Inc.), p62, and Actin (Santa Cruz Biotechnology, Inc., CA, USA) at
4°C for overnight. The blots were developed using an enhanced
chemiluminescence (ECL) kit (Amersham Biosciences, Buckinghamshire,
UK). The protein levels were normalized by a comparison with the
actin levels.
Flow cytometric analysis
The cells were stained using an Alexa
Fluor® 488 Annexin V/Dead Cell apoptosis kit
(Invitrogen, Eugene, Oregon, USA) for apoptosis analysis according
to the manufacturer’s instructions. Data acquisition and analysis
were carried out by a Cell Lab Quanta™ SC flow cytometer (Beckman
Coulter Inc., Miami, FL, USA) and software.
Detection of acidic vesicular
organelles
Autophagy is characterized by the formation and
promotion of acidic vesicular organelles (AVOs). To detect the
development of AVOs, the cells were stained with acridine orange as
previously described (20). In
acridine orange-stained cells, the cytoplasm and nucleus show
bright green fluorescence, whereas the acidic compartments exhibit
bright red fluorescence (autophagic cells). Briefly, the treated
cells were stained with acridine orange (1 μg/ml, Sigma-Aldrich)
for 15 min. The samples were observed under fluorescence microscopy
(Olympus, Centre Valley, PA, USA). To quantify the development of
AVOs, the stained cells were analyzed by FACScan flow cytometer
(Beckman Coulter Inc.) with CellQuest software.
Monodansylcadaverine staining
The autofluorescent agent monodansylcadaverine (MDC;
Sigma-Aldrich) was used to evaluate the abundance of autophagic
vacuoles in cells. Cells were stained with MDC at a final
concentration of 0.05 mmol/l, for 10 min at 37°C. The cells were
washed with phosphate-buffered saline and observed under
fluorescence microscopy (Olympus).
siRNA transfection
The siRNA oligonucleotides for IGF-1R were
synthesized by Bioneer (Daejeon, Korea), and the non-targeting
control siRNA (NC. siRNA) was used as the negative control. The
IGF-1R-siRNA sequence used was: sense,
5′-CGACAGGCCUCGUGUAUGA(UU)-3′, and anti-sense,
5′-UCAUACACGAGGCCUGUCG(UU)-3′ (21). The cells were transfected with 100
nM siRNA for 24 h using Lipofectamine Plus reagent (Invitrogen).
The cells were harvested for protein analysis 3 days after
transfection. Western blot analysis was used to validate silencing
of the protein expression.
Statistical analysis
Data are presented as the mean ± SD of at least
three individual experiments. Statistical comparisons between the
groups were performed using a two-tailed Student’s t-test (Excel,
Microsoft). Statistical significance differences were set at
P<0.05, P<0.01 and P<0.01.
Results
Apicidin inhibits cell proliferation and
HDAC activity
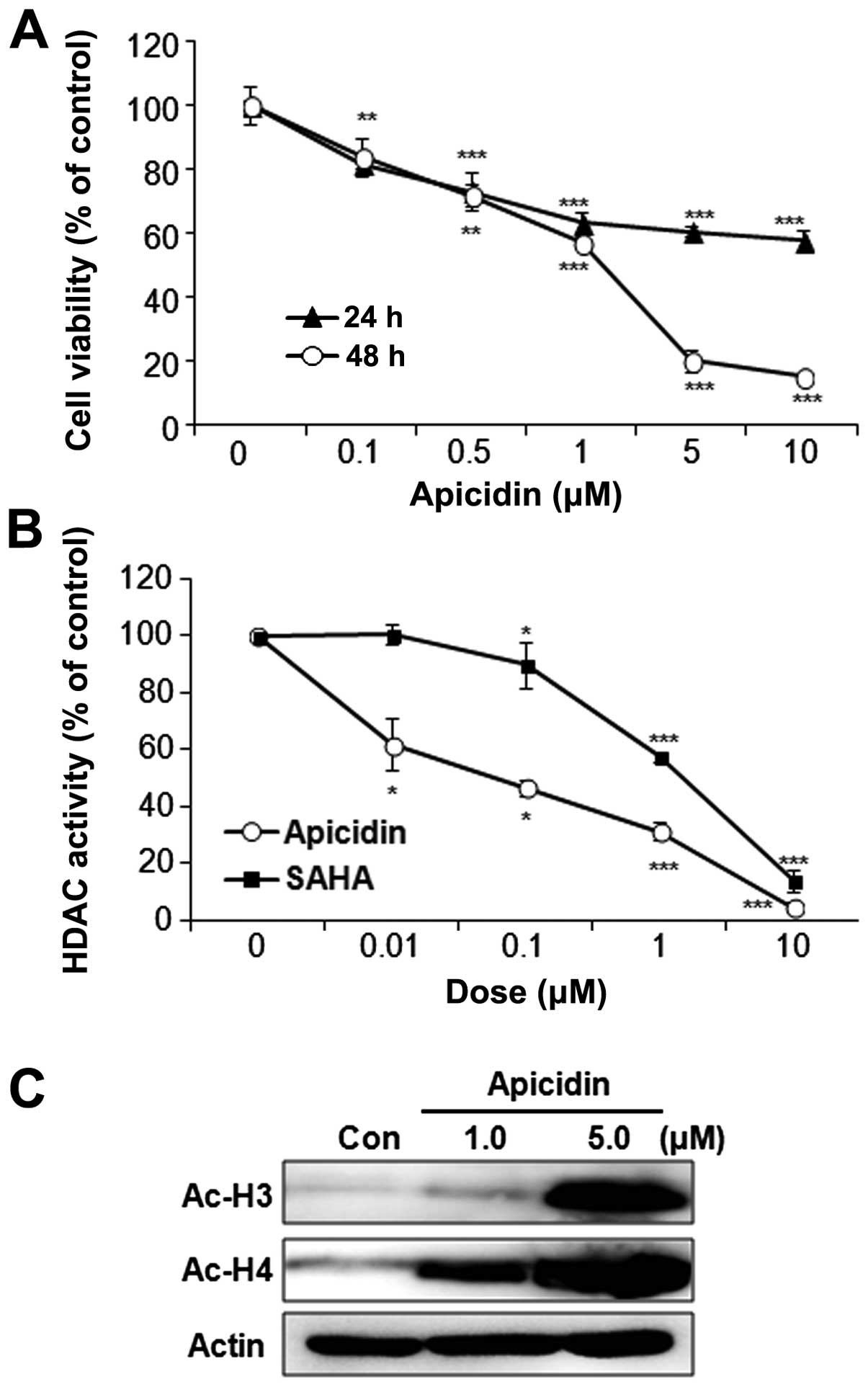
The effect of apicidin on MEC cell proliferation was
first examined using the MTT assay. The cells were treated with
various doses of apicidin until 48 h. Apicidin significantly
inhibited the cell proliferation of YD-15 cells in a dose- and
time-dependent manner (Fig. 1A).
Specifically, cell viability was markedly decreased when the cells
were treated with 5.0 μM apicidin for 48 h. The enzyme activity and
the expression of acetylated histones were next examined to
determine the HDAC inhibitory effect by apicidin. The HDAC activity
was markedly inhibited by apicidin in YD-15 cells, which reduced
the HDAC activity more effectively as compared to SAHA, the first
HDAC inhibitor to obtain FDA approval (Fig. 1B). In addition, apicidin
significantly increased the levels of acetylated histone H3 and H4
(Fig. 1C).
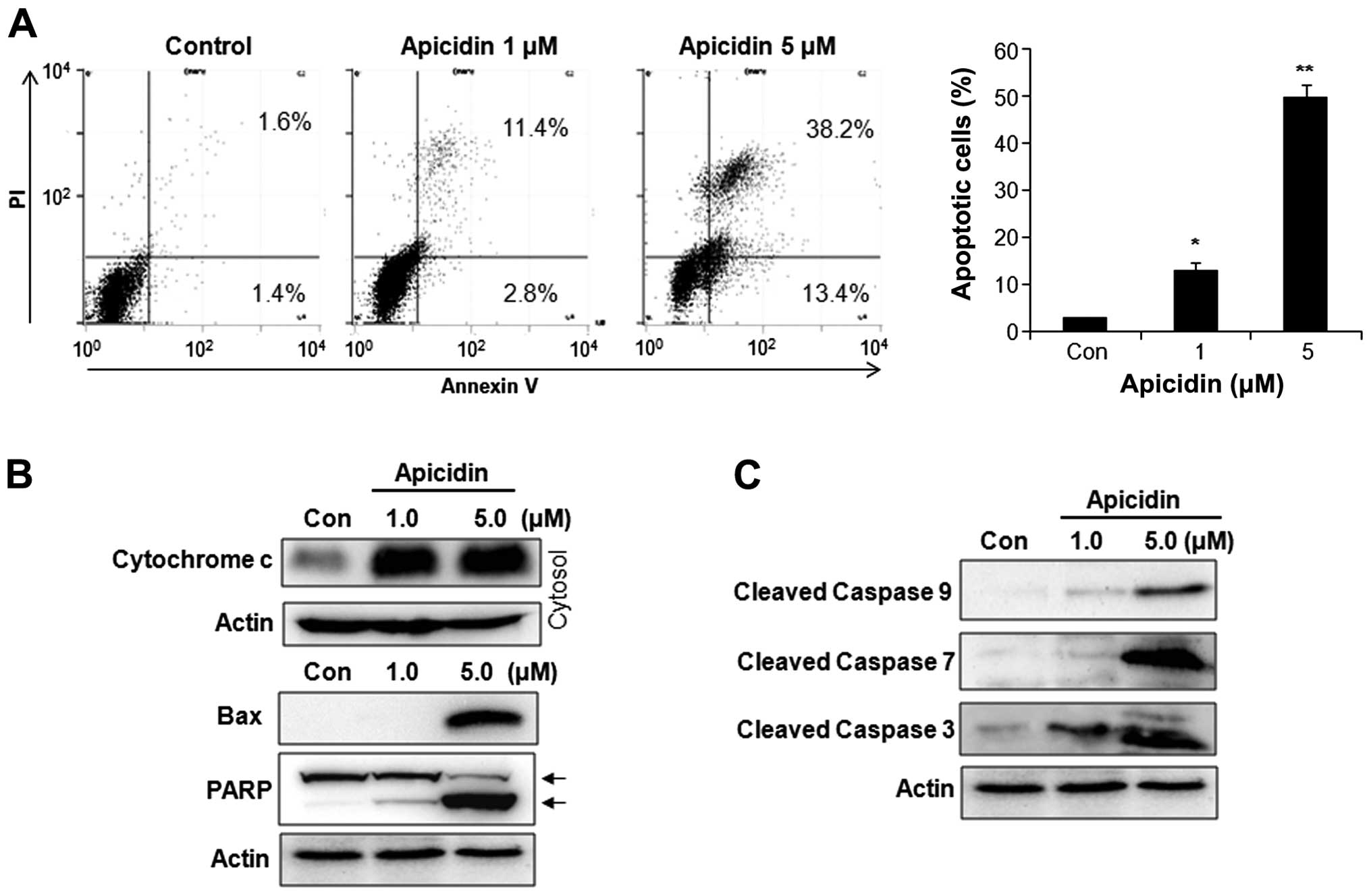
Induction of apoptosis by apicidin
The apoptotic effect of apicidin was examined by
flow cytometry and western blotting. Apicidin significantly
increased the number of Annexin V-positive apoptotic cells in a
dose-dependent manner (Fig. 2A). As
shown in Fig. 2B, cytochrome
c levels were increased in the cytosol fraction, and Bax and
cleaved PARP were also induced in YD-15 cells after apicidin
treatment. Apicidin significantly induced the activation of
caspase-9, -7 and -3 in YD-15 cells (Fig. 2C). These results showed that
apicidin induced caspase-dependent apoptosis via the
mitochondria-mediated intrinsic pathway in YD-15 MEC cells.
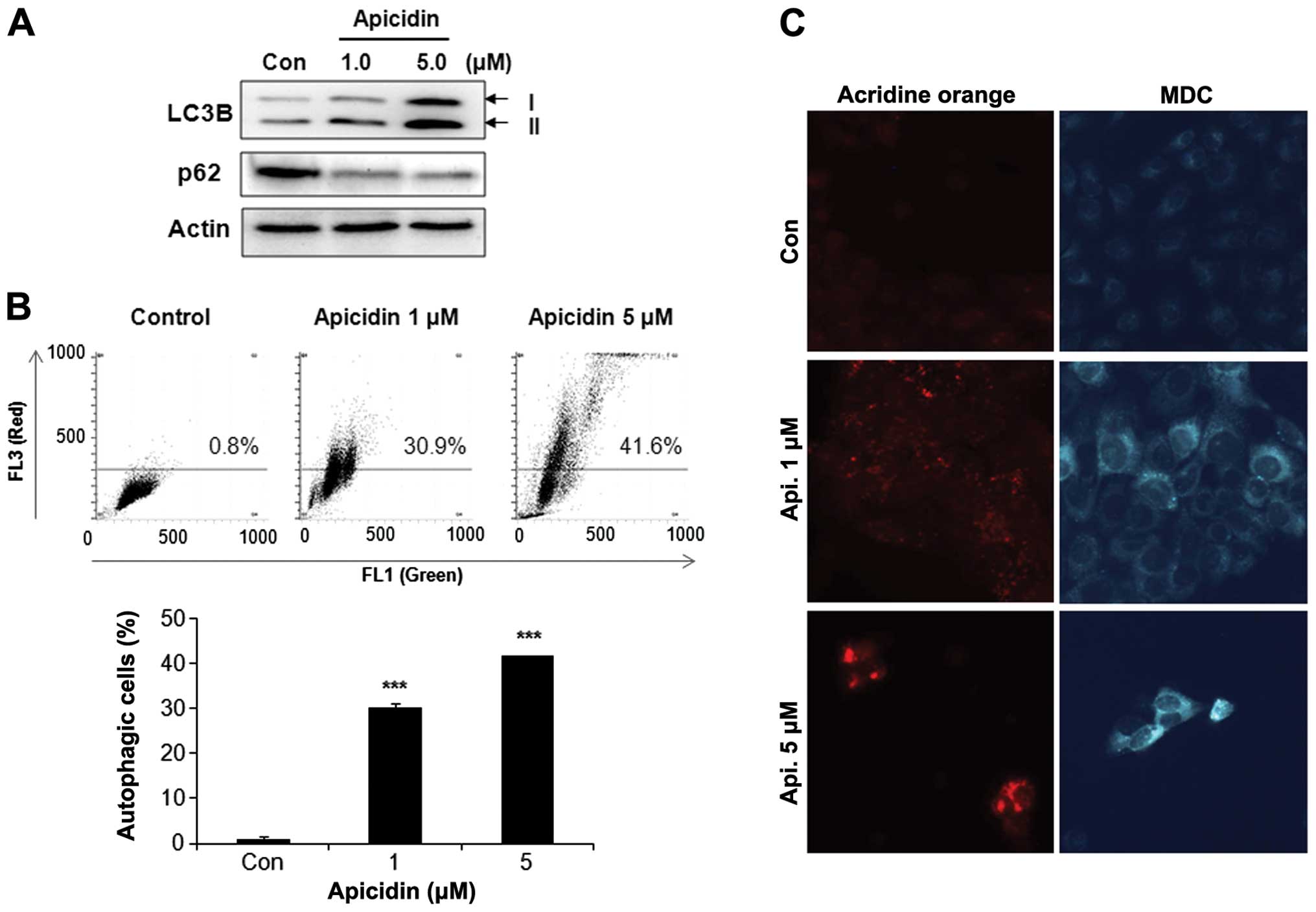
Induction of autophagy by apicidin
The levels of the microtubule-associated protein 1
LC3-II, a marker for autophagic vesicles and p62 as a marker for
autophagic flux were determined to investigate the possibility of
apicidin-inducing autophagy in YD-15 cells. As shown in Fig. 3A, the level of LC3B-II increased by
apicidin treatment in a dose-dependent manner, whereas p62 was
decreased. The autophagy response by apicidin was examined using
acridine orange and MDC staining. Flow cytometry using acridine
orange staining showed that apicidin induced the number of acidic
vesicles (red signal) in a dose-dependent manner, as confirmed by
fluorescence microscopy (Fig. 3B and
C). MDC staining also showed increased AVOs in apicidin-treated
cells compared to the control (Fig.
3C). These results showed that apicidin induced autophagy in
YD-15 cells.
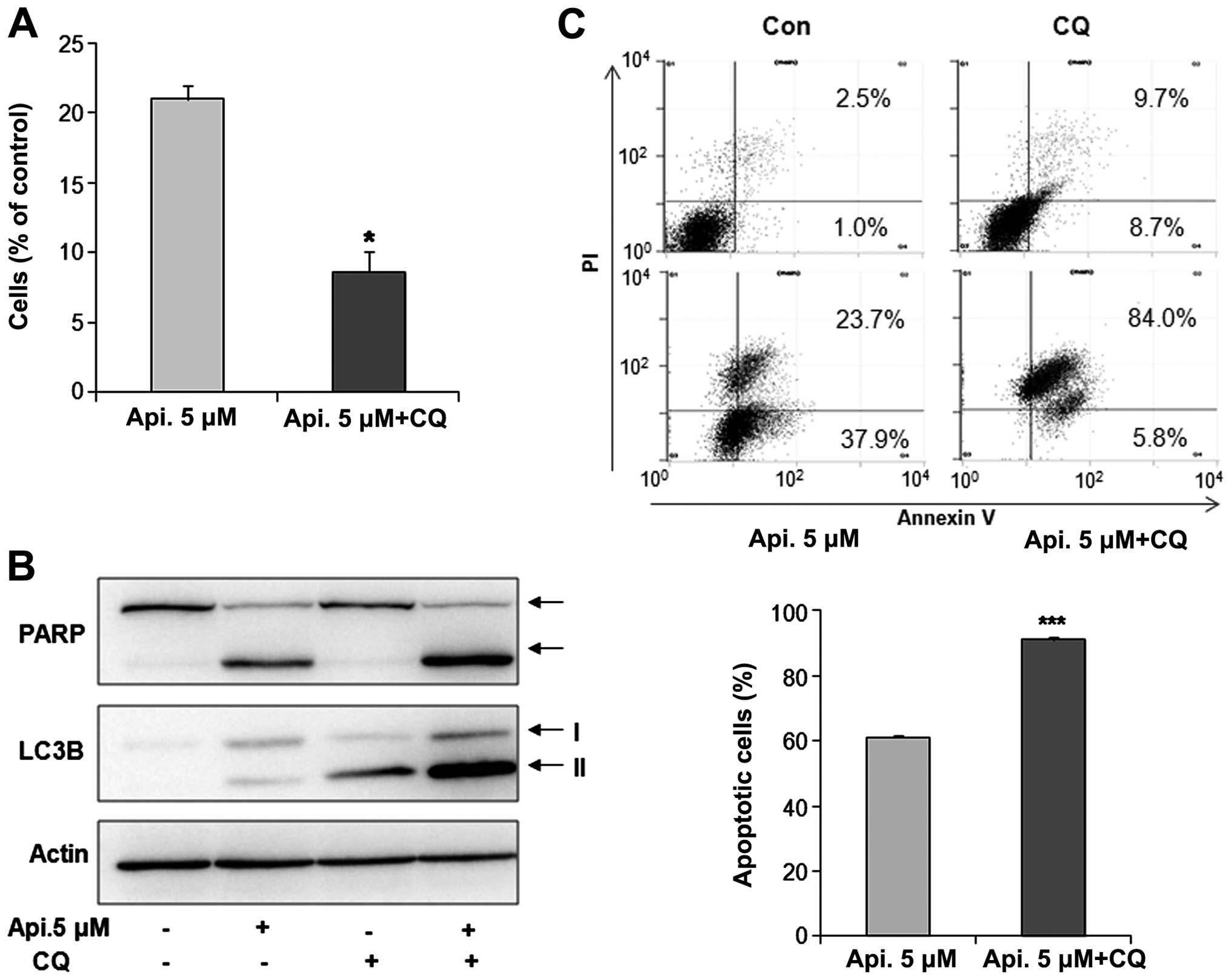
Function of autophagy and interaction
between autophagy and apoptosis
To investigate the function of autophagy by apicidin
and interaction between apoptosis and autophagy, YD-15 cells were
co-treated with the specific autophagy inhibitor, chloroquine (CQ),
and apicidin. Cell viability was examined using the Trypan blue
exclusion assay. As shown in Fig.
4A, co-treatment with apicidin and CQ reduced cell viability
compared to apicidin treatment alone. The levels of PARP and
LC3B-II expression were examined by western blotting. Apicidin with
CQ increased the levels of PARP cleavage and LC3B-II arrest by
inhibiting autophagic degradation compared to apicidin-alone
treatment (Fig. 4B). These results
indicated that the inhibition of autophagy enhanced
apicidin-induced apoptosis (Fig.
4B). Increased apoptosis by autophagy inhibition was confirmed
by flow cytometry. The percentage of apoptotic cells was
significantly increased in the co-treated cells compared to
apicidin alone-treated cells (Fig.
4C). These results suggested that autophagy has a pro-survival
function in YD-15 cells.
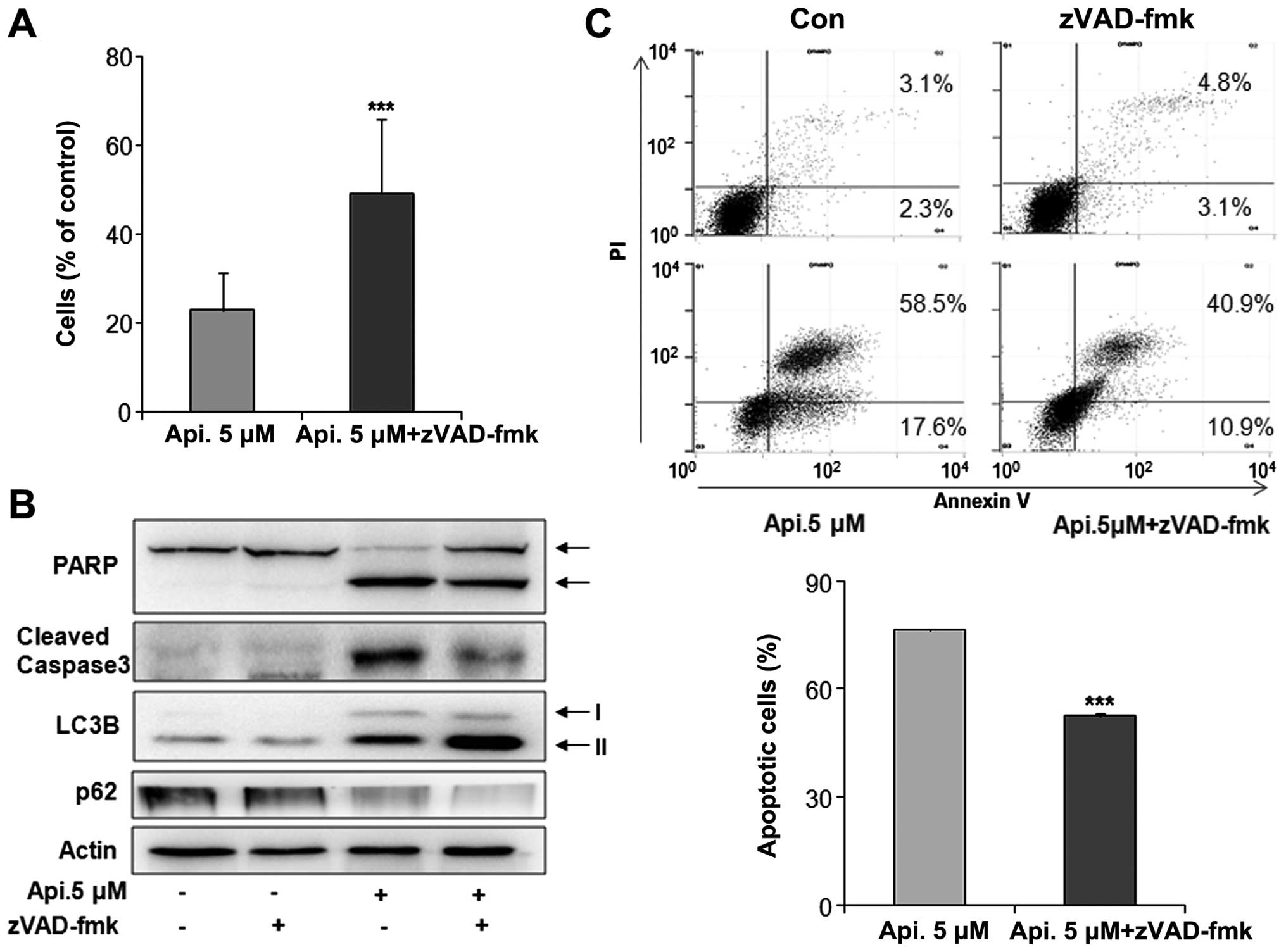
Apicidin-induced apoptosis was oppositely inhibited
by co-treatment with a caspase pan-inhibitor, z-VAD-fmk, and the
induction of apoptosis and autophagy was observed. Apicidin with
z-VAD-fmk increased the cell viability and reduced the levels of
cleaved PARP and caspase-3 compared to apicidin-alone treatment
(Fig. 5A and B). Reduced apoptosis
by co-treatment was confirmed by flow cytometry (Fig. 5C). However, apicidin treatment with
z-VAD-fmk increased the level of LC3B-II expression and reduced p62
expression compared to apicidin treatment (Fig. 5B). These results showed that the
inhibition of apoptosis enhanced apicidin-induced autophagy in
YD-15 cells.
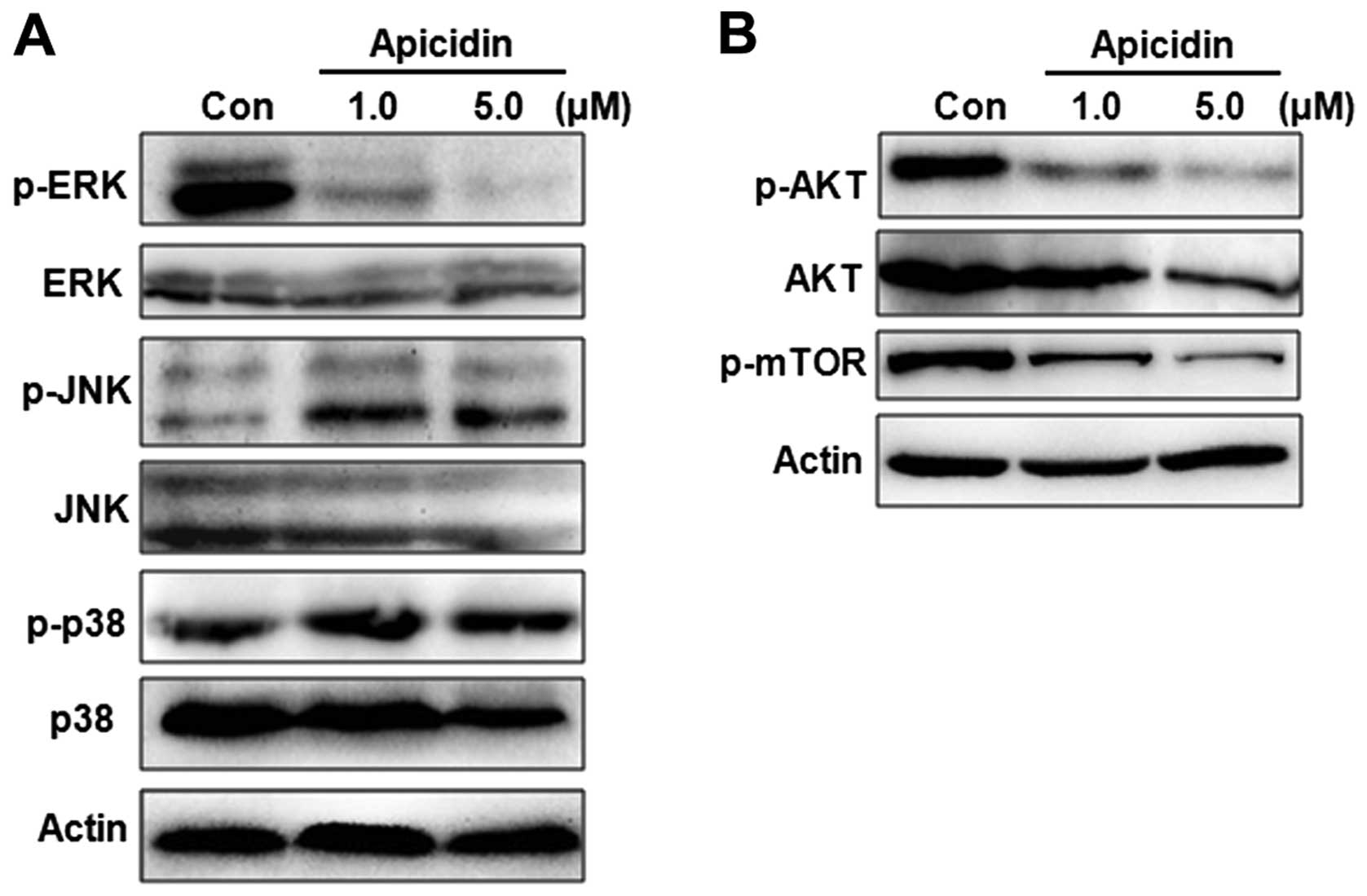
Regulation of MAPK and AKT/mTOR pathway
by apicidin in YD-15 cells
To evaluate the molecular mechanism of apoptosis or
autophagy by apicidin, the regulation of MAPK and AKT/mTOR pathways
was monitored in apicidin-treated cells. These signaling pathways
are important in cell survival and proliferation events and are
well-established mediators of the malignant phenotype. The MAPK
family mainly consists of ERK, JNK, and p38 MAPK kinase members
(22). The effects of apicidin on
the activation of ERK, JNK and p38 MAP kinases were first examined
by western blotting. Apicidin decreased the level of ERK
phosphorylation and increased the level of JNK phosphorylation, but
had no effect on the level of p38 phosphorylation (Fig. 6A). The phosphorylation of AKT and
mTOR as the main downstream mediator of AKT was measured in
apicidin-treated cells. Apicidin markedly inhibited the
phosphorylation of AKT and mTOR in YD-15 cells (Fig. 6B). These results suggested that
apicidin inhibited cell proliferation by regulating ERK, JNK and
AKT/mTOR pathways.
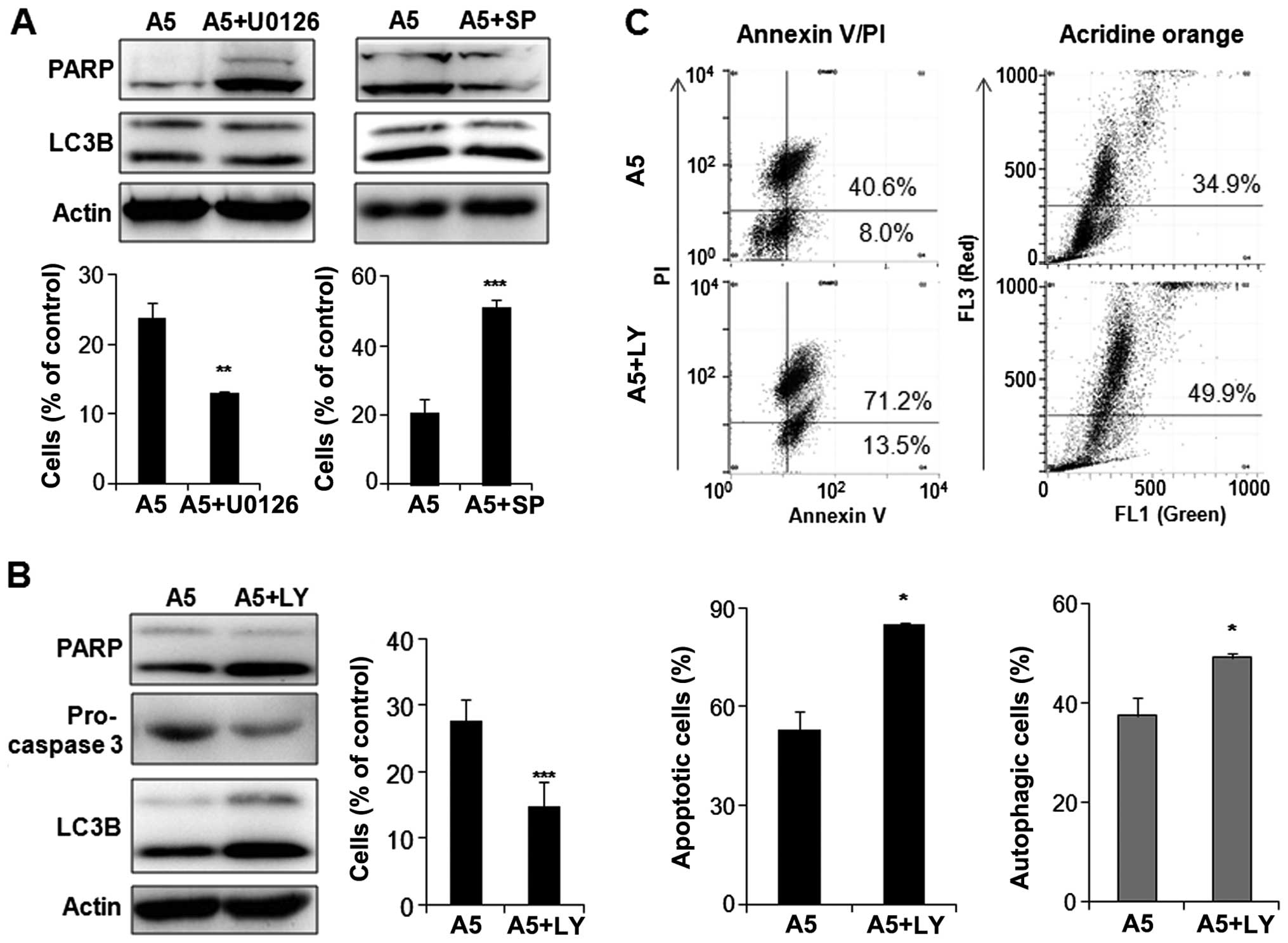
Involvement of the MAPK and AKT/mTOR
pathways in apicidin-induced apoptosis and autophagy
In order to determine which pathway is involved in
apoptosis or autophagy in apicidin-treated YD-15 cells, the cells
were co-treated with apicidin and specific MAPK or AKT/mTOR pathway
kinase inhibitors, including U0126 (ERK inhibitor), SP600125 (JNK
inhibitor) and LY294002 (PI3K/AKT inhibitor). As shown in Fig. 7A, enhanced ERK inhibition by
apicidin with U0126 increased the cleaved PARP and decreased the
cell viability of the co-treated cells compared to apicidin
alone-treated cells. On the other hand, there were no significant
differences in LC3B-II expression. The inhibition of
apicidin-induced JNK phosphorylation by SP600125 reduced cleaved
PARP, but not LC3B-II expression, and increased the cell viability
in the co-treated cells compared to the apicidin-treated cells.
This result suggested that the inhibition of ERK and induction of
JNK signals by apicidin mediated apoptosis in YD-15 cells, but not
autophagy. Enhanced AKT inhibition by apicidin with LY294002
induced the increase of cleaved PARP and the decrease of
pro-caspase-3, indicating the induction of apoptosis. In addition,
the level of LC3B-II expression was higher in the co-treated cells
than the apicidin-treated cells (Fig.
7B). These results suggested that the inhibition of AKT/mTOR
signal by apicidin mediated apoptosis and autophagy simultaneously
in YD-15 cells. Induction of apoptosis and autophagy through
enhanced AKT inhibition was confirmed by flow cytometric analysis
using Annexin V/PI and acridine orange staining in co-treated YD-15
cells (Fig. 7C).
Inhibition of IGF-1R was associated with
apoptosis and autophagy by apicidin
IGF-1R is commonly overexpressed in cancer cells and
stimulates two major downstream proliferating signaling pathways,
the MAPK and AKT/mTOR pathways (23). We examined whether apicidin
inhibited the expression of IGF-1R inducing the inhibition of MAPK
and AKT/mTOR pathways in YD-15 cells. As shown in Fig. 8A, apicidin significantly inhibited
the level of IGF-1R expression in a dose-dependent manner. To
determine whether the inhibition of IGF-1R by apicidin was
associated with the induction of apoptosis and autophagy, IGF-1R
was knocked down by treatment with a specific IGF-1R siRNA
(siIGF-1R) in YD-15 cells. Knockdown of IGF-1R inhibited cell
proliferation and induced Annexin V-positive apoptotic cells and
AVO formation, as indicated by the autophagic marker by flow
cytometric analysis (Fig. 8B and
C). The expression levels of cleaved PARP and LC3B-II were also
increased in siIGF-1R-transfected YD-15 cells (Fig. 8D). These results showed that IGF-1R
knockdown induced apoptosis and autophagy in YD-15 cells. We also
examined whether the effect of IGF-1R knockdown on apoptosis and
autophagy was associated with the regulation of ERK, JNK and
AKT/mTOR signals by western blotting. As shown in Fig. 8E, knockdown of IGF-1R markedly
inhibited ERK phosphorylation and induced slight JNK
phosphorylation in YD-15 cells. The inhibition of AKT and mTOR
phosphorlyation was also observed in siIGF-1R-treated cells. These
effects of IGF-1R knockdown in siIGF-1R-transfected YD-15 cells
showed a similar pattern to that observed in apicidin-treated YD-15
cells. Therefore, the inhibition of IGF-1R in YD-15 cells by
apicidin was strongly mediated by the induction of apoptosis and
autophagy through regulation of the ERK, JNK and AKT/mTOR
pathways.
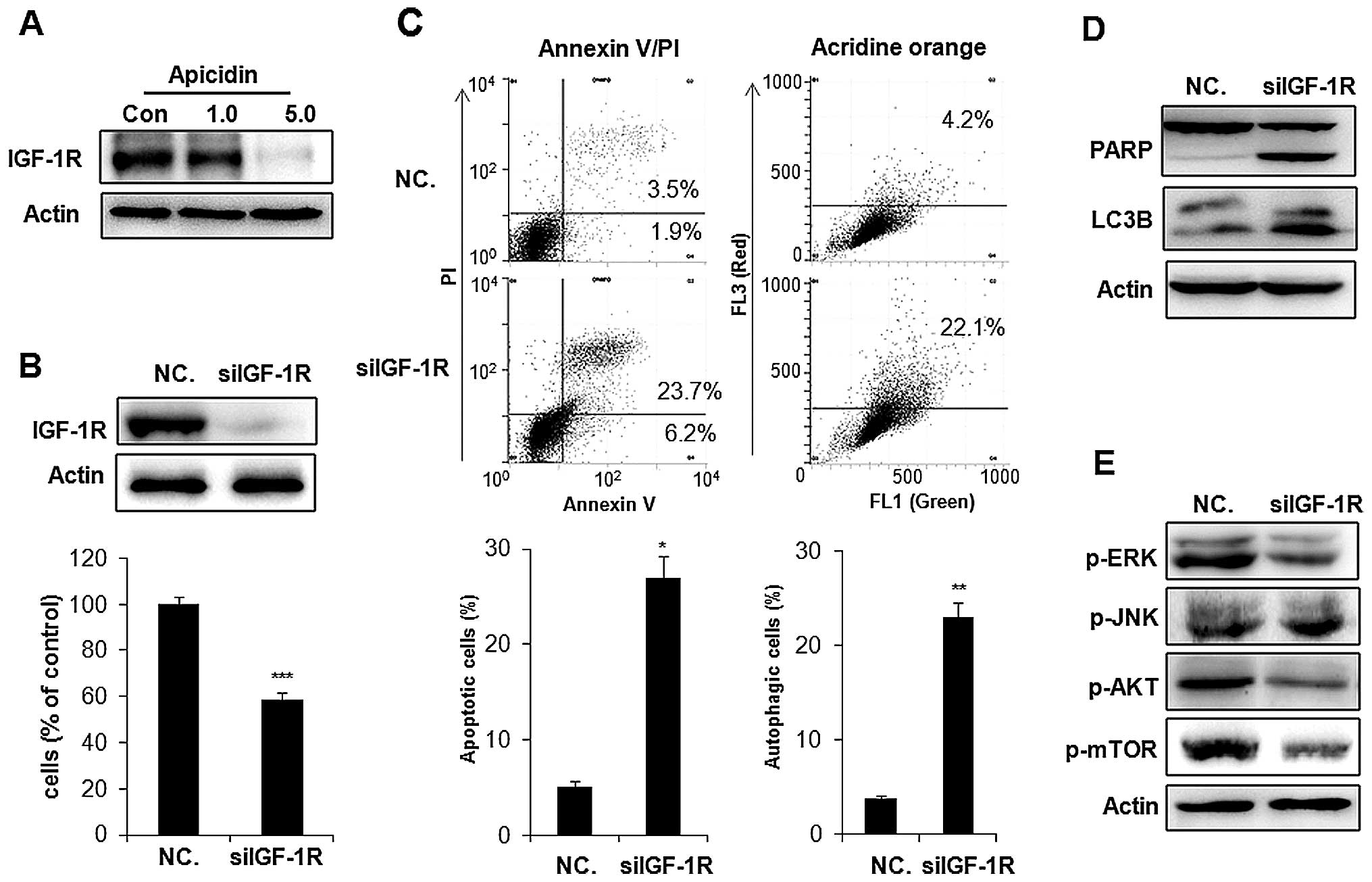 | Figure 8Inhibitory effect of IGF-1R by
apicidin and IGF-1R siRNA transfection in YD-15 cells. (A)
Expression of IGF-1R in apicidin-treated YD-15 cells. The cells
were treated with apicidin (1.0 and 5.0 μM) for 48 h. Cell lysates
were subjected to western blot analysis using an antibody against
IGF-1R. The protein levels were normalized by by a comparison with
the actin levels. (B) The cells were transfected with IGF-1R siRNA
(100 nM) and NC. siRNA (100 nM). After 24 h transfection, the
medium was changed to remove the transfection complexes and the
cells were harvested after 48 h. Western blot analysis was
performed using IGF-1R. The protein levels were normalized by a
comparison with the actin levels. Cell viability was determined
using a trypan blue exclusion assay. (C) Apoptosis was determined
by flow cytometry for Annexin V-FITC and PI dual labeling.
Autophagy was determined by flow cytometry using acridine orange
staining. The data are presented as the mean ± SD of three
independent experiments. **P<0.01,
***P<0.001, compared to the untreated control. (D)
Western blot analysis was performed using PARP and LC3B. The
protein levels were normalized by comparing to the actin levels.
(E) Western blot analysis was performed using p-ERK, p-JNK, p-AKT
and p-mTOR. The protein levels were normalized by a comparison with
the actin levels. The representative results from three independent
experiments are shown. IGF-1R, insulin-like growth factor 1
receptor; PARP, poly ADP ribose polymerase; LC3B, light change 3B;
ERK, extracellular signal-regulated kinase; JNK, c-Jun NH2-terminal
kinase; PI, propidium iodide. |
Discussion
Salivary gland cancers account for ~5% of all head
and neck malignancies (24). MEC is
the most common primary malignancy in the salivary gland, however,
few clinical trial data have addressed the role of systemic therapy
specific to this type of tumor (25). HDAC inhibitors are a relatively new
class of anticancer agents that play an important role in inducing
apoptosis, and cell cycle arrest in several cancer cells (1,8,9).
However, the antitumor effect of HDAC inhibitor on MEC has yet to
be determined. The present study first examined the antitumor
efficacy of apicidin against human MEC cells. The data showed that
apicidin inhibited cell proliferation and HDAC activity in YD-15
cells. Inhibition of cell growth by apicidin was also confirmed
with MC-3 human MEC cells (data not shown). Our previous study
reported for the first time that apicidin induced both apoptosis
and autophagy in OSCC cells (18).
Consistent with this observation, apicidin induced apoptosis as
well as autophagy in YD-15 cells.
Several studies have described autophagy in tumor
cell lines treated with chemotherapeutic agents including HDAC
inhibitor. However, whether this role is protective or toxic to
cancer cells has to be answered differently depending on the model
used (1,26,27).
Autophagy offers protection against cancer by promoting autophagic
cell death or contributing to cancer by promoting the survival of
nutrient-starved cells (28).
Moreover, the relationship between apoptosis and autophagy is
different in cancer cells depending on the type of cell stress
induced; in some cases apoptosis and autophagy act
antagonistically, while in other cases autophagy enables apoptosis
(29–31). To determine the function of
autophagy and the relationship between apoptosis and autophagy, an
autophagy inhibitor, CQ, and caspase inhibitor, z-VAD-fmk, were
treated with apicidin. The inhibition of autophagy by CQ reduced
the cell viability through enhanced apicidin-induced apoptotic cell
death, similar to previous studies on apicidin-treated OSCC cells
(18). By contrast, the inhibition
of apoptosis by z-VAD-fmk increased apicidin-induced autophagy,
resulting in an increase in cell survival. These results suggest
that apoptosis and autophagy by apicidin act antagonistically and
autophagy exerts a protective role towards the activation of
apoptosis by apicidin treatment in human MEC cells.
Although apicidin induces apoptosis and autophagy in
cancer cells, the molecular mechanisms of apicidin remain unclear.
The MAPK and PI3K/AKT/mTOR pathways are important signaling
cascades in human cancer as a result of genetic alterations
(32). The present results showed
that apicidin inhibited the activation of ERK and AKT/mTOR but also
induced JNK activation in YD-15 cells. Although JNK and p38 MAPK
activities are involved in pro-apoptotic events, activation of the
ERK pathway is involved in numerous cell responses, such as cell
proliferation and survival (33).
Similarly, the activated AKT pathway is involved in survival and
proliferation (34). Inhibition of
the MEK/ERK and AKT signaling pathways potentiates HDAC
inhibitor-induced cell death in cancer cells (35,36).
Therefore, we suggest that apicidin inhibits cell proliferation
through the inactivation of ERK, AKT signals and activation of JNK
signal. It was confirmed that ERK and AKT inhibitors enhanced
apicidin-induced cytotoxicity, whereas JNK inhibitors reduced this
cytotoxicity. This result suggests that apicidin-induced cell death
is mediated by the reduction of ERK and AKT, and induction of JNK
pathways in YD-15 cells.
The present study aimed to identify which signaling
pathway is involved in apicidin-induced apoptosis and autophagy.
The MAPK and PI3K/AKT/mTOR signaling pathways are well
characterized with regard to the transmission of anti-apoptotic
signals in cell survival and determining the effect of apoptotic
responses on anticancer drugs (37,38).
Disruption of the PI3K/AKT signaling pathway, culminating in the
inhibition of AKT has been consistently associated with indicators
of autophagy in cancer cells (39,40).
Inhibition of AKT signaling reduces the activity of mTOR, which is
a key negative regulator of autophagy. The MAPK/ERK signaling
pathway has been associated with the regulation of autophagy. In
particular, the upregulation of ERK1/2 activity is imperative for
induction of autophagy in cancer cells (41). Our results show that the inhibition
of ERK and JNK activation oppositely regulated apicidin-induced
apoptosis, although the autophagic effect was unaffected. By
contrast, the inhibition of AKT activation enhanced
apicidin-induced apoptosis and autophagic effects. Therefore, the
data suggest that the apoptotic effect of apicidin is mediated by
the ERK, JNK and AKT/mTOR pathways, whereas the autophagic response
of apicidin is mediated by the AKT/mTOR pathways.
We also aimed to identify a potential upstream
regulator of MAPK and AKT/mTOR pathways modulated by apicidin. The
present study focused on IGF-1R, which is one of the upstream
receptor tyrosine kinase responsible for activating the two major
cascades, PI3K/AKT and MAPK (22).
IGF-1R is frequently overexpressed in several cancers and the
tumor-genetic salivary gland epithelial cells model and is now an
attractive anticancer treatment target (42,43).
Previous studies have shown that HDAC inhibitors inhibited IGF-1R
expression in cancer cells (44,45).
The present data also show that apicidin markedly inhibited the
level of IGF-1R expression in YD-15 cells. Inhibition of IGF-1R
expression by apicidin was also confirmed with MC-3 human MEC cells
(data not shown). To further examine whether the inhibition of
IGF-1R regulates apoptosis and autophagy through MAPK and AKT/mTOR
pathway, IGF-1R was silenced by siRNA transfection in YD-15 cells.
The data show that silencing of IGF-1R with siRNA results in
apoptosis and autophagy through the action of downstream kinases,
and is similar to the results obtained by apicidin treatment in
YD-15 cells. These results suggest that apicidin exerts its
antiproliferative properties through the suppression of IGF-1R in
MEC cells.
In conclusion, apicidin induces apoptotic cell death
by inactivating the ERK and AKT/mTOR signaling pathways and
activating JNK signaling, whereas apicidin promoted pro-survival
autophagy by inactivating the AKT/mTOR signaling pathways in YD-15
cells. These two events are mediated by the inhibition of IGF-1R.
Therefore, the present study suggests that apicidin is an
attractive antitumor agent for MEC treatment and a good candidate
for targeting IGF-1R for cancer therapy. In addition, autophagy
inhibitor enhances the anti-therapeutic effects of apicidin against
human MEC.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(NRF-2011-0023907, NRF-2013R1A2A2A01067254).
References
|
1
|
Kim HJ and Bae SC: Histone deacetylase
inhibitors: molecular mechanisms of action and clinical trials as
anti-cancer drugs. Am J Transl Res. 3:166–179. 2011.PubMed/NCBI
|
|
2
|
Grunstein M: Histone acetylation in
chromatin structure and transcription. Nature. 389:349–352. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miller CP, Singh MM, Rivera-Del Valle N,
Manton CA and Chandra J: Therapeutic strategies to enhance the
anti-cancer efficacy of histone deacetylase inhibitors. J Biomed
Biotechnol. 2011:5142612011. View Article : Google Scholar
|
|
4
|
Carew JS, Giles FJ and Nawrocki ST:
Histone deacetylase inhibitors: mechanisms of cell death and
promise in combination cancer therapy. Cancer Lett. 269:7–17. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Spiegel S, Milstien S and Grant S:
Endogenous modulators and pharmacological inhibitors of histone
deacetylases in cancer therapy. Oncogene. 31:537–551. 2012.
|
|
6
|
Jung M: Inhibitors of histone deacetylase
as new anticancer agents. Curr Med Chem. 8:1505–1511. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Darkin-Rattray SJ, Gurnett AM, Myers RW,
et al: Apicidin: a novel antiprotozoal agent that inhibits parasite
histone deacetylase. Proc Natl Acad Sci USA. 93:13143–13147. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ahn MY, Na YJ, Lee JW, Lee BM and Kim HS:
Apicidin induces apoptosis via Cytochrome c-mediated intrinsic
pathway in human ovarian cancer cells. Biomol Ther. 17:17–24. 2009.
View Article : Google Scholar
|
|
9
|
Im JY, Park H, Kang KW, Choi WS and Kim
HS: Modulation of cell cycles and apoptosis by apicidin in estrogen
receptor (ER)-positive and-negative human breast cancer cells. Chem
Biol Interact. 172:235–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahn MY, Lee J, Na YJ, Choi WS, Lee BM,
Kang KW and Kim HS: Mechanism of apicidin-induced cell cycle arrest
and apoptosis in Ishikawa human endometrial cancer cells. Chem Biol
Interact. 179:169–177. 2009. View Article : Google Scholar
|
|
11
|
Choi KH, Shim JH, Huong LD, Cho NP and Cho
SD: Inhibition of myeloid cell leukemia-1 by tolfenamic acid
induces apoptosis in mucoepidermoid carcinoma. Oral Dis.
17:469–475. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen S, Kepp O, Michaud M, et al:
Association and dissociation of autophagy, apoptosis and necrosis
by systematic chemical study. Oncogene. 30:4544–4556. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Portt L, Norman G, Clapp C, Greenwood M
and Greenwood MT: Anti-apoptosis and cell survival: a review.
Biochim Biophys Acta. 1813:238–259. 2011. View Article : Google Scholar
|
|
14
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carew JS, Nawrocki ST and Cleveland JL:
Modulating autophagy for therapeutic benefit. Autophagy. 3:464–467.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rikiish H: Autophagic and apoptotic
effects of HDAC inhibitors on cancer cells. J Biomed Biotechnol.
2011:8302602011.
|
|
17
|
Amaravadi RK and Thompson DB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahn MY, Ahn SG and Yoon JH: Apicidin, a
histone deaceylase inhibitor, induces both apoptosis and autophagy
in human oral squamous carcinoma cells. Oral Oncol. 47:1032–1038.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee EJ, Kim J, Lee SA, Kim EJ, Chun YC,
Ryu MH and Yook JI: Characterization of newly established oral
cancer cell lines derived from six squamous cell carcinoma and two
mucoepidermoid carcinoma cells. Exp Mol Med. 37:379–390. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanematsu S, Uehara N, Miki H, Yoshizawa
K, Kawanaka A, Yuri T and Tsubura A: Autophagy inhibition enhances
sulforaphane-induced apoptosis in human breast cancer cells.
Anticancer Res. 30:3381–3390. 2010.PubMed/NCBI
|
|
21
|
Warnken M, Reitzenstein U, Sommer A, et
al: Characterization of proliferative effects of insulin, insulin
analogues and insulin-like growth factor-1 (IGF-1) in human lung
fibroblasts. Naunyn Schmiedebergs Arch Pharmacol. 382:511–524.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brunet A and Pouyssegur J: Mammalian MAP
kinase modules: how to transduce specific signals. Essays Biochem.
32:1–16. 1997.PubMed/NCBI
|
|
23
|
LeRoith D and Roberts CT Jr: The
insulin-like growth factor system and cancer. Cancer Lett.
195:127–137. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lalami Y, Vereecken P, Dequanter D,
Lothaire P and Awada A: Salivary gland carcinomas, paranasal sinus
cancers and melanoma of the head and neck: an update about rare but
challenging tumors. Curr Opin Oncol. 18:258–265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McHugh CH, Roberts DB, El-Naggar AK, et
al: Prognostic factors in mucoepidermoid carcinoma of the salivary
glands. Cancer. 118:3928–3936. 2012. View Article : Google Scholar
|
|
26
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kond Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR
and Chem CC: Autophagy potentiates the anti-cancer effects of the
histone deacetylase inhibitors in hepatocellular carcinoma.
Autophagy. 6:1057–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amaravadi RK, Yu D, Lum JJ, et al:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boya P, Gonzalez-Polo RA, Casares N, et
al: Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol.
25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan CH, Liang ZQ, Gu ZL, Yang YP, Reid P
and Qin ZH: Contributions of autophagic and apoptotic mechanisms to
CrTX-induced death of K562 cells. Toxicon. 47:521–530. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Luca A, Maiello MR, D’Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signalling pathways: role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16:S17–27.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu C, Friday BB, Lai JP, McCollum A,
Atadja P, Roberts LR and Adjei AA: Abrogation of MAPK and Akt
signaling by AEE788 synergistically potentiates histone deacetylase
inhibitor-induced apoptosis through reactive oxygen species
generation. Clin Cancer Res. 13:1140–1148. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sarker D, Reid AH, Yap TA and de Bono JS:
Targeting the PI3K/AKT pathway for the treatment of prostate
cancer. Clin Cancer Res. 15:4799–4805. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jang ER, Kim YJ, Myung SC, Kim W and Lee
CS: Different effect of protein kinase B/Akt and extracellular
signal-regulated kinase inhibition on trichostatin A-induced
apoptosis in epithelial ovarian carcinoma cell lines. Mol Cell
Biochem. 353:1–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nishioka C, Ikezoe T, Yang J, Koeffler HP
and Yokoyama A: Inhibition of MEK/ERK signaling synergistically
potentiates histone deacetylase inhibitor-induced growth arrest,
apoptosis and acetylation of histone H3 on p21waf1 promoter in
acute myelogenous leukemia cell. Leukemia. 22:1449–1452. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kennedy SG, Wagner AJ, Conzen SD, Jordan
J, Bellacosa A, Tsichlis PN and Hay N: The PI 3-kinase/Akt
signaling pathway delivers an antiapoptotic signal. Genes Dev.
11:701–713. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Widmann C, Gibson S, Jarpe MB and Johnson
GL: Mitogen-activated protein kinase: conservation of a
three-kinase module from yeast to human. Physiol Rev. 79:143–180.
1999.PubMed/NCBI
|
|
39
|
Takeuchi H, Kanzawa T, Kondo Y and Kondo
S: Inhibition of platelet-derived growth factor signaling induces
autophagy in malignant glioma cells. Br J Cancer. 90:1069–1075.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mochizuki T, Asai A, Saito N, et al: Akt
protein kinase inhibits non-apoptotic programmed cell death induced
by ceramide. J Biol Chem. 277:2790–2797. 2002. View Article : Google Scholar
|
|
41
|
Ellington AA, Berhow MA and Singletary KW:
Inhibition of Akt signaling and enhanced ERK1/2 activity are
involved in induction of macroautophagy by triterpenoid B-group
soyasaponins in colon cancer cells. Carcinogenesis. 27:298–306.
2006. View Article : Google Scholar
|
|
42
|
Riedemann J and Macaulay VM: IGF1R
signalling and its inhibition. Endocr Relat Cancer. 13:S33–43.
2006. View Article : Google Scholar
|
|
43
|
Obara K, Ide F, Mishima K, Inoue H, Yamada
H, Hayashi Y and Saito I: Biological and oncogenic properties of
p53-deficient salivary gland epithelial cells with particular
emphasis on stromal-epithelial interactions in tumorigenesis.
Pathobiology. 73:261–270. 2006. View Article : Google Scholar
|
|
44
|
Mitsiades CS, Mitsiades NS, McMullan CJ,
et al: Transcriptional signature of histone deacetylase inhibition
in multiple myeloma: biological and clinical implications. Proc
Natl Acad Sci USA. 101:540–545. 2004. View Article : Google Scholar :
|
|
45
|
Kim SO, Choi BT, Choi IW, et al:
Anti-invasive activity of histone deacetylase inhibitors via the
induction of Egr-1 and the modulation of tight junction-related
proteins in human hepatocarcinoma cells. BMB Rep. 42:655–660. 2009.
View Article : Google Scholar : PubMed/NCBI
|