Introduction
High-throughput approaches have revealed numerous
overlapping transcripts. Approximately 50% of the human genome was
found to be bidirectionally transcribed and the transcripts from
the opposite strand of a functional gene were denoted natural
antisense transcripts (NATs) (1,2). The
study of gene regulation by antisense transcription is particularly
noteworthy, as their genomic arrangement immediately indicates that
they may act on each other. NATs, most of which are antisense long
non-coding RNAs (AS lncRNAs), play versatile roles in refined
regulation in almost all stages of gene expression, including DNA
methylation, transitional interference, RNA splicing, editing,
stabilization, localization, and translation (3–6).
Recent findings have shown that several AS lncRNAs are dysregulated
in human cancers, and their aberrant expression is associated with
tumorigenesis or metastasis (7–9).
However, compared with sense protein-coding genes, the
characteristics of their AS transcripts remain to be
determined.
Sirtuins are NAD+-dependent deacetylases
and major factors in the response to cellular stresses. The
importance of sirtuins is manifested by their roles in several
major human disorders, such as cancer, cardiovascular diseases,
muscular atrophy and neurodegenerative diseases (10–13).
SIRT1 is the best-characterized sirtuin in the seven-member family
(SIRT1-7), which is involved in cell survival, the cellular stress
response, energy metabolism and cell apoptosis (14–16).
Silencing SIRT1 in cancer cell lines has been reported to restrain
cancer cell proliferation and result in cell cycle arrest and
apoptosis (17). Dozens of
substrates of SIRT1 have been identified and well-characterized,
including p53, p73, FOXO3a, PGC-1α and PPAR (18). There has been a focus on ncRNAs,
which modulate SIRT1 expression and function in the development and
progression of human cancer. It has been demonstrated that human
miR-29c suppresses SIRT1 by binding to the 3′UTR region of SIRT1
causing translational inhibition in hepatocellular carcinoma (HCC)
cells (19). Other ncRNAs such as
miR-34a, miR-195, miR-132 and Let-7g were also proven to target the
SIRT1 gene. However, to the best of our knowledge, no report
identifying that lncRNA selectively regulates SIRT1 is
available.
In the present study, we identified a single-exon
NAT transcribed from the oncogenic SIRT1 antisense strand,
designated as SIRT1-AS. SIRT1-AS masked the miR-29c binding site on
the SIRT1 3′UTR and stabilized SIRT1 mRNA. Wild-type SIRT1-AS had a
promoting effect on HCC cell proliferation in vitro. A
single-nucleotide mutation (622U>C) was identified in SIRT1-AS,
which caused its inability to stabilize SIRT1 mRNA and decreased
the risk of HCC.
Materials and methods
Cell culture
The HCC-9903 cell line was presented by Professor
Liu Chunsheng from the Qilu Hospital of Shandong University
Bioengineering Center (Shandong, China). The cells were taken from
liquid nitrogen and then thawed in a 37°C water bath. After
centrifugation at 1,000 × g for 5 min, the cells were suspended in
RPMI-1640 complemented with 10% FBS (Invitrogen-Life Technologies,
Carlsbad, CA, USA) and seeded in 6-well plates at a density of
1×105/cm2.
Strand-specific RT-PCR and qPCR
Extracted RNA was treated with DNase I (Fermentas,
Thermo Fisher Scientific, Waltham, MA, USA) to exclude the
possibility of DNA contamination. The RNA concentration in each
sample was quantified using a spectrophotometer at 260 nm and the
purity of RNA was assessed by measuring the OD260/280 ratio
(1.85–1.96). The integrity of RNA was checked by electrophoresis on
a 1.0% agarose gel with ethidium bromide staining. Subsequently, 2
µg of DNase I-treated RNA was applied in the reverse
transcription reaction with 2 pM specific RT primer and SuperScript
III reverse transcriptase (Invitrogen-Life Technologies). PCR
reactions were performed on 1 µl of strand-specific cDNA by
mixing 2X Thermo Pol Taq buffer (New England Biolabs, Ipswich, MA,
USA), 0.25 mM forward primer, 0.25 mM reverse primer, 0.2 mM dNTPs
(Amersham, Buckinghamshire, UK), and 1.25 units Taq polymerase (New
England Biolabs). The PCR conditions used were: 95°C for 5 min
followed by 30 cycles of 95°C for 30 sec, 48°C for 30 sec and 72°C
for 45 sec with a final incubation at 72°C for 10 min. The product
was checked by electrophoresis on a 1.5% agarose gel with ethidium
bromide staining.
RT-qPCR reactions were carried out in a final volume
of 25 µl, using SYBR Premix Ex Taq (Takara), 0.4 mM of each
primer, and 200 ng of cDNA template. Each individual sample was run
in triplicate wells. PCR amplification cycles were performed using
the iQ™5 Multicolor Real-Time PCR detection system (Bio-Rad,
Hercules, CA, USA) and SYBR Premix Ex Taq II kit (Takara, Dalian,
China). The reactions were initially denatured at 95°C for 3 min
followed by 40 cycles of 95°C for 10 sec, 50°C for 30 sec, 72°C for
20 sec. The change in transcript abundance of all tested genes was
calculated using the 2−ΔΔCt method. Gene mRNA amounts
were normalized to β-actin.
Northern blotting
Northern blotting was performed according to a
previous study (?). Specific hybridization probe complementary to
SIRT1-AS was designed and synthesized by GenScript Co. (Nanjing,
China). Total RNA (20 µg) was analyzed on a 12%
polyacrylamide denaturing gel containing 7.5 M urea and transferred
onto a Hybond N+ nylon membrane (Amersham). The
membranes were cross-linked using ultraviolet light for 30 sec at
1,200 mJ/cm2. Hybridization was performed according to
the manufacturer's instructions. Following washing, the membranes
were exposed for 20–40 h to Kodak XAR5 films (Sigma-Aldrich, St.
Louis, MO, USA). As a control, the membranes were hybridized with a
human U6 snRNA probe. The probes used were: SIRT1-AS,
5′-TAACACCAAATCCTCCCAAT-3′; SIRT1 V1,
5′-AGGCAGTTGGAAGATGGCGGACGAGG-3′; SIRT1 V2,
5′-CCTGAGGTTGAGGGCGGCTGGG-3′; and U6,
5′-GCAGGGGCCATGCTAATCTTCTCTGTATCG-3′.
Cell proliferation and viability
testing
Proliferation of HCC-9903 was determined using an
Apollo® 567 EdU staining kit (Roche, Basel, Switzerland)
according to the manufacturer's instructions, and the nuclei were
stained by Hoechst 33342.
Cell viability was performed using a CellTiter-Blue
H cell viability assay kit (Promega, Promega, WI, USA) according to
the manufacturer's instructions.
Plasmid construction and
transfection
The full-length cds sequence of SIRT1 mRNA, WT
SIRT1-AS and the mutant SIRT1-AS were amplified by PCR using
primers containing KpnI (in forward primer) and XhoI
(in reverse primer) sites. The primers used were: SIRT1 forward,
5′-CGGGGTACCTATGCTATGAACAATGGAAG-3′ and reverse,
5′-CCGCTCGAGTTGCCTGTTGAGGATTTGGT-3′; and SIRT1-AS forward,
5′-CGGGGTACCAGTCAAATGACAATTTTAATAGAC-3′ and reverse,
5′-CCGCTCGAGTTAGTGCCTGCCTGGA-3′. The products were cleaved and
ligated onto corresponding sites of the pcDNA3.1 plasmid, which was
confirmed by sequencing. The constructs and miR-29c mimic were
transfected with X-tremeGENE DNA transfection reagents (Roche)
according to the manufacturer's instructions.
Western blotting
The cells were lysed in lysis buffer (Beyotime Co.,
Shanghai, China) supplemented with 1 mM PMSF. Protein concentration
was determined with the BCA protein assay kit (Tiangen). Protein
(20 µg) of each sample were separated by 12% SDS-PAGE and
electro-transferred to a PVDF membrane (Millipore, Billerica, MA,
USA) for immunoblot analysis. The primary antibodies used were:
anti-SIRT1 (1:300), anti-p53 (acetyl K382, 1:200) and anti-p53
(1:400) (all from Abcam, Cambridge, MA, USA), and anti-β-actin
(1:800; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) which
was used as the internal reference. After incubation with the
appropriate HRP-conjugated secondary antibody, the proteins were
detected using a ChemiDoc XRS imaging system and analysis software
Quantity One (Bio-Rad).
RNA stability assay
To detect the impact of SIRT1-AS on SIRT1 mRNA
half-life, after 48 h of transfection of each constructs or miR-29c
mimic, HCC-9903 cells were treated with 2 µg/ml actinomycin
D (Sigma-Aldrich), which had an inhibitory effect on transcription.
The cells were collected at 0, 0.5, 1, 2, 4, 6, 8, 10 and 12 h
after treatment, and then total cell RNA was isolated to measure
the residual mRNAs by RT-qPCR. GAPDH mRNA was applied as an
internal control, which was relatively stable within 32 h.
In vitro ribonuclease protection assay
(RPA)
According to the sequencing result of SIRT1-AS,
full-length WT and mutant SIRT1-AS were synthesized, both of which
contained the complimentary sequence of the miR-29c binding site
and the no. 622 mutation site. Additionally, 1 µg of each
fragment was incubated with synthetic SIRT1 mRNA (a part
complementary to SIRT1-AS) at 37°C for 3 h under simulated
physiological conditions (10 mM Tris-HCl pH 7.4, 15 mM NaCl). Then,
20 U RPA-grade RNase A (Applied Biosystems) was added to each
mixture to remove the single-strand RNAs. Four microliters of each
residue was added to the 20 µl volume double-strand reverse
transcription reaction with 5 pM random primers adding SuperScript
III reverse transcriptase (both from Invitrogen-Life Technologies).
The system was incubated at 60°C for 60 min and terminated at 75°C
for 15 min, and the resultant double-strand cDNAs were amplified in
a 25 µl PCR reaction system as templates. The primers used
were: forward, 5′-ATT CTG CTA TTA CAA GTT-3′ and reverse, AAG TAC
ATG TCT CCC AT. After a 35-cycle amplification, the product was
checked by electrophoresis on a 1.5 % agarose gel with ethidium
bromide staining.
Population investigated
In the in vivo study of the association
between SIRT1-AS level with HCC risk, 52 patients (26 male and 26
female; average age 49.2±18.3 years) were invited to participate in
the study. The patients were diagnosed as primary HCC and treated
with tumor resection in our hospital from May, 2012 to June, 2014.
The patients did not suffer any complications. Their tumor sizes
ranged from 3 to 6 cm. There was no angiogenesis and invasion to
peripheral tissues. Twenty-six male and 26 female healthy
volunteers (average age 49.4±18.6 years) also participated in the
investigation. Each individual in the control group was gender- and
age-matched with the patients in a one-on-one manner.
The study protocol was approved by the Ethics
Committee of The Affiliated Hospital of Inner Mongolia Medical
College. The subjects provided written informed consent prior to
their inclusion in the study.
Calculation of odds ratio (OR)
The odds ratio was used as a precise estimation for
the risk of 622U>C mutation in SIRT1-AS contributing to HCC in
this study. The 2,000 primary HCC patients and 1,500 volunteers
were from The Inner Mongolia Autonomous Region and Northeast China.
Male and female subjects were equally divided, and had an age
ranged of 22–58 years. Among the 2,000 HCC patients, the proportion
of each stage (HCC staging according to the TNM Standard published
by the Union for International Cancer Control in 2003) was
approximately the same. The odd ratio was calculated as: Odd ratio
T = (number of patients possessing allele T x number of volunteers
possessing allele C)/(number of patients possessing allele C x
number of volunteers possessing allele T), and the odd ratio C =
(number of patients possessing allele C x number of volunteers
possessing allele T)/(number of patients possessing allele T x
number of volunteers possessing allele C).
Website references
SIRT1-AS was predicted using information obtained
from the Natural Antisense Transcript Database (http://natsdb.cbi.pku.edu.cn). SIRT1 mRNA variant
sequences were obtained from GenBank (http://www.ncbi.nlm.nih.gov) and UCSC Genome
(http://genome.ucsc.edu/index.html).
The Coding Potential Calculator (http://cpc.cbi.pku.edu.cn/) was used to calculate the
coding potential of the sequences. The secondary structures of WT
SIRT1-AS and the mutant were predicted by the mFold online server
(http://mfold.rna.albany.edu/?q=mfold).
Statistical analysis
Data were obtained from at least three independent
experiments. Values were presented as means ± SEM. Statistics were
calculated using SPSS statistics v19.0 software. Multiple
comparisons were assessed by one-way ANOVA followed by Dunnett's
tests. The difference between groups was considered statistically
significant when P<0.05.
Results
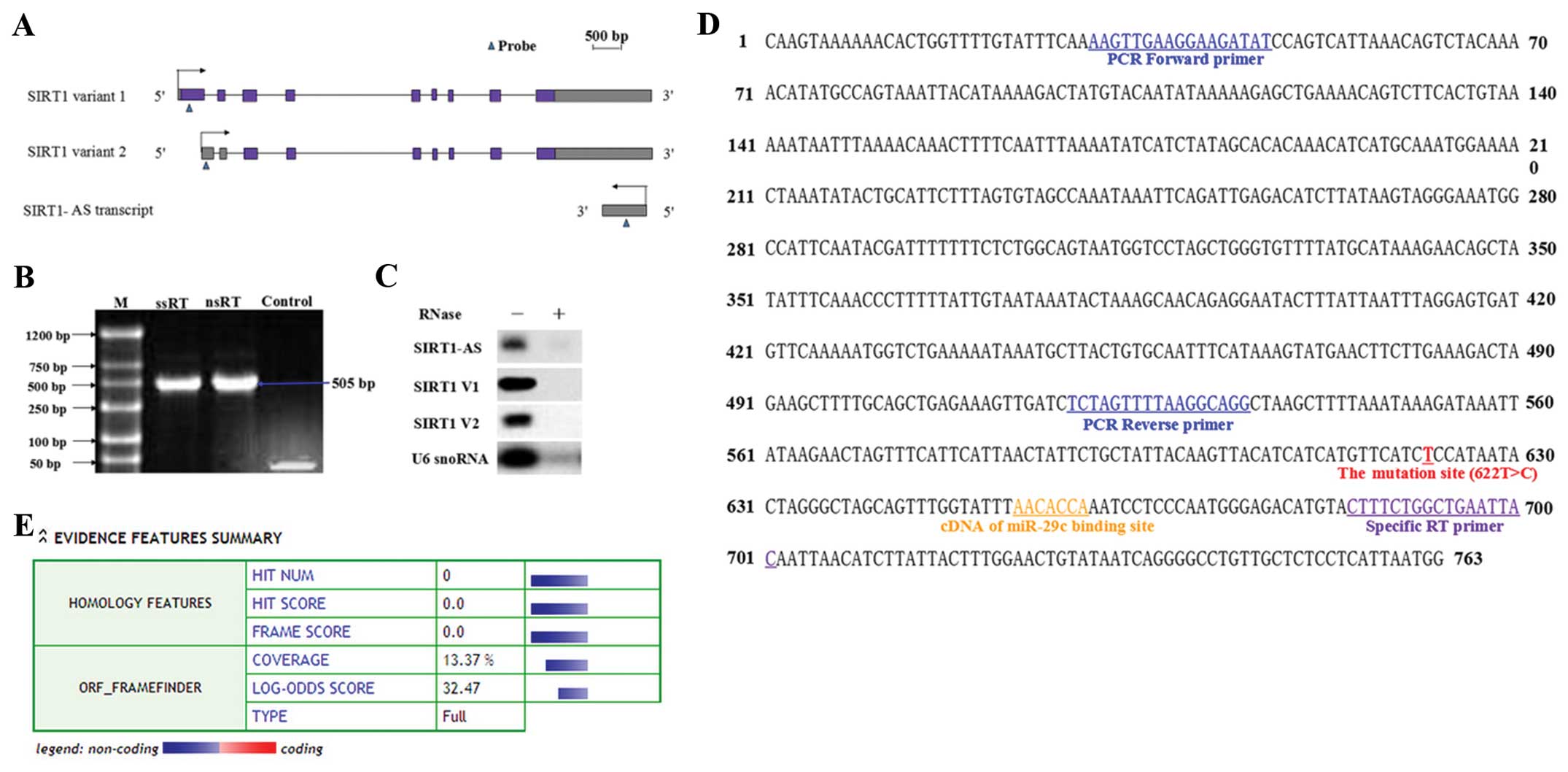
Identification of single-exon SIRT1-AS
overlapping with SIRT1 3′UTR
We detected part of a transcript derived from the
antisense strand of the SIRT1 3′UTR region by specific-strand
RT-PCR in RNA samples that were extracted from the HCC-9903 cell
line (Fig. 1B). Then, a specific
probe was applied in the northern blotting to assay the transcript
and a single band was obtained (Fig. 1A
and C). The probed AS transcript was extracted and sent to BGI
(Shenzhen, China) for high-precision sequencing. The sequencing
results revealed the single-exon AS transcript with a size of 763
nt, designated as SIRT1-AS (Fig.
1D). The sequence was inserted into a coding potential
calculator to calculate its coding potential. The outputs suggested
that SIRT1-AS was probably non-protein-coding, because no available
ORF structure was identified in the sequence (Fig. 1E). Therefore, we identified a
single-exon AS transcript that overlapped with SIRT1 3′UTR and it
was probably an AS lncRNA.
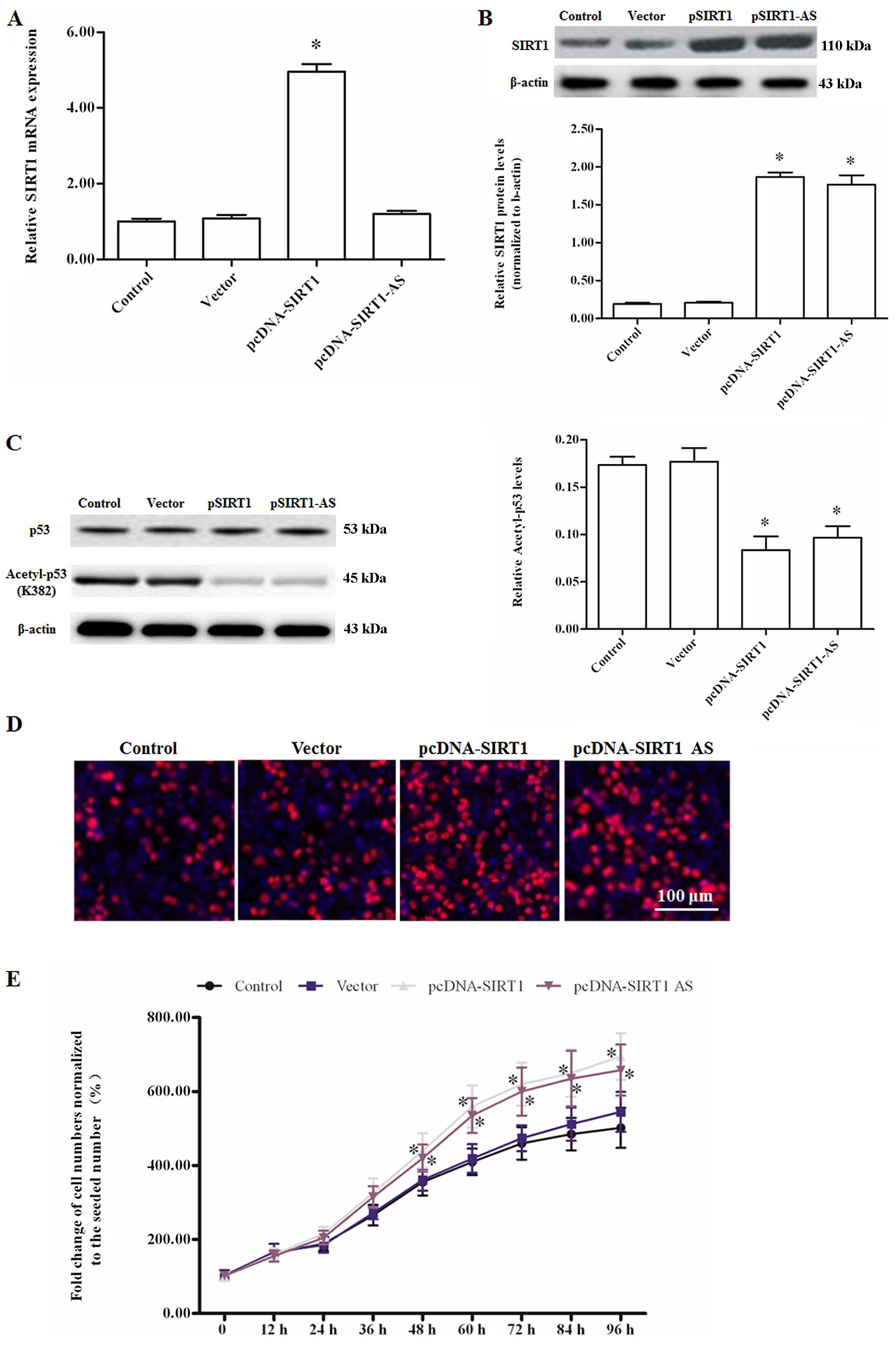
SIRT1-AS promotes the proliferation of
HCC-9903 cells by upregulating the SIRT1 protein level
The pcDNA-SIRT1-AS or pcDNA-SIRT1 expression vectors
were transfected into HCC-9903 cells to assess the role of SIRT1-AS
in HCC cell proliferation. The specific-strand RT-qPCR and western
blotting results showed that SIRT1-AS overexpression did not
influence SIRT1 mRNA expression, but markedly elevated the SIRT1
protein level (Fig. 2A and B). As a
result, the downstream anti-oncogene p53 was deacetylated by SIRT1
and SIRT1-AS overexpression (Fig.
2C). Edu staining and cell viability detection by the
CellTiter-Blue H cell viability assay kit revealed that SIRT1 and
SIRT1-AS overexpression promoted the proliferation of HCC-9903
cells (Fig. 2D and E). The results
suggested that SIRT1-AS functioned positively in the regulation of
SIRT1 expression at the post-transcriptional level.
The 622C single-nucleotide mutation
disables SIRT1-AS to stabilize SIRT1 mRNA
In clinical diagnosis, our team identified a
single-nucleotide mutation in a small number of HCC patient's
SIRT1-AS by gene sequencing assistant diagnosis. The no. 622 base T
(also U in RNA) was substituted by C in the minority of the
patients (the site was marked in Fig.
1D). To verify the performance of the mutant SIRT1-AS, a
pcDNA-SIRT1 mutant was additionally established. WT or mutant
SIRT1-AS expression constructs were transfected into HCC-9903
cells. After an incubation for 48 h, western blot assay showed that
the SIRT1 protein level was elevated in the WT transfection group,
but did not significantly change in the group of mutant SIRT1-AS
transfection (Fig. 3A).
Additionally, as a proven SIRT1-targeting miRNA, miR-29c mimics
reduced the protein level that was elevated by WT SIRT1-AS
(Fig. 3A). The transfected cells
were treated with 2 µg/ml actinomycin D to block the
transcription activity in the cells. After treatment for 0, 0.5, 1,
2, 4, 6, 8, 10 and 12 h, the cells were collected and used to
extract total RNA separately. The residual SIRT1 mRNA was
quantified to evaluate the variation of the half-life between the
groups. The results revealed that the half-life of SIRT1 mRNA was
~8 h under normal circumstances, which was not influenced by the
mutant. As expected, the miR-29c mimic significantly shortened the
half-life of SIRT1 mRNA (to <6 h, Fig. 3B). Notably, the WT SIRT1-AS greatly
extended the period to ~11 h (Fig.
3B). In particular, WT SIRT1-AS was likely to antagonize the
negative regulation of miR-29c.
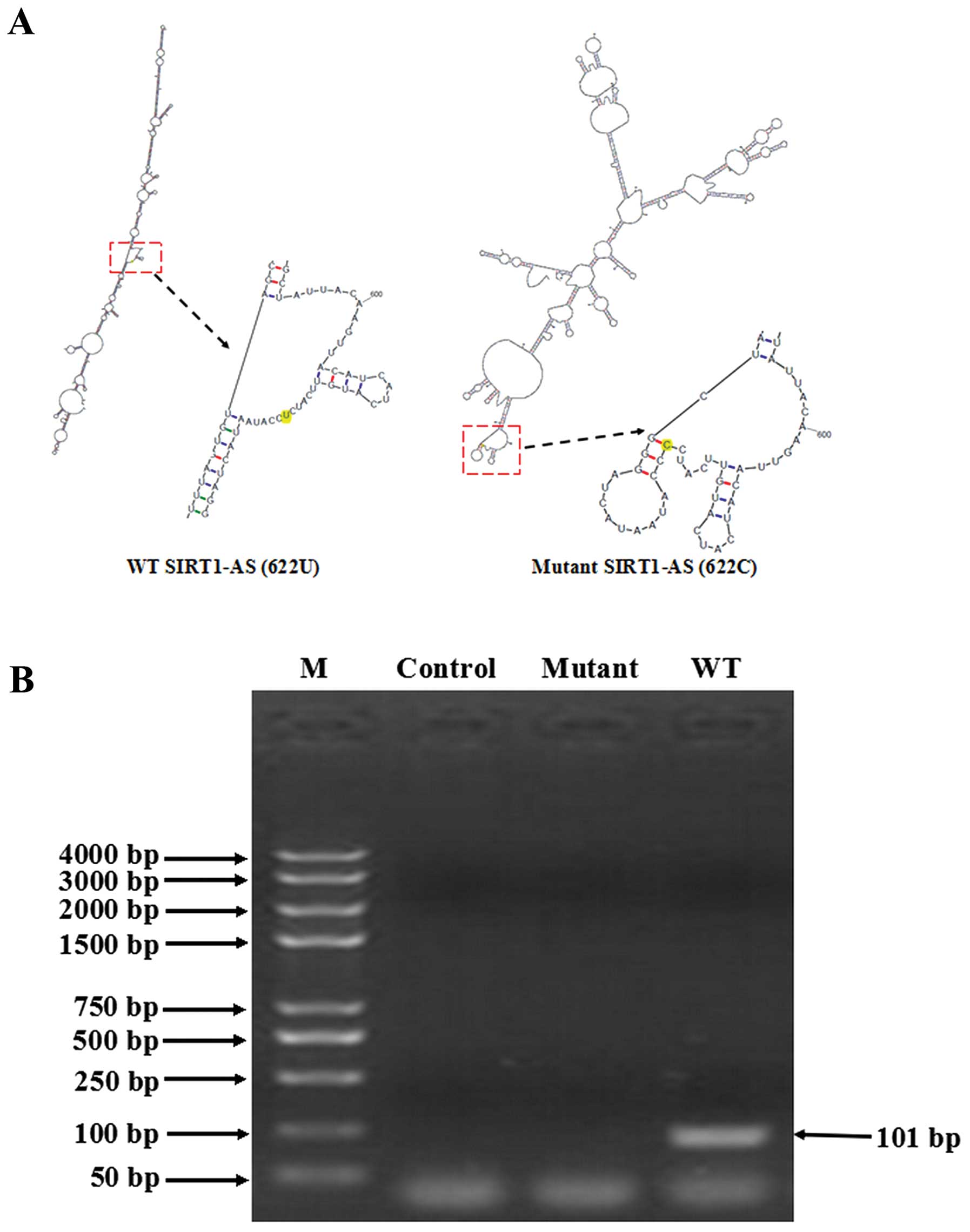
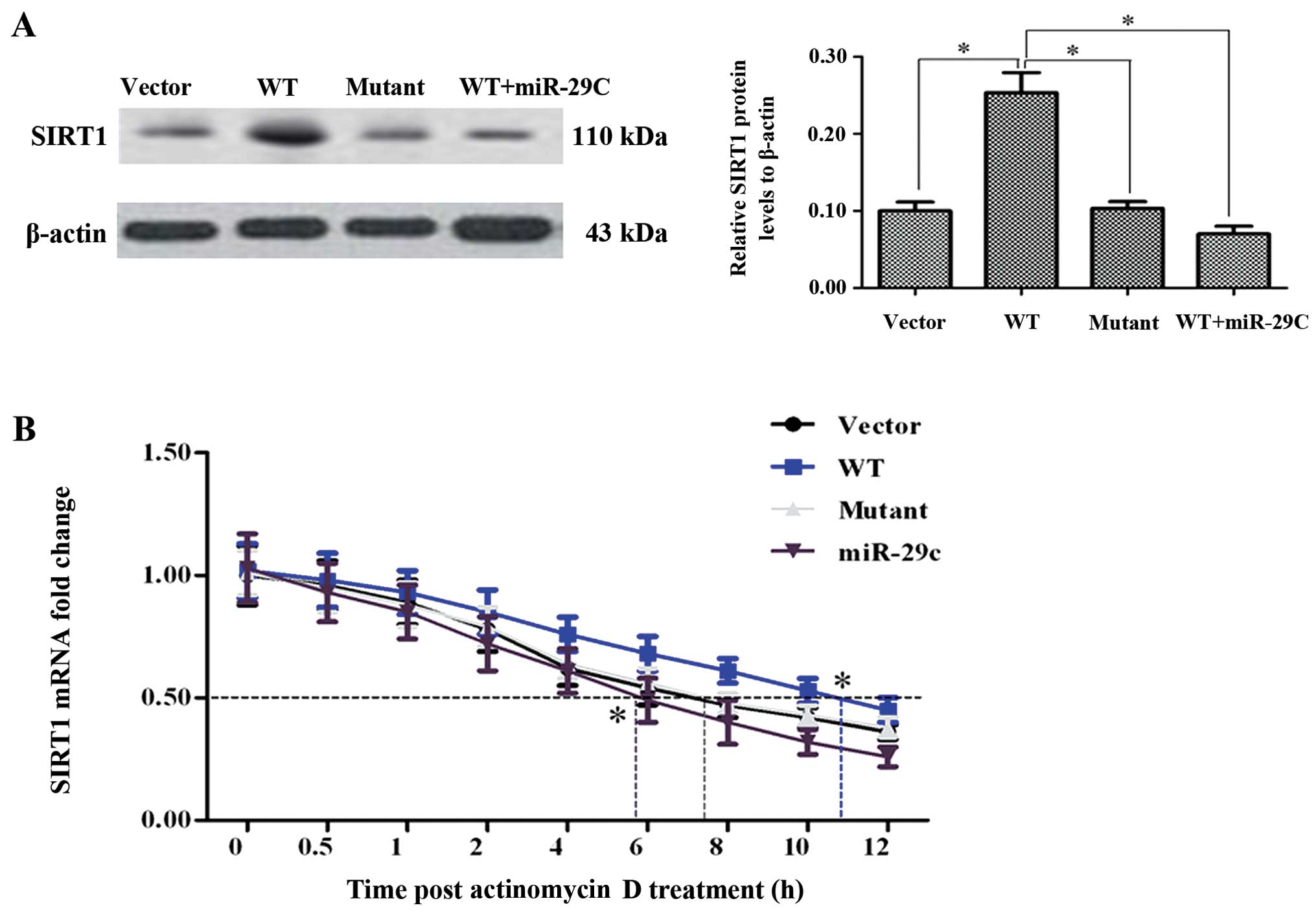 | Figure 3The 622C mutation disables SIRT1-AS to
stabilize SIRT1 mRNA. (A)No. 622U>C mutant SIRT1-AS did not
elevate the SIRT1 protein level. HCC-9903 cell were transfected
with negative vector, pcDNA-WT-SIRT1-AS, pcDNA-mutant-SIRT1-AS, or
pcDNA-WT-SIRT1-AS and miR-29c mimic for 48 h. The expression of
SIRT1 protein was then examined. (B) WT SIRT1-AS prolonged the
half-life of SIRT1 mRNA although the mutant did not. After 48 h of
transfection of each construct or miR-29c mimic, HCC-9903 cells
were treated with 2 µg/ml actinomycin D to inhibit
transcription. The cells were harvested at 0, 0.5, 1, 2, 4, 6, 8,
10 and 12 h after treatment, and the residual mRNAs were measured
by RT-qPCR. GAPDH mRNA was applied as an internal control, which
was relatively stable within 32 h. Stabilization of SIRT1 mRNA was
weight by the time of half-life. *P<0.05. HCC,
hepatocellular carcinoma. |
WT SIRT1-AS masked the miR-29c binding
site on the 3′UTR of SIRT1 mRNA whereas the mutant did not mask the
binding site
As the miR-29c binding site was within the
overlapping region of SIRT1-AS and SIRT1 mRNA, we hypothesized that
WT SIRT1-AS was able to bind to SIRT1 mRNA, thus miR-29c was unable
to target SIRT1 mRNA, whereas the mutant was not able to bind to
SIRT1 mRNA. The sequences of WT SIRT1-AS and the mutant were,
respectively, entered into the mFold web server to calculate the
most probable secondary structures. The outputs showed that their
secondary structures were entirely different (Fig. 4A), suggesting at their distinct
ability to combine with SIRT1 mRNA. WT or the mutant synthetic
SIRT1-AS was incubated with SIRT1 mRNA under simulated
physiological conditions for 3 h, and then the mixture was digested
with RPA-grade RNase A. RT-PCR was subsequently performed to detect
whether an RNA duplex was formed. A 101-bp band, which included the
mutant site and cDNA of the miR-29c binding site, was only detected
in the residue of the WT SIRT1-AS/mRNA mixture. The RPA results
showed that only WT SIRT1-AS formed a sense-antisense RNA duplex
(Fig. 4B), which also supported our
hypothesis.
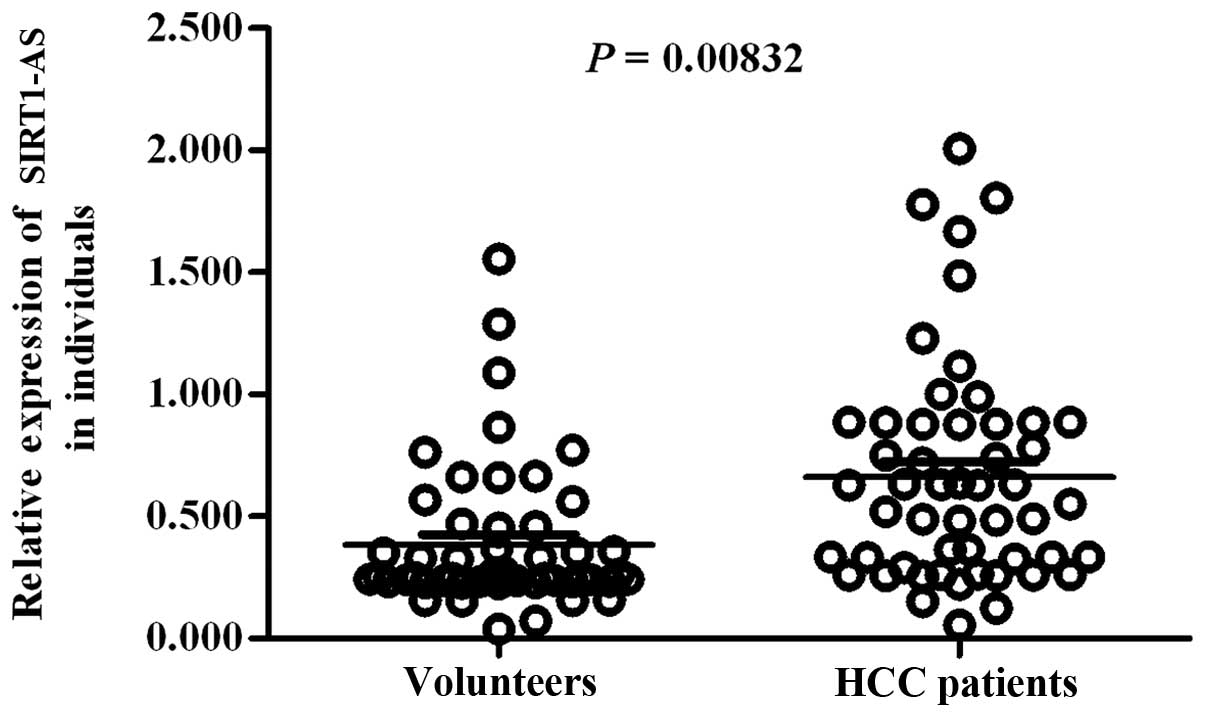
Mutation decreases the risk of HCC
The above results suggested that the mutation
622U>C 're-liberated' the miR-29c binding site via WT SIRT1-AS
and downregulated SIRT1 expression at the post-transcriptional
level. We randomly detected total SIRT1-AS levels in the liver
tissues of 52 HCC patients and 52 non-hepatopathic volunteers. The
strand-specific RT-qPCR results showed that SIRT1-AS was more
highly expressed in HCC patients (Fig.
5). A survey was performed to assess the relationship between
622C and the risk of HCC. Among 2,000 HCC patients, the mutant
ratio was 4.8% smaller than that among 1,500 non-hepatopathic
volunteers (up to 16.3%). The statistical analysis showed that the
OR value of 622C was only 0.253 (<1), which was lower than that
of 622T (3.958>1) (Table I).
These results suggested that the mutant C allele reduced the risk
of HCC.
 | Table IInvestigation of different genotypes
in HCC patients and non-hepatopathy volunteers. |
Table I
Investigation of different genotypes
in HCC patients and non-hepatopathy volunteers.
| Variables | Genotype
| Mutation ratio
(%) |
|---|
| WT (622T) | Mutant (622 C) |
|---|
| HCC patients | 1906 | 94 | 4.8 |
| Volunteers | 1255 | 245 | 16.3 |
| Odds ratio | 3.958 | 0.253 | |
Discussion
In the present study, we characterized the AS lncRNA
SIRT1-AS which suppressed the miRNA-induced translational
repression of SIRT1 mRNA by masking the miR-29c binding site on the
SIRT1 3′UTR. In vitro studies showed that SIRT1-AS
positively regulates HCC proliferation, and the comparison between
the HCC patients and non-hepatopathic volunteers indicated that
high-level expression of the WT SIRT1-AS increases the risk of HCC.
Additionally, we identified a 622U>C mutant of SIRT1-AS in the
clinical diagnosis of HCC. The in vitro experiments and
statistical analysis of sequences strongly suggested that the
mutation 're-liberated' the masked miR-29c binding site and
contributed to decreasing the risk of HCC.
Few lncRNAs have been found to be dysfunctional in
HCC; however, to the best of our knowledge, none of them were
transcribed from the AS strand of a functional gene (20). Thus, our results present the first
identification of a p53-repressing AS lncRNA acting as a
carcinogenic factor. Half of the human genomic loci lead to NATs;
however, NATs associated with HCC or other liver cancers have
rarely been reported. It is expected that NATs associated with HCC
or other liver cancer types are a promising field in liver cancer
biology.
Gene regulation by AS transcription is complicated.
AS lncRNAs that share apparently identical mechanisms often have
opposite functions. BACE1-AS lncRNA and tie-1 AS lncRNA formed a
sense/antisense RNA duplex with their mRNAs. Of note, BACE-AS
lncRNA masked miR-485–5p binding site in the mRNA and stabilized
BACE1 mRNA, while tie-1 AS lncRNA negatively regulated tie-1
expression by downregulating tie-1 mRNA (21,22). A
negative or positive response may depend on the site where the S-AS
is bound and the binding length of the two transcripts. In the
present study, SIRT1-AS functioned in a similar manner to BACE1-AS
in their sense gene regulation. The RPA results exhibited the
binding region of WT SIRT1-AS, and SIRT1 mRNA included the binding
site of the SIRT1 suppressor miR-29c. The mutation resulted in
SIRT1-AS being unable to bind to SIRT1 mRNA and mask the miR-29c
binding site. Therefore, the mutant SIRT1-AS played a negative role
in SIRT1 translation.
In the clinical context, there is a need for the
verification of therapeutic targets for HCC. The mutation 622C in
SIRT1-AS decreased the risk of HCC, thus the mutant may be regarded
as a potential target for gene therapy for HCC or other diseases
associated with SIRT1 dysregulation.
In conclusion, we characterized a carcinogenic NAT
(also known as AS lncRNA) SIRT1-AS and identified a benign mutation
622U>C in its sequence. An in vitro study and statistical
analysis indicated that the mutation minimized the risk of HCC. The
findings of this study provide a new therapeutic target for HCC and
improve our understanding of the mechanisms of AS lncRNAs.
References
|
1
|
Werner A and Berdal A: Natural antisense
transcripts: Sound or silence? Physiol Genomics. 23:125–131. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He Y, Vogelstein B, Velculescu VE,
Papadopoulos N and Kinzler KW: The antisense transcriptomes of
human cells. Science. 322:1855–1857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Katayama S, Tomaru Y, Kasukawa T, Waki K,
Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et
al FANTOM Consortium: Antisense transcription in the mammalian
transcriptome. Science. 309:1564–1566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Faghihi MA and Wahlestedt C: Regulatory
roles of natural antisense transcripts. Nat Rev Mol Cell Biol.
10:637–643. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pelechano V and Steinmetz LM: Gene
regulation by antisense transcription. Nat Rev Genet. 14:880–893.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Magistri M, Faghihi MA, St Laurent G III
and Wahlestedt C: Regulation of chromatin structure by long
noncoding RNAs: Focus on natural antisense transcripts. Trends
Genet. 28:389–396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bertozzi D, Iurlaro R, Sordet O, Marinello
J, Zaffaroni N and Capranico G: Characterization of novel antisense
HIF-1α transcripts in human cancers. Cell Cycle. 10:3189–3197.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bhan A, Hussain I, Ansari KI, Kasiri S,
Bashyal A and Mandal SS: Antisense transcript long noncoding RNA
(lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol
Biol. 425:3707–3722. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Su WY, Li JT, Cui Y, Hong J, Du W, Wang
YC, Lin YW, Xiong H, Wang JL, Kong X, et al: Bidirectional
regulation be tween WDR83 and its natural antisense transcript DHPS
in gastric cancer. Cell Res. 22:1374–1389. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haigis MC and Guarente LP: Mammalian
sirtuins - emerging roles in physiology, aging, and calorie
restriction. Genes Dev. 20:2913–2921. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Michan S and Sinclair D: Sirtuins in
mammals: Insights into their biological function. Biochem J.
404:1–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Finkel T, Deng CX and Mostoslavsky R:
Recent progress in the biology and physiology of sirtuins. Nature.
460:587–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang HC and Guarente L: SIRT1 and other
sirtuins in metabolism. Trends Endocrinol Metab. 25:138–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lagouge M, Argmann C, Gerhart-Hines Z,
Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P,
Elliott P, et al: Resveratrol improves mitochondrial function and
protects against metabolic disease by activating SIRT1 and PGC-1α.
Cell. 127:1109–1122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD,
Jones DR, Frye RA and Mayo MW: Modulation of NF-kappaB-dependent
transcription and cell survival by the SIRT1 deacetylase. EMBO J.
23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bai L, Pang WJ, Yang YJ and Yang GS:
Modulation of Sirt1 by resveratrol and nicotinamide alters
proliferation and differentiation of pig preadipocytes. Mol Cell
Biochem. 307:129–140. 2008. View Article : Google Scholar
|
|
17
|
Cheng HL, Mostoslavsky R, Saito S, Manis
JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW and Chua KF:
Developmental defects and p53 hyperacetylation in Sir2 homolog
(SIRT1)-deficient mice. Proc Natl Acad Sci USA. 100:10794–10799.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee JT and Gu W: SIRT1: Regulator of p53
deacetylation. Genes Cancer. 4:112–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bae HJ, Noh JH, Kim JK, Eun JW, Jung KH,
Kim MG, Chang YG, Shen Q, Kim SJ, Park WS, et al: MicroRNA-29c
functions as a tumor suppressor by direct targeting oncogenic SIRT1
in hepatocellular carcinoma. Oncogene. 33:2557–2567. 2014.
View Article : Google Scholar
|
|
20
|
He Y, Meng XM, Huang C, Wu BM, Zhang L, Lv
XW and Li J: Long noncoding RNAs: Novel insights into
hepatocellular carcinoma. Cancer Lett. 344:20–27. 2014. View Article : Google Scholar
|
|
21
|
Faghihi MA, Modarresi F, Khalil AM, Wood
DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G III, Kenny PJ and
Wahlestedt C: Expression of a noncoding RNA is elevated in
Alzheimer's disease and drives rapid feed-forward regulation of
β-secretase. Nat Med. 14:723–730. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li K, Blum Y, Verma A, Liu Z, Pramanik K,
Leigh NR, Chun CZ, Samant GV, Zhao B, Garnaas MK, et al: A
noncoding antisense RNA in tie-1 locus regulates tie-1 function in
vivo. Blood. 115:133–139. 2010. View Article : Google Scholar :
|