Introduction
There is a strong rationale for the 'complete'
blockade of the malignant metabolic phenotype as antitumor therapy.
It is currently widely accepted that the 'malignant metabolic
phenotype' is another hallmark of cancer and that it results from a
number of gain-of-function mutations in oncogenes and
loss-of-function of tumor-suppressor genes (1,2). This
malignant metabolic phenotype is characterized by higher rates of
glycolysis, higher rates of glutaminolysis and an increased de
novo synthesis of fatty acids (FAs) or lipogenic phenotype.
Malignant cell proliferation requires energy in the
form of ATP as well as synthesis of macromolecules including
nucleotides, amino acids and lipids. For most mammalian cells in
culture, the only two molecules catabolized in appreciable
quantities are glucose and glutamine. This means that glucose and
glutamine supply most of the carbon and nitrogen for the synthesis
of macromolecules, energy and reducing equivalents necessary to
support cell growth (3–5).
A third metabolic characteristic of the malignant
phenotype is increased lipogenesis and this process requires
glucose and glutamine. Most adult tissues take up circulating FAs
instead of using de novo synthesis (6,7).
During malignant transformation, cells have been shown to
upregulate FA synthesis instead of increasing the uptake of
exogenous FAs to meet the increasing demand for biomass production.
This process is thought to be part of a general metabolic
remodeling from a non-proliferative catabolic phenotype to a
proliferative anabolic phenotype. During this process, glucose is
first converted to acetyl-CoA in the mitochondrial matrix and used
to synthesize citrate in the tricarboxylic acid (TCA) cycle.
Glutamine appears to be critical for lipid synthesis as it supplies
carbon in the form of mitochondrial oxaloacetate to maintain
citrate production in the first step of the TCA cycle. Thus, the
metabolism of glutamine and glucose is orchestrated to support the
production of acetyl-CoA and NADPH needed for FA synthesis
(8,9).
Despite the strong rationale for developing a
combination of drugs to simultaneously target these three key
processes for malignant cells, there is no experimental evidence
available thus far to support this hypothesis despite the
availability of drugs known to inhibit glycolysis, glutaminolysis
and the de novo FA synthesis. In this regard, among
anti-glycolytic drugs, lonidamine, an inhibitor of hexokinase-2
(HK-2) was clinically used but was deemed ineffective (10). Similarly, 6-diazo-5-oxo-L-norleucine
(DON) is a well-known inhibitor of the enzyme glutaminase (GLS)
that was also clinically tested and deemed to be ineffective
(11). Regarding FA synthesis, a
number of experimental compounds have been developed, none of which
has been utilized in the clinic (12). Of these, orlistat has been
extensively evaluated in vitro and in vivo and has
shown promising activity in a number of malignancies due to its
ability to inhibit FA synthase, the gene product of fatty acid
synthase (FASN), responsible for the de novo synthesis of
FAs (13–22).
These above mentioned findings provide strong
support to determine whether a combination of inhibitors of these
three key pathways of cancer cell metabolism exert higher antitumor
effects as compared to inhibition of each pathway individually. The
results of this study showed that the combination of lonidamine,
DON and orlistat, inhibitors of glycolysis, glutaminolysis and
de novo synthesis of FAs, respectively, exhibits significant
efficacy in vitro and is tolerable in vivo.
Materials and methods
Cell lines, drugs and antibodies
Thirteen human malignant cell lines were used for
the drug treatments: Prostate (DU-145 and PC-3), ovarian (SK-OV-3)
and breast (MDA-MB-231 and MCF7) cancer, osteosarcoma (U-2OS),
cervical (HeLa), gastric (AGS), glioma (D54), bladder (T24),
melanoma (A-375), pancreatic (PANC-1) and colon (SW480) cancer. The
cell lines were obtained from the American Type Culture Collection
(ATCC; Manassas, VA, USA). Primary lung fibroblasts from a
non-cancer individual were kindly provided by Dr Moisés Selman
(National Institute for Respiratory Diseases, Mexico City, Mexico)
(23). The cells were cultured in
Dulbecco's modified Eagle's medium-F12 (cat. no. 12400-016)
supplemented with 10% fetal bovine serum (FBS) (cat. no. 16000-044)
with the exception of fibroblasts which were cultured in F-12
medium (cat. no. 21127-022) (all from Gibco-Life Technologies,
Grand Island, NY, USA) supplemented with 5% FBS. The cells were
maintained with 5% CO2 in a humidified incubator at
37°C. Lonidamine (cat. no. 1646) and DON (cat. no. D2145-25MG) were
purchased from Tocris Bioscience (Bristol, UK) and Sigma-Aldrich
(St. Louis, MO, USA), respectively. Orlistat was a gift from
Psicofarma, S.A. de C.V. (Mexico City, Mexico).
Drug solutions
Lonidamine was dissolved in dimethyl sulfoxide
(DMSO; cat. no. D2650), DON in culture medium without serum and
orlistat in ethanol (cat. no. E7023) (all from Sigma-Aldrich).
Stock solutions were prepared and stored at 4°C for up to one week.
Appropriate dilutions from the stock solutions were produced in
medium before adding drugs to the experimental cultures. The final
concentration of DMSO and ethanol in growth medium for all the drug
treatments was maintained at <0.1%.
Cell line treatments
For the cell viability assays, 1×105
cells were seeded in 6-well plates and allowed to attach to the
bottom overnight and then treated with 500 µM DON, 100
µM lonidamine, 25 µM orlistat and a combination of
the three drugs at the same concentration as the one used for
single drug treatment for 48 h. The concentrations for the three
drugs were selected on the information that these were clinically
achievable according to the literature. Thus, all the cell lines
were treated with the same concentration of the drugs.
vehicle-treated cells were used as controls. Treated cells were
stained with 0.4% trypan blue solution and counted under an
inverted microscope with a Neubauer chamber. Any cells that
excluded the dye were considered as viable. Experiments were
performed in triplicate, with duplicates for each experiment.
RT-qPCR. Total RNA isolation from untreated cell lines was
carried out using TRIzol (cat. no. 15596-018; Invitrogen Life
Technologies, Carlsbad, CA, USA) and reverse transcription was
performed from 1 µg of total RNA using random primers and
MultiScribe Reverse Transcriptase (cat. no. 4304134; Applied
Biosystems, Foster City, CA, USA), according to the manufacturer's
instructions. 2 µg cDNA was used for the amplification of
HK-2, GLS and FASN by RT-PCR with Bio-Rad iCycler iQ Real-Time PCR
detection system (Bio-Rad, Hercules, CA, USA). β-actin mRNA levels
were used for normalization of the interest genes. HK-2, GLS and
FASN qPCRs were run using primers and iQ SYBR-Green Supermix (cat.
no. 170-8882; Bio-Rad), whereas β-actin qPCR was run using TaqMAn
probe and 2X TaqMan Universal PCR Master mix (cat. no. 4304437;
Applied Biosystems). PCR reactions were run in triplicate and
repeated in three independent experiments. The standard curve
method for absolute quantification was used to calculate the gene
copy number relative to β-actin mRNA expression levels. The final
primer concentration for HK-2 was 0.5 µM for GLS was 0.4
µM for FASN was 0.5 µM and for β-actin probe was 0.1
µM. PCR conditions used were: AmpErase UNG incubation for 2
min at 50°C (β-actin); polymerase activation for 10 min at 95°C
(HK-2, GLS, FASN and β-actin) and denaturation for 30 sec at 95°C
(HK-2, GLS and FASN) and 15 sec at 95°C (β-actin); annealing for 30
sec at 60°C (HK-2, GLS and FASN) for 1 min at 60°C (β-actin).
Extension and cycle numbers were carried out as follows: 40 sec at
72°C, 40 cycles (GLS and FASN); 40 sec at 72°C, 35 cycles (HK-2)
and 40 cycles (β-actin). The primer pairs used were: GLS forward,
5′-GGAC AGAGGCATTCTACTG-3′ and reverse, 5′ATCTTAGTCCAC TCGGCTC-3′;
HK-2 forward, 5′-GAGAGGGGACTTTGA TATC-3′ and reverse,
5′GGCGTTGCTGCCCGTGCC-3′; FASN forward, 5′-CATCCAGATAGGCCTCATAGAC-3′
and reverse, 5′-CTCCATGAAGTAGGAGTGGAA-3′. The probe for β-actin was
selected from Life Technology pre-designed probes, Hs 01060665_g1
TaqMan Probe.
Immunofluorescence (IF)
SW480 cells were seeded in 4-well chambers (Lab-Tek
II Chamber Slide system; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and allowed to adhere overnight at 37°C with 5%
CO2. The cells were fixed with 2% paraformaldehyde for
20 min and permeabilized with 0.01% Triton X-100 for 30 sec and
blocked with power block universal blocking reagent 10X (cat. no.
BS-1310-25; Biogenex, Fremont, CA, USA) for 1 h. Incubation was
performed overnight at 4°C with the following antibodies and
concentrations: mouse monoclonal raised against recombinant GLS-K
of human origin (cat. no. IJ-2) at 1,000 ng/ml; goat polyclonal
raised against a peptide mapping at the C-terminal of HK-2 of human
origin (cat. no. C-14) at 800 ng/ml; or rabbit polyclonal antibody
raised against amino acids 2205–2504 mapping at the C-terminal of
FASN of human origin (cat. no. H-300) at 400 ng/ml. DyLight
488-conjugated affinipure bovine antigoat (1:200) was used to
reveal HK-2; DyLight 649-conjugated affinipure donkey anti-rabbit
(1:300) to detect FASN; and alexa fluor 594 goat anti-mouse IgG
(H+L) antibody (1:200) for GLS. Antibodies were diluted in 1% BSA
in PBS. Secondary antibody incubation was for 1 h. UltraCruz
Mounting Medium (Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
was used for nuclei staining after secondary antibody incubation
and it contained 4′,6-diamidino-2-phenylindole (DAPI). The slides
were mounted with a coverslip. After each step, washing was
performed with 0.05% PBS Tween-20. The slides were analyzed using a
zeiss Axioplan Epifluorescence microscope (Carl Zeiss, Inc.,
Göettingen, Ger many). The image density was measured by ImageJ
1.440. At least 30 cells were measured per experiment. The primary
antibodies against HK-2, GLS-K and FASN were purchased from Santa
Cruz Biotechnology, Inc. DyLight 488-conjugated affinipure bovine
anti-goat and 649-conjugated affinipure donkey anti-rabbit were
purchased from Jackson Immuno Research Laboratories (West Grove,
PA, USA). Alexa Fluor 594 goat anti-mouse IgG (H+L) antibody was
purchased from Invitrogen Life Technologies.
siRNA transfection
Specific siRNAs were used to knock down the
expression of FASN, GLS and HK-2 genes. SW480 cells
(1×105) were seeded in 6-well plates to achieve ~30%
confluence at the time of transfection. Then, 100 pmol Silencer
select RNAi (HK-2 cat. no. 4390824 ID s6562, GLS cat. no. 4392420
ID s5838 and FASN cat. no. 4390824 ID s5032) were diluted in 200
µl Opti-MEM I reduced serum medium (cat. no. 31985-070) (all
from Ambion, Austin, TX, USA). Lipofectamine RNAiMAX transfection
reagent (2 µl) was diluted in 200 µl Opti-MEM I
reduced serum medium. The two mixes were pooled and incubated for
20 min at room temperature. Silencer select negative control no. 1
siRNA was used as the negative control and had no significant
similarity to human gene sequences and no significant effect on
cell viability. The mix was added to each well containing the cells
in 2 ml Opti-MEM I reduced serum medium without serum and replaced
after 8 h incubation by new fresh media or media plus drugs for the
experiments combining siRNA and the drugs. The siRNA final
concentration for the three genes and the negative control was 50
nM. The cells were collected for RNA extraction after counting
using trypan blue dye exclusion.
Quantification of the synergism of
lonidamine, DON and orlistat
SW480 (1×106) cells were seeded in a
6-well plate and treated for 48 h with different concentrations of
lonidamine, DON and orlistat as monotherapy to establish their
IC50 and in triple combination (lonidamine, DON and
orlistat) at doses determined by the IC50 of each drug,
while maintaining the constant ratio of 4:1:2 for lonidamine, DON
and orlistat, respectively. Trypan blue dye exclusion assay was
used to assess cell viability. To quantify the interaction of the
three drugs, analyses were based on the combination index (CI). A
CI of 1 indicated an additive effect between the agents, whereas a
CI <1 or >1 indicated synergism or antagonism, respectively.
The Chou-Talalay method was used to analyze synergism, using
CalcuSyn software (Biosoft, Cambridge, UK).
Administration of the triple combination
in vivo
To determine whether the triple combination was
tolerable in vivo, 6-week-old BALB/c female mice were
obtained from Harlan Laboratories, Inc. (Indianapolis, IN, USA).
Six mice/cage were housed with access to food and water ad
libitum in a 12:12 light-dark cycle. Prior to treatment, the
animals were allowed to acclimatize for 1 week. The animal studies
were in compliance with policies of the Institutional Research
Ethics Board and Animal Care Committee of the Instituto Nacional de
Cancerología, Mexico (permit nos. CA006/CB595/10 and
INCAN/CC/010/10). Six mice were injected with lonidamine (0.125
mg/kg) and orlistat (240 mg/kg) daily and DON (0.250 mg/kg) on days
1, 4 and 8 for 3 weeks by intraperitoneal route (DON only three
injections in the 21-day cycle). A control vehicle group of 6
animals was treated in an identical schedule. Animal injections
were performed inside a laminar flow cabinet. Changes in body
weight were used as a parameter to measure toxic effects. Each
mouse was clinically inspected and weighed on days 0, 7, 14 and 21.
At week 4, the mice were sacrificed in a CO2 chamber.
All the experimental procedures performed aimed to reduce suffering
and are in strict accordance with the Regional Animal Ethics
Committee approval.
Statistical analysis
Data are presented as means ± standard deviations
(SD) from three determinations. Statistical differences between two
groups were evaluated using the Student's t-test. The Pearson
correlation test was used to analyze the correlation between mRNA
and protein expression of the three enzymes. Statistical analyses
were conducted using Excel 2008 for Mac (Microsoft Corp., Redmond,
WA, USA) for the Student's t-test and SPSS statistics (SPSS, Inc.,
Chicago, IL, USA) for Pearson correlation
Results
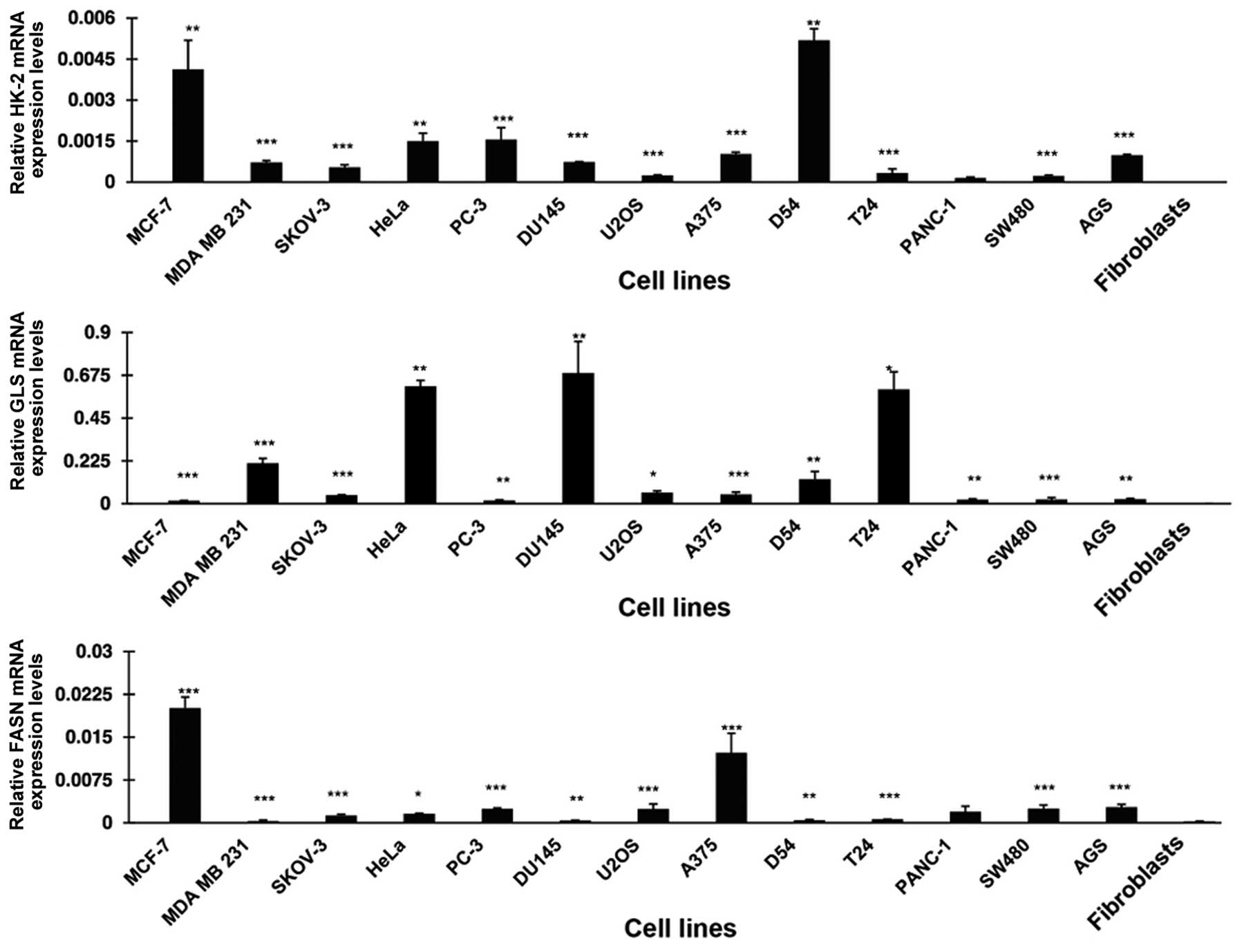
Expression of the mRNA of HK-2, GLS and
FASN
The metabolic phenotype characterized by increased
glycolysis, glutaminolysis and de novo synthesis of FAs is
common to cancer cells. Thus HK-2, GLS and FASN, which are at least
partly responsible for such phenotypes, must be expressed in most
cancer cells. To demonstrate this, we used RT-qPCR to determine the
expression levels of these genes in a panel of cancer cell lines.
Fig. 1 shows that all the cell
lines overexpressed HK-2 at variable degrees in comparison to
normal primary fibroblasts. These differences were statistically
significant, with exception of the pancreatic cancer cells
(PANC-1). MCF7 and D54 had the highest expression. For GLS, all 13
malignant cell lines were overexpressed compared to fibroblasts.
HeLa, DU 145 and T24 had the highest expression whereas MCF7, PC-3,
PANC-1, SW480 and AGS had a lower expression with differences being
statistically significant. FASN was also statistically
significantly overexpressed in all the cell lines analyzed. Each of
the three genes was minimally expressed in primary fibroblasts.
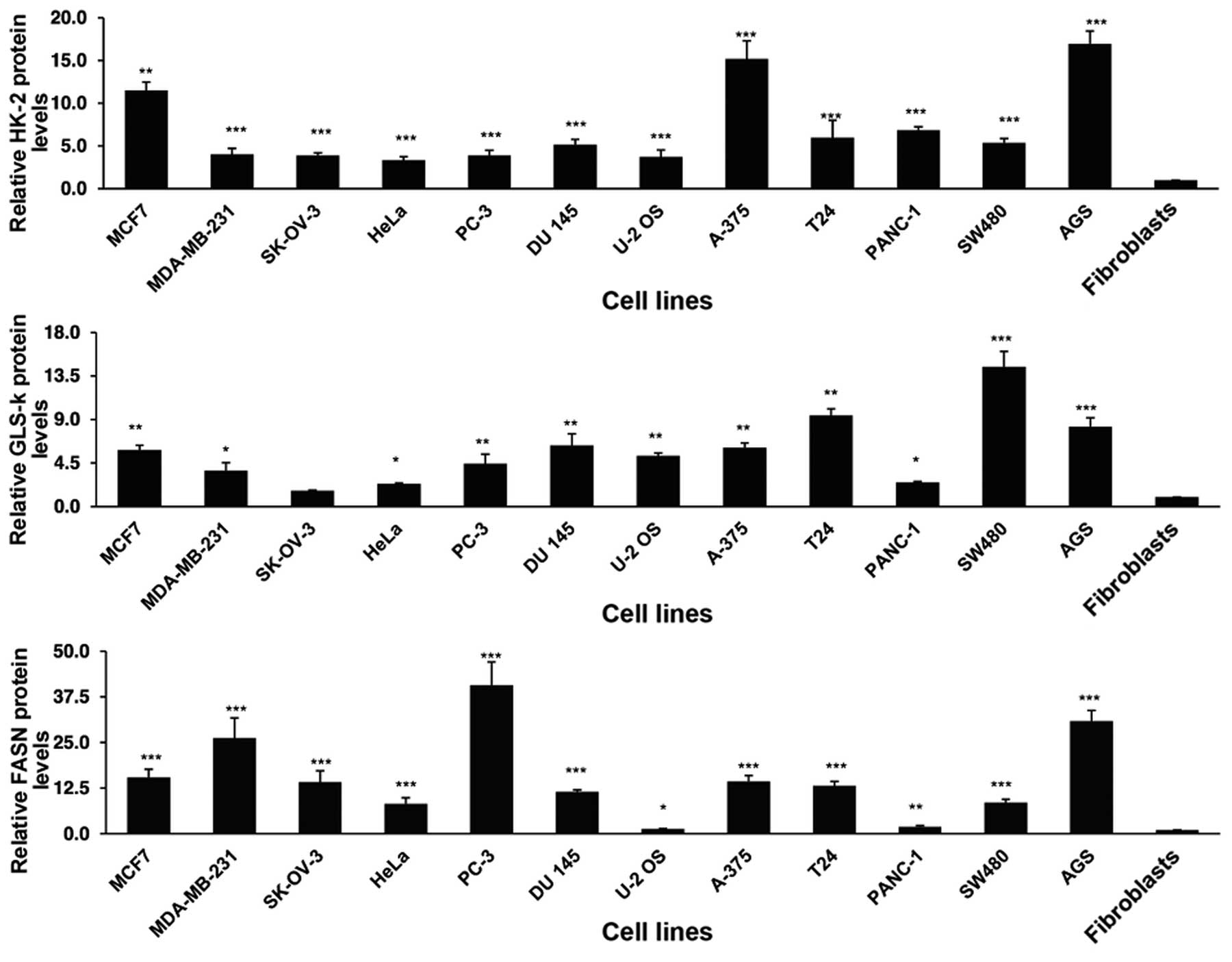
Protein expression of HK-2, GLS-K and
FASN
To investigate whether there was overexpression at
protein level, the cells were analyzed by IF imaging. As shown in
Fig. 2, there was a variable
overexpression for all three gene products in the whole cell lines
in comparison with the primary fibroblasts. The differences were
statistically significant with the exception of GLS in the ovarian
SK-OV-3 cell line compared to fibroblasts.
Correlation between mRNA and protein
expression levels of target genes
To determine whether there was a correlation between
the levels of mRNA and protein in the malignant cell lines, a
correlation analysis was performed. For the three enzymes there was
no correlation between their protein and mRNA expression, GLS
(R=0.055, P=0.86), HK-2 (R=0.487, P=0.91) and FASN (R=0.56,
P=0.056).
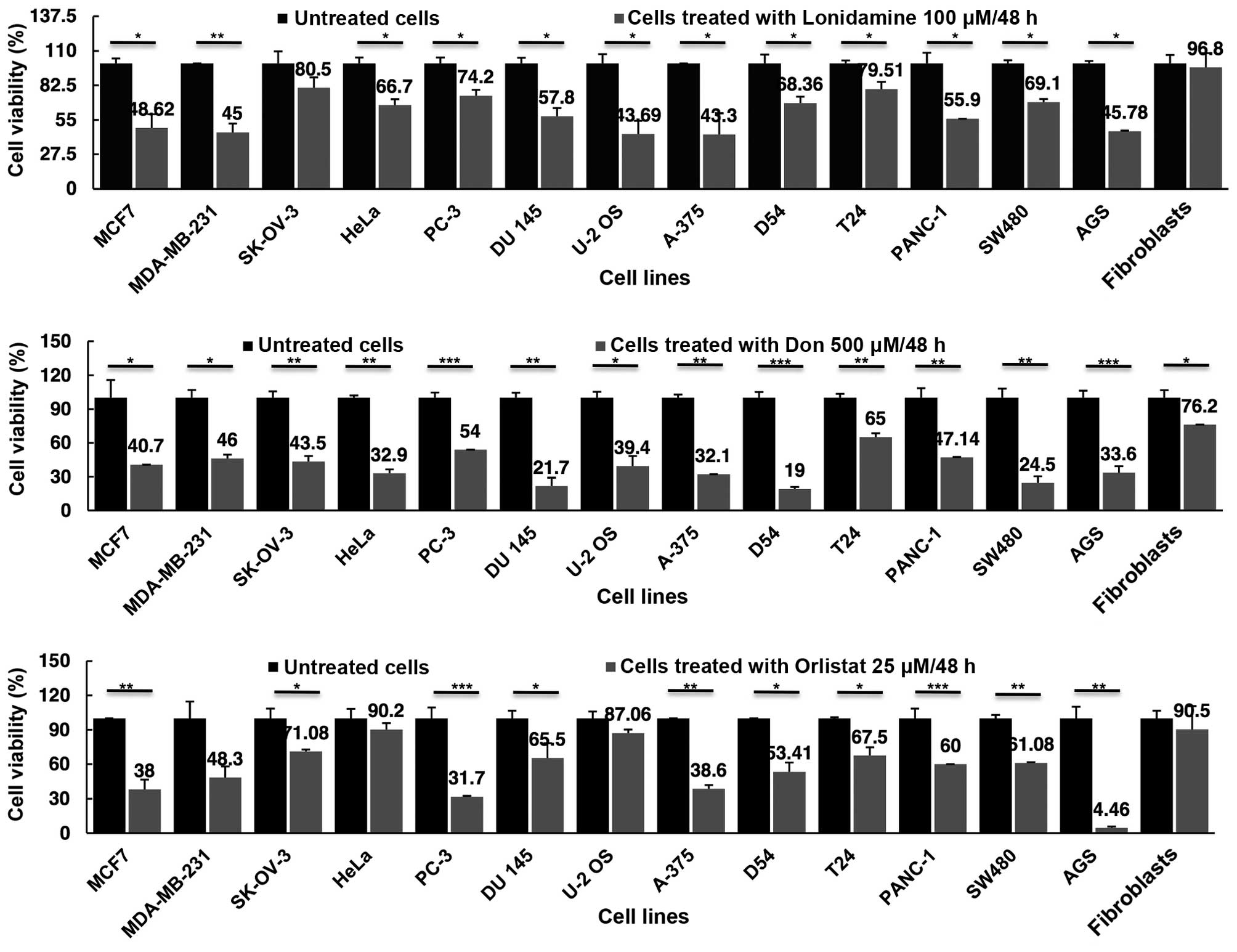
Cell viability inhibition by lonidamine,
DON and orlistat as single drugs
Once demonstrated that all cell lines expressed the
target genes at RNA and protein levels, cell viability assays after
treatments were performed and the results analyzed at 48 h.
Fig. 3 shows that, as compared with
primary fibroblasts, lonidamine led to a reduction in cell
viability ranging from 56.7% for A-375 melanoma cells to 19.5% in
SK-OV-3 ovarian cancer cells. The ovarian cancer cell line was the
only one that did not show any statistically significant difference
(P=0.16). Essentially no cell viability inhibition was observed for
the primary fibroblasts. DON appeared to reduce viability to a
greater extent as compared to lonidamine. For DON, the greatest
inhibition was observed for D54 (81%) and the lowest for T24 (35%),
with all values being statistically significant in comparison to
fibroblasts. Of note DON caused a moderate but significant
viability reduction in the primary fibroblasts (23.8%). Orlistat
was able to decrease cell viability in all the cell lines, although
the difference was not statistically significant for MDA-MB-231,
HeLa and U-2OS cell lines. The less sensitive cells (HeLa) had an
almost identical reduction in viability as compared to 9.8 and 9.5%
in fibroblasts, respectively. It was noteworthy that, orlistat
reached almost a total cell viability inhibition of AGS gastric
cells, 95.5% (P=0.006).
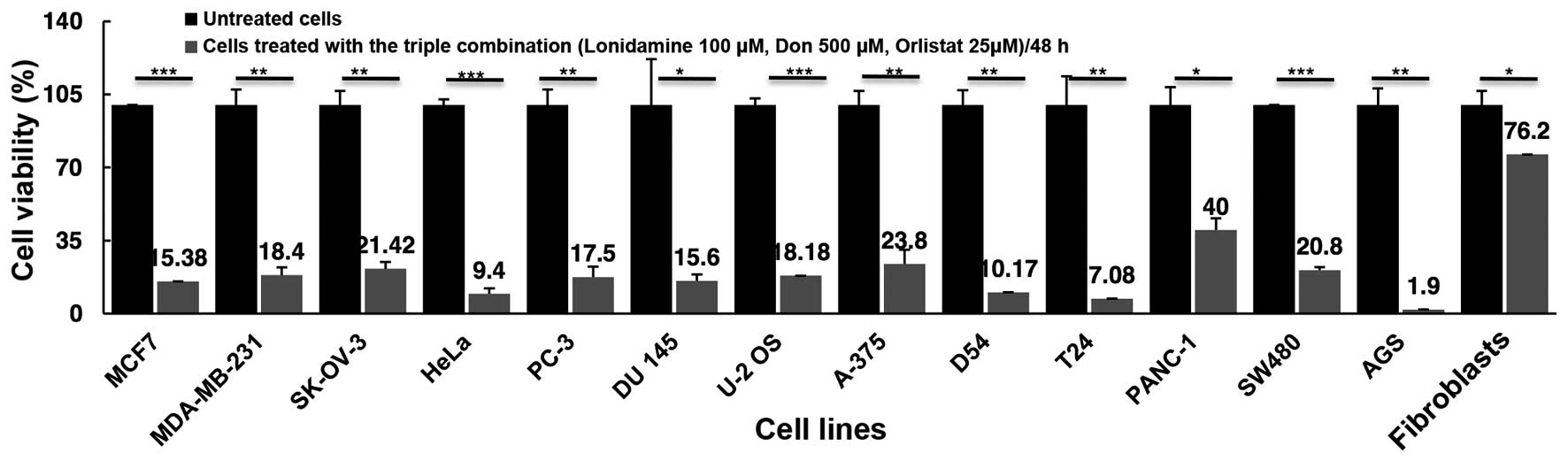
Cell viability inhibition using the
triple combination
As shown in Fig. 4,
the combination of the three drugs led to a marked reduction in
cell viability exceeding 75% inhibition in all but the pancreatic
cancer cell line PANC-1, in which only a 60% reduction was
observed. The triple drug combination treatment had a higher effect
than treatment with any single drug, with the exception of cell
lines, such as AGS (1.9 vs. 4.46% with orlistat) and SW480 (20.08
vs. 24.5% with DON) where the effect of one drug was almost the
same as that of the triple combination effect. By contrast, T24
exerted a much higher cytotoxic effect with the combination of 93%,
compared with DON 35%, lonidamine 21% and orlistat 32.3%. Of utmost
importance, blocking these three 'basic' metabolic pathways under
our experimental conditions showed a minimal but statistically
significant effect upon the viability of primary fibroblasts, with
only a 23.8% (P=0.04) reduction, suggesting that using a 'full
blockade' with these drugs can be feasible in vivo.
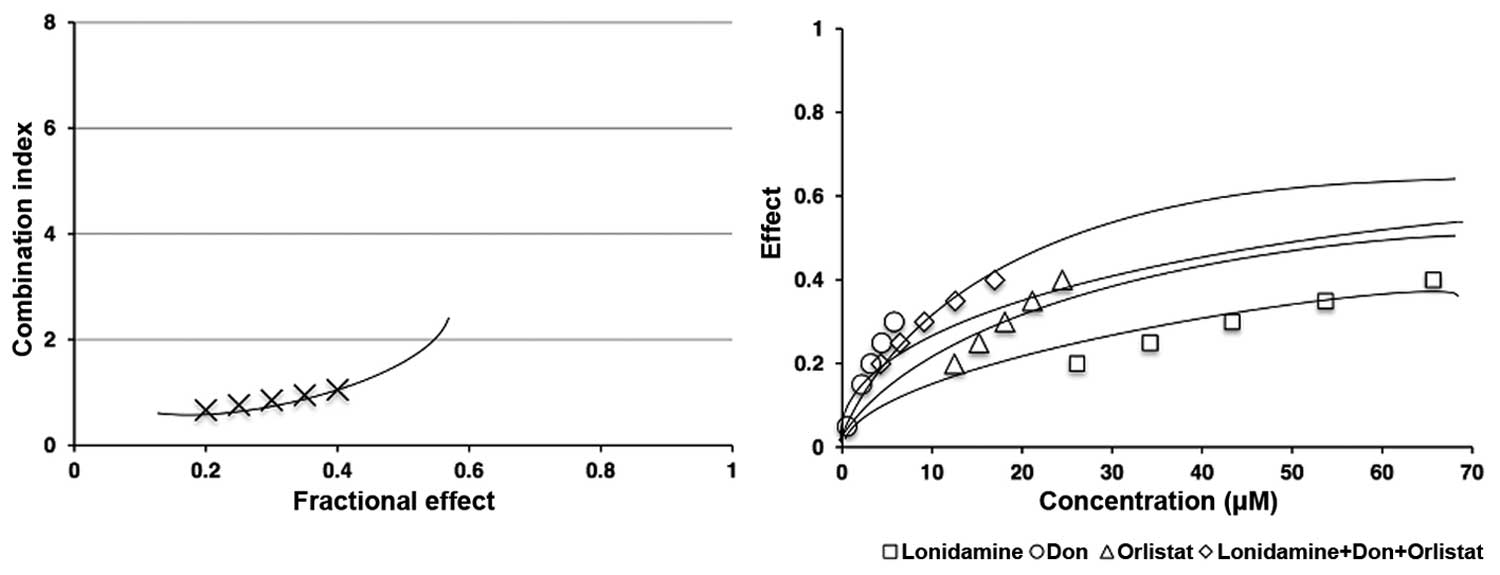
Synergy of the triple combination
We determined whether the triple combination of
lonidamine, DON and orlistat exerted a synergistic, antagonistic,
or additive effect using the Chou-Talalay method. For this purpose,
the SW480 cell line was treated with different concentrations of
each drug (Table I) and the triple
combination, always under the IC50 of the drugs. As shown in
Fig. 5 left panel, lonidamine, DON
and orlistat in combination had a synergistic effect (CI <1)
measured based on cell viability after 48-h treatment. The right
panel shows the comparison between the single and triple
combination treatments. All of our experiments had a linear
correlation coefficient (r) of the median-effect plot >0.90,
which indicates good conformity of the data.
 | Table IEffects of the monotherapy treatment
with lonidamine, DON or orlistat on the SW480 colon cancer cell
line. |
Table I
Effects of the monotherapy treatment
with lonidamine, DON or orlistat on the SW480 colon cancer cell
line.
| Cell line | Lonidamine
(µM) | Fa
(Lonidamine) | DON
(µM) | Fa (DON) | Orlistat
(µM) | Fa (orlistat) |
|---|
| SW480 | 50 | 0.4069 | 15 | 0.4492 | 17.5 | 0.2629 |
| 100 | 0.4257 | 30 | 0.7286 |
35 | 0.6382 |
| 200 | 0.6336 | 60 | 0.7860 |
70 | 0.6841 |
| 300 | 0.7519 | 90 | 0.8156 | 105 | 0.8027 |
| 400 | 0.8662 | 120 | 0.8695 | 140 | 0.9241 |
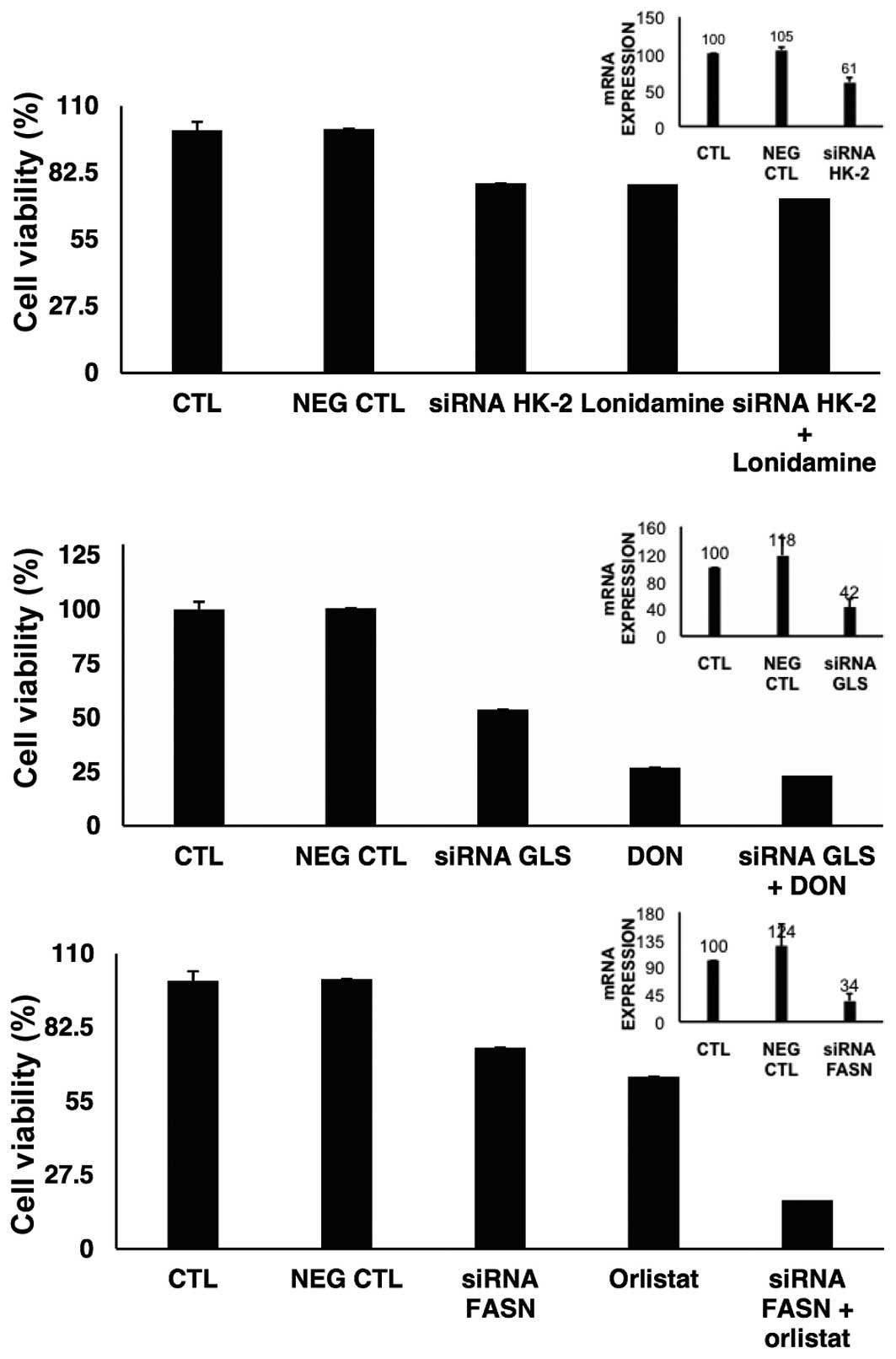
siRNA silencing of target genes
To investigate to what extent the knockdown of HK-2,
GLS or FASN led to cell viability inhibition, the gene expression
was individually suppressed. Results in Fig. 6 show that for HK-2 we were able to
downregulate almost by half the expression of this gene, which led
to 21.8% reduction on viability. The results of the treatment with
lonidamine on viability were very similar to the effects of the
HK-2 knocked down cells. For GLS gene, the suppression was >50%
and the reduction on viability was 46.3%. However, DON led to an
important reduction in cell viability with and without GLS
depletion. FASN was also downregulated >65% and induced only a
small reduction on cell viability. The effect of orlistat was
slightly lower than the effect of depleted mRNA of FASN. However,
cell viability was decreased when orlistat was added to
FASN-depleted cells.
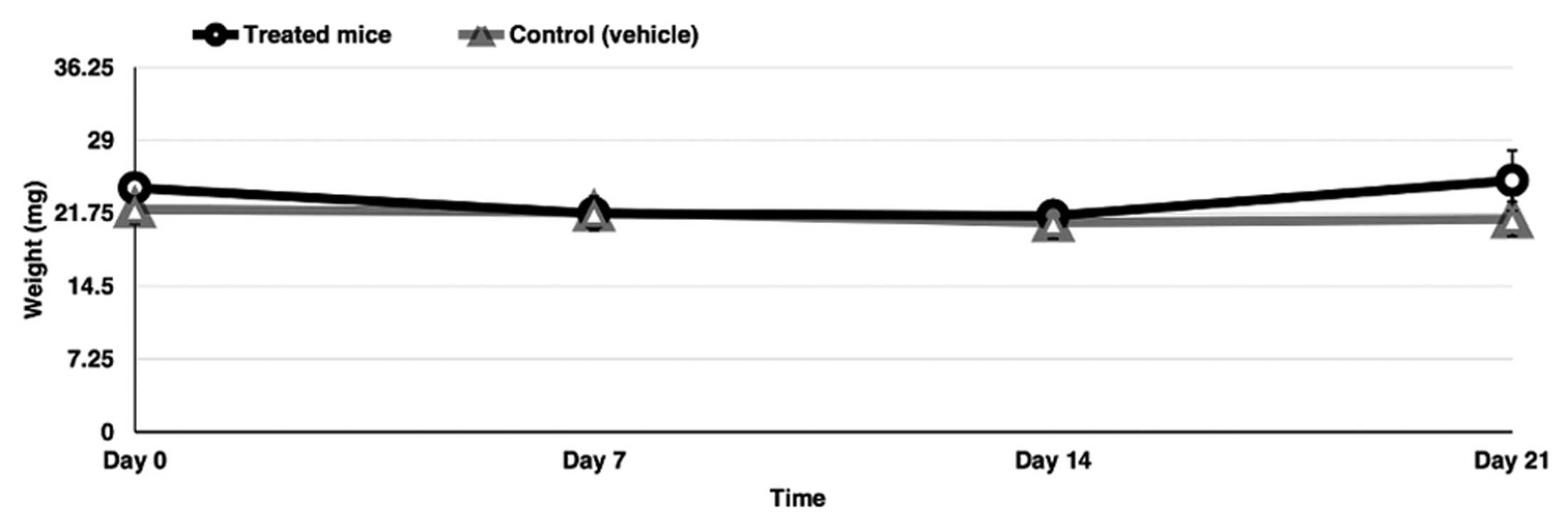
Administration of the triple combination
in vivo
The blockage of glycolysis, glutaminolysis and de
novo FA synthesis may affect normal cells in vivo. To
demonstrate the feasibility of using a triple combination in
vivo and based on the small effect on cell viability on primary
fibroblasts, six BALB/c healthy female mice were treated with the
triple combination of drugs at doses clinically achievable for 3
weeks. Notably, the mice had no significant clinical deterioration
and the weight was only moderately reduced during the first and
second week and then maintained or recuperated at week 4 (Fig. 7).
Discussion
The results of this study show that cancer cells as
compared to primary fibroblasts have overexpression of key genes
whose products are involved in the malignant metabolic phenotype.
Additionally, the use of drugs targeting glycolysis, glutaminolysis
and de novo synthesis of FAs or the downregulation of these
target genes with siRNAs leads to a decrease on cell viability. Of
note, the use of the three drugs in combination markedly increased
the growth inhibitory effect on cancer cells and exerted a clear
synergistic effect in vitro. As predicted by the relatively
low inhibition of primary fibroblasts, the triple combination is
well-tolerated in mice.
In this era of molecular-targeted therapy, most
approved drugs are genotype-driven (24). Thus, drugs are directed to the
product of a single genetic abnormality, while other drugs are
directed to the cancer phenotype. Thus, most classical cytotoxic
drugs as well as current angiogenic inhibitors are directed against
the proliferative (25) and
neoangiogenic phenotypes, respectively (26). The metabolic alterations of cancer
cells were previously described but only recently have they been
particularly investigated. Thus, the malignant metabolic phenotype
is now considered another cancer hallmark (27) and many preclinical studies focusing
on the development of anti-metabolic cancer agents are
underway.
The three most commonly investigated metabolic
alterations of cancer cells are glycolysis, glutaminolysis and the
de novo synthesis of FAs. The increased activities of these
pathways are therefore, natural targets to attack the malignant
metabolic phenotype (28). A number
of preclinical studies using drugs to target either of these
pathways have demonstrated that they are effective. Among
glycolytic inhibitors a high number of this drug class is being
evaluated in experimental systems, reviewed in refs. 29,30.
However, only lonidamine, 2-deoxy-D-glucose and dichloracetate have
reached clinical trials with modest results as single agents or in
combination with chemotherapy or radiation (10,31,32).
In particular, lonidamine the HK-2 inhibitor used in this study,
has been widely investigated for the treatment of solid tumors with
encouraging results in phase II-III trials for the treatment of
advanced breast, ovarian and lung cancer. A review by Di Cosimo
et al (10) concluded that
data are insufficient to draw a firm conclusion on the efficacy of
lonidamine. Thus, it seems clear that this agent merits further
clinical development. Regarding glutaminolysis inhibitors,
azaserine, DON and azotomycin, the three diazo analogs of
L-glutamine that showed preclinical antitumor activity (33), remain unstudied as glutaminolytic
agents, with the exception of a recent study in which DON was used
together with recombinant GLS, showing encouraging results
(34). Instead, newer selective
agents against GLS are being developed. One of these new agents has
recently entered into clinical phases (35). No clinical trials in cancer have
been undertaken with FASN inhibitors, despite a number of these
agents showed encouraging activity in several solid and
hematological malignancies (13–22).
To the best of our knowledge, no preclinical studies
have been performed with this triple drug combination. The most
closely related study was undertaken in 1993, in which the
combination of DON and 2-deoxy-D-glucose led to marked inhibition
of both glutamine oxidation and glycolysis which was accompanied by
increased cytotoxicity against the human TPH-1 myeloid cell line
and freshly cultured myeloid blast cultures obtained from a patient
(36).
The results clearly demonstrate that despite the
metabolic plasticity of malignancies, most cancer cells need to
overexpress these enzymes as a source of energy and precursors for
macromolecule synthesis (glucose and glutamine) as well as the key
enzyme to synthesize FAs, in other words, to become anabolic. In
addition, the relevance of such a pattern of gene expression is
shown by the fact that their pharmacological or genetic
downregulation, even individually, results in growth inhibition.
The variable effect of adding lonidamine, DON or orlistat on cells
with depletion of the gene-coding enzyme can be explained by the
fact that as expected, most drugs have off-target effects and
complex diseases such as cancer may require combinatorial
therapeutic approaches (37). This
is most notable for orlistat where the inhibition of viability is
higher when orlistat is added to FASN-downregulated cells.
Nevertheless, the results of these experiments must be considered
with caution because the efficiency of downregulation by siRNA was
limited. The doses used for each individual drug to assay drug
inhibition were those used in other studies and/or taken from
clinical studies of DON and lonidamine where the plasma
concentration in patients was determined (10,33).
However, the most interesting fact is that the assays used to
determine the pharmacological interaction among them showed that
they are highly synergistic, particularly with lower doses of each
of them. Nevertheless, these are in vitro assays thus an
in vivo pharmacological interaction should be performed to
confirm the results.
The results are of relevance in the field of drug
repositioning for cancer therapy. As stated above, DON and
lonidamine were previously clinically tested, mostly when knowledge
on the metabolic alterations of cancers was limited (10,33).
In fact, these drugs were used as a single agent or in combination
with cytotoxic drugs and overall, they were well tolerated.
Orlistat, an oral drug, has never been used systemically in humans
but data from literature indicate that its systemic absorption is
almost none. In a study, it was found that long-term oral
administration of this drug reaches <10 ng/ml or 0.02 µM
in plasma (38). A case report on a
child who ingested a massive dose of oral orlistat showed no
systemic toxicity (39), suggesting
that its systemic administration was tolerable. Our results of the
cultured cells indicated that in general, primary fibroblasts are
less inhibited than cancer cell lines with any of these drugs or
the combination. In addition, the triple combination administered
in normal BALB/c mice at doses clinically achievable do not induce
major weight loss, suggesting that the repositioning of these drugs
for cancer treatment as a combination is feasible.
Although in this study we did not analyze enzymatic
inhibition of these drugs, its inhibitory effect on each of the
enzymes mentioned has been previously demonstrated. Therefore, we
can state that a 'full blockade' of the three most characterized
metabolic pathways observed in cancer has promising antitumor
activity in vitro and the treatment is well tolerated in
vivo. As cancer cells are able to reprogramme their metabolism
depending on nutrient availability (40,41),
more studies are needed on how cells respond to metabolic pathway
inhibition. In addition, antitumor in vivo models of the
combination should be undertaken.
In conclusion, to the best of our knowledge, our
results demonstrate, for the first time, that cancer cells in
general simultaneously overexpressed three key glycolytic,
glutaminolytic and FA synthesis enzymes and that their concurrent
inhibition in vitro has potent antitumor activity and
synergy, suggesting that a full blockade of these metabolic
alterations may be feasible and should be pursued with re-purposed
or novel drugs.
Acknowledgments
Diana Cervantes-Madrid was a PhD student in the
Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional
Autónoma de México, and received a scholarship from Consejo
Nacional de Ciencia y Tecnología, Conacyt, México (245314), from
Programa de Apoyo a los Estudios de Posgrado (PAEPUNAM) and the
Programa de Movilidad Internacional de Estudiantes of the Dirección
General de Estudios de Posgrado (DGEP-UNAM). This work was
supported by CONACyT grant no. 140654. We also like to thank Dr
victor Ruiz-López and Dr Marcela Lizano-Soberon for their support
and guidance in this project.
References
|
1
|
Chen JQ and Russo J: Dysregulation of
glucose transport, glycolysis, TCA cycle and glutaminolysis by
oncogenes and tumor suppressors in cancer cells. Biochim Biophys
Acta. 1826:370–384. 2012.PubMed/NCBI
|
|
2
|
Levine AJ and Puzio-Kuter AM: The control
of the metabolic switch in cancers by oncogenes and tumor
suppressor genes. Science. 330:1340–1344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barger JF and Plas DR: Balancing
biosynthesis and bioenergetics: Metabolic programs in oncogenesis.
Endocr Relat Cancer. 17:R287–R304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Newsholme EA, Crabtree B and Ardawi MS:
The role of high rates of glycolysis and glutamine utilization in
rapidly dividing cells. Biosci Rep. 5:393–400. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McKeehan WL: Glycolysis, glutaminolysis
and cell proliferation. Cell Biol Int Rep. 6:635–650. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Menendez JA, Ropero S, Mehmi I, Atlas E,
Colomer R and Lupu R: Overexpression and hyperactivity of breast
cancer-associated fatty acid synthase (oncogenic antigen-519) is
insensitive to normal arachidonic fatty acid-induced suppression in
lipogenic tissues but it is selectively inhibited by tumoricidal
α-linolenic and γ-linolenic fatty acids: A novel mechanism by which
dietary fat can alter mammary tumorigenesis. Int J Oncol.
24:1369–1383. 2004.PubMed/NCBI
|
|
7
|
Wang Y, Jones Voy B, Urs S, Kim S,
Soltani-Bejnood M, Quigley N, Heo YR, Standridge M, Andersen B,
Dhar M, et al: The human fatty acid synthase gene and de novo
lipogenesis are coordinately regulated in human adipose tissue. J
Nutr. 134:1032–1038. 2004.PubMed/NCBI
|
|
8
|
Little JL and Kridel SJ: Fatty acid
synthase activity in tumor cells. Subcell Biochem. 49:169–194.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Di Cosimo S, Ferretti G, Papaldo P,
Carlini P, Fabi A and Cognetti F: Lonidamine: Efficacy and safety
in clinical trials for the treatment of solid tumors. Drugs Today.
39:157–174. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kisner DL, Catane R and Muggia FM: The
rediscovery of DON (6-diazo-5-oxo-L-norleucine). Recent Results
Cancer Res. 74:258–263. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lupu R and Menendez JA: Pharmacological
inhibitors of Fatty Acid Synthase (FASN) - catalyzed endogenous
fatty acid biogenesis: A new family of anti-cancer agents? Curr
Pharm Biotechnol. 7:483–493. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Menendez JA, Vellon L and Lupu R:
Antitumoral actions of the anti-obesity drug orlistat (Xenical™) in
breast cancer cells: Blockade of cell cycle progression, promotion
of apoptotic cell death and PEA3-mediated transcriptional
repression of Her2/neu (erbB-2) oncogene. Ann Oncol. 16:1253–1267.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang CS, Matsuura K, Huang NJ, Robeson AC,
Huang B, Zhang L and Kornbluth S: Fatty acid synthase inhibition
engages a novel caspase-2 regulatory mechanism to induce ovarian
cancer cell death. Oncogene. 34:3264–3272. 2015. View Article : Google Scholar
|
|
15
|
Fujiwara J, Sowa Y, Horinaka M, Koyama M,
Wakada M, Miki T and Sakai T: The anti-obesity drug orlistat
promotes sensitivity to TRAIL by two different pathways in
hormone-refractory prostate cancer cells. Int J Oncol.
40:1483–1491. 2012.PubMed/NCBI
|
|
16
|
Zecchin KG, Rossato FA, Raposo HF, Melo
DR, Alberici LC, Oliveira HC, Castilho RF, Coletta RD, Vercesi AE
and Graner E: Inhibition of fatty acid synthase in melanoma cells
activates the intrinsic pathway of apoptosis. Lab Invest.
91:232–240. 2011. View Article : Google Scholar
|
|
17
|
Samudio I, Harmancey R, Fiegl M,
Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W,
Duvvuri S, Taegtmeyer H, et al: Pharmacologic inhibition of fatty
acid oxidation sensitizes human leukemia cells to apoptosis
induction. J Clin Invest. 120:142–156. 2010. View Article : Google Scholar :
|
|
18
|
Kant S, Kumar A and Singh SM: Tumor growth
retardation and chemosensitizing action of fatty acid synthase
inhibitor orlistat on T cell lymphoma: Implication of reconstituted
tumor microenvironment and multidrug resistance phenotype. Biochim
Biophys Acta. 1840:294–302. 2014. View Article : Google Scholar
|
|
19
|
Tirado-Vélez JM, Joumady I, Sáez-Benito A,
Cózar-Castellano I and Perdomo G; Tirado-Vélez JM: Inhibition of
fatty acid metabolism reduces human myeloma cells proliferation.
PLoS One. 7:e464842012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Olsen AM, Eisenberg BL, Kuemmerle NB,
Flanagan AJ, Morganelli PM, Lombardo PS, Swinnen JV and Kinlaw WB:
Fatty acid synthesis is a therapeutic target in human liposarcoma.
Int J Oncol. 36:1309–1314. 2010.PubMed/NCBI
|
|
21
|
Dowling S, Cox J and Cenedella RJ:
Inhibition of fatty acid synthase by Orlistat accelerates gastric
tumor cell apoptosis in culture and increases survival rates in
gastric tumor bearing mice in vivo. Lipids. 44:489–498. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chuang HY, Chang YF and Hwang JJ:
Antitumor effect of orlistat, a fatty acid synthase inhibitor, is
via activation of caspase-3 on human colorectal carcinoma-bearing
animal. Biomed Pharmacother. 65:286–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pardo A, Selman M, Ramírez R, Ramos C,
Montaño M, Stricklin G and Raghu G: Production of collagenase and
tissue inhibitor of metalloproteinases by fibroblasts derived from
normal and fibrotic human lungs. Chest. 102:1085–1089. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang RS and Ratain MJ: Pharmacogenetics
and pharmacogenomics of anticancer agents. CA Cancer J Clin.
59:42–55. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fernandes DJ, Sur P, Kute TE and Capizzi
RL: Proliferation-dependent cytotoxicity of methotrexate in murine
L5178Y leukemia. Cancer Res. 48:5638–5644. 1988.PubMed/NCBI
|
|
26
|
Gatto B and Cavalli M: From proteins to
nucleic acid-based drugs: The role of biotech in anti-VEGF therapy.
Anticancer Agents Med Chem. 6:287–301. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao Y, Liu H, Riker AI, Fodstad O, Ledoux
SP, Wilson GL and Tan M: Emerging metabolic targets in cancer
therapy. Front Biosci. 16:1844–1860. 2011. View Article : Google Scholar
|
|
29
|
Ganapathy-Kanniappan S and Geschwind JF:
Tumor glycolysis as a target for cancer therapy: Progress and
prospects. Mol Cancer. 12:1522013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Granchi C and Minutolo F: Anticancer
agents that counteract tumor glycolysis. ChemMedChem. 7:1318–1350.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dwarakanath BS, Singh D, Banerji AK, Sarin
R, Venkataramana NK, Jalali R, Vishwanath PN, Mohanti BK, Tripathi
RP, Kalia VK, et al: Clinical studies for improving radiotherapy
with 2-deoxy-D-glucose: Present status and future prospects. J
Cancer Res Ther. 5(Suppl 1): S21–S26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garon EB, Christofk HR, Hosmer W, Britten
CD, Bahng A, Crabtree MJ, Hong CS, Kamranpour N, Pitts S,
Kabbinavar F, et al: Dichloroacetate should be considered with
platinum-based chemotherapy in hypoxic tumors rather than as a
single agent in advanced non-small cell lung cancer. J Cancer Res
Clin Oncol. 140:443–452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Catane R, Von Hoff DD, Glaubiger DL and
Muggia FM: Azaserine, DON and azotomycin: Three diazo analogs of
L-glutamine with clinical antitumor activity. Cancer Treat Rep.
63:1033–1038. 1979.PubMed/NCBI
|
|
34
|
Unger C, Mueller C, Bausch MP, et al: A
phase I schedule optimization study of pegylatedglutaminase
(PEG-PGA) plus 6-diazo-5-oxo-l-norleucine (DON) in patients (pts)
with advanced solid tumors. J Clin Oncol. 29:abs. 3049. 2011.
|
|
35
|
Strickland DK: Study of the glutaminase
inhibitor CB-839 in solid tumors. Clinical Trials Identifier:
NCT02071862. February. 2014, http://clinicaltrials.gov/ct2/results?term=glutaminasecancer&Search=Search.
Last updated March 9, 2015.
|
|
36
|
Griffths M, Keast D, Patrick G, Crawford M
and Palmer TN: The role of glutamine and glucose analogues in
metabolic inhibition of human myeloid leukaemia in vitro. Int J
Biochem. 25:1749–1755. 1993. View Article : Google Scholar
|
|
37
|
Anighoro A, Bajorath J and Rastelli G:
Polypharmacology: Challenges and opportunities in drug discovery. J
Med Chem. 57:7874–7887. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhi J, Mulligan TE and Hauptman JB:
Long-term systemic exposure of orlistat, a lipase inhibitor and its
metabolites in obese patients. J Clin Pharmacol. 39:41–46. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
O'Connor MB: An orlistat 'overdose' in a
child. Ir J Med Sci. 179:3152010. View Article : Google Scholar
|
|
40
|
Young CD and Anderson SM: Sugar and fat -
that's where it's at: Metabolic changes in tumors. Breast Cancer
Res. 10:2022008. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sankaranarayanapillai M, Zhang N, Baggerly
KA and Gelovani JG: Metabolic shifts induced by fatty acid synthase
inhibitor orlistat in non-small cell lung carcinoma cells provide
novel pharmacodynamic biomarkers for positron emission tomography
and magnetic resonance spectroscopy. Mol Imaging Biol. 15:136–147.
2013. View Article : Google Scholar :
|