Introduction
Adult T-cell leukemia-lymphoma (ATL), an aggressive
peripheral T-cell neoplasm, results from long-term infection with
human T-cell leukemia virus-1 (HTLV-1) and is associated with a
poor prognosis (1,2). DNA-damaging chemotherapy drugs, such
as etoposide and anthracyclines, are the first-line treatment for
ATL. However, drug resistance is a challenge for the management of
ATL patients (3,4). Most cancer chemotherapeutic agents
induce cell death by producing DNA strand breaks and DNA
replication fork collapse (5).
However, cells have evolved complicated DNA integrity surveillance
systems including DNA damage response (DDR) and repair networks,
which jointly function to maximize survival and minimize the gene
mutation rate (6). Accumulating
evidence has shown that enhanced DNA damage repair plays a critical
role in the resistance of cancer cells to chemotherapy (7). Therefore, targeting DNA repair
pathways is a promising strategy to overcome ATL resistance to
chemotherapy.
Most DNA-damaging chemotherapeutic agents directly
or indirectly cause DNA double-strand breaks (DSBs), which are
highly lethal lesions that kill cells by inactivating essential
genes or, in metazoans, by triggering apoptosis (8,9).
Although replication-associated DSBs are repaired by homologous
recombination (HR) and related replication repair pathways, DSBs
are mainly repaired by non-homologous end joining (NHEJ), which
comprises the Ku70 and Ku80 heterodimer, DNA-dependent protein
kinase (DNA-PKcs), DNA ligase IV and scaffold Xrcc4. The Ku70 and
Ku80 heterodimer recognizes and binds to DNA ends, and activates
DNA-PKcs by stabilizing its interaction with the DNA ends, while
DNA ligase IV and the scaffold Xrcc4 complex are involved in the
final ligation step (10).
Deregulation of the NHEJ pathway affects the efficiency of
DNA-damaging drugs, thereby leading to escape and survival of
leukemia cells following chemotherapy.
SIRT1 is a multifaceted, NAD+-dependent
protein deacetylase, and a key regulator of lifespan mediated by
caloric restriction (11). SIRT1
also plays an important role in anti-aging and tumorigenesis
(12). One of the mechanisms by
which SIRT1 prolongs lifespan and suppresses tumorigenesis is the
modification of numerous DNA damage repair proteins, including Ku70
(13), FOXOs (14,15),
Nijmegen breakage syndrome protein (NBSP) (16), WRN protein (17), xeroderma pigmentosum C protein
(18) and NF-κB (19). Recent findings have shown that SIRT1
plays a key role in leukemogenesis and resistance to leukemia
treatment presumably by facilitating DNA repair in leukemia cells
(20–22). SIRT1 has been shown to be
consistently overexpressed in primary ATL samples and the
pharmacologic inhibition or shRNA-mediated knockdown of
SIRT1-induced apoptosis in ATL (23). However, the underlying molecular
mechanism and whether inhibition of SIRT1 sensitizes ATL cells to
DNA damage drugs remain to be determined.
In the present study, we knocked down SIRT1 by shRNA
in Jurkat cells, one of the most representative cell lines of ATL,
and assessed apoptosis, cell cycle arrest and proliferation
following etoposide treatment in cell culture, and leukemia blast
and the survival time of Jurkat-xenografted mice. We further
assayed DNA damage repair capacity and examined the underlying
mechanism after SIRT1 silencing in response to etoposide
treatment.
Materials and methods
Cell lines and peripheral blood
mononuclear cells
HL-60, THP-1, HEL, Daudi, Karpas 299, K562, Namalwa,
Su-DHL-4, Jurkat and 293T/17 cell lines were purchased from the
Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, China).
The lentivirus packaging cell line 293T/17 was cultured in
Dulbecco's modified Eagle's medium (DMEM) and the remaining cells
were maintained in RPMI-1640 medium and Iscove's modified
Dulbecco's medium (IMDM). The above media were supplemented with
100 U/ml of penicillin, 0.1 mg/ml of streptomycin, 2 mM L-glutamine
and 10% fetal bovine serum (FBS). The cells were maintained at 37°C
in a humidified atmosphere of 5% CO2. The cells in the
logarithmic phase were used for subsequent experiments. Four
peripheral blood (PB) samples were obtained from four healthy
volunteers. Written informed consent for participation in the
present study was obtained, and laboratory experiments were
approved by the ethics committee of the Tongji Hospital of Tongji
University (Shanghai, China). Peripheral blood mononuclear cells
(PBMCs) were obtained from peripheral blood by separation on the
Lymphoprep (Axis-Shield, Oslo, Norway) density gradient with
centrifugation at 400 x g for 30 min. The fresh PBMCs were used for
reverse-transcriptase PCR (RT-PCR).
Reagents
Etoposide (Sigma-Alrich, St. Louis, MO, USA), also
known as VP16, was used at the concentration of 20 µM to
treat cells seeded at 1×105/ml in 6-well plates. After
incubation for 4 h, cells were washed twice in PBS and maintained
in fully supplemented RPMI-1640 medium for 48 h before further
analysis. Primary antibodies against SIRT1, Ku70 (Abcam, Cambridge,
UK), phospho-Histone H2AX (Ser139), acetylated-lysine, GAPDH (Cell
Signaling Technology, Danvers, MA, USA), acetylated FOXO1 (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were used.
Horseradish peroxidase-conjugated secondary antibodies were
purchased from Cell Signaling Technology.
Gene knockdown
The shRNA targeting SIRT1 (sequence as
5′-GAAGTGCCTCAGATATTAA-3′) and negative control (sequence as
5′-TTCTCCGAACGTGTCACGT-3′) were cloned into pLVX-shRNA1 vector
(Clontech Laboratories) and designated as shSIRT1 and SCR.
Lentiviral particles were produced by co-transfection of 293T/17
cells with shSIRT1 or SCR and the lentiviral packaging plasmid at a
ratio of 4:3:2 using the calcium phosphate precipitation method.
Transduction was carried out in the presence of 5 µg/ml
polybrene. Following transduction, 1.5 µg/ml puromycin
(Sigma-Aldrich) was added for positive selection. Subsequent
experiments were performed 72 h after transduction.
Measurement of DNA repair capacity
Plasmids containing NHEJ, HR reporter cassettes and
pDsRed-N1 as the internal controls were kindly provided by Dr
Zhiyong Mao from the School of Life Science and Technology of
Tongji University (Shanghai, China). NHEJ or HR reporter cassettes
containing plasmids were first linearized by I-SceI
restriction enzymes and purified using the Qiagen QIAEX II
purification kit (20021; Qiagen, Valencia, CA, USA). A 0.5
µg of the NHEJ reporter construct, or 2 µg of the HR
reporter constructs, and 0.1 µg of pDsRed-N1 were
transfected into exponentially growing cells. Transfections were
performed using the Amaxa Nucleofector (Walkersville, MD, USA).
Jurkat cells were transfected using Cell Line Nucleofector kit V
(Amaxa VPA-1003) and program X001. Cells were analyzed by FACS 72 h
after the transfection.
RNA extraction and qPCR
Total RNA was extracted from cell lines and PBMCs
using TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA)
according to the manufacturer's instructions. First-strand cDNA was
synthesized in 10 µl reaction volume using PrimeScript RT
Master Mix (Takara, Dalian, China). Relative mRNA levels of target
gene and β-actin were detected by RT-qPCR in Applied Biosystems
7500 Fast Real-Time PCR systems (Life Technologies) with SYBR
Premix Ex Taq™ (Takara). The primers were used: SIRT1, forward
5′-ATACCCAGAACATAGACAC GCT-3′ and reverse 5′-CGT
ACAGCTTCACAGTCAACTT-3′; β-actin, forward 5′-GAA
CGGTGAAGGTGACAGCAG-3′ and reverse 5′-GTGGAC TTGGGAGAGGACTGG-3′.
Data were analyzed using the 2−ΔΔCt method, where ΔCt =
(Cttarget gene − Ctβ-actin).
Immunoblotting and
immunoprecipitation
The cells were lysed in Cell Lysis Buffer (Thermo
Fisher Scientific, Waltham, MA, USA) supplemented with 1 mM PMSF
(Beyotime Institute of Biotechnology, Nantong, China). Cell lysates
were incubated on ice for 30 min and the supernatants were
collected for immunoblotting by centrifuging at 13,300 rpm for 30
min at 4°C. BCA assay (Beyotime Institute of Biotechnology) was
used to determine the protein concentration. The proteins were
subjected to sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE; Beyotime Institute of Biotechnology).
The separated proteins were electrophoretically transferred to PVDF
membranes (Millipore, Billerica, MA, USA). After blocking in 5%
non-fat milk in Tris-buffered saline/Tween-20 (TBST) for 1 h, the
membranes were incubated independently with the primary antibodies
against specific proteins in 5% BSA TBST overnight at 4°C. For the
detection of acetylated Ku70, the cell lysates were first
immunoprecipitated by anti-Ku70 antibody overnight at 4°C with
gentle rotation. The immunoprecipitants were then incubated with 30
µl protein G agarose beads (Santa Cruz Biotechnology, Inc.)
for 4 h and resolved by SDS-PAGE after four washes with lysis
buffer. The immunoprecipitated samples were subjected to immunoblot
analysis using an anti-acetylated antibody. The membranes were
incubated with an anti-rabbit or anti-mouse IgG, HRP-linked
antibody (Cell Signaling Technology) for 2 h at room temperature
and detected with ECL Plus (Millipore) followed by LAS-4000
scanning (Fujifilm, Tokyo, Japan).
Apoptosis and cell cycle analysis
Apoptosis was analyzed by staining with Annexin
V-FITC and propidium iodide (BD Biosciences Franklin Lakes, NJ,
USA) according to the manufacturer's instructions. Flow cytometry
was then performed within 1 h. Data was analyzed through FlowJo
software (Tree Star, Ashland, OR, USA). For the cell cycle
analysis, the cells were collected and washed twice with PBS.
Subsequently, the cells were fixed with cold ethanol and incubated
at −20°C for 2 h. After centrifugation, the cells were treated with
PI/RNase staining buffer (BD Biosciences), and then incubated for
15 min at room temperature followed by flow cytometry.
Soft agar colony formation assay
For the clonogenic assay, a standard two-layer soft
agar culture was performed with bottom 0.6% agarose and top 0.3%
agarose. The cells were seeded at 105/ml in 24-well
plates with soft agar as described above, and colonies were scored
after 7 days.
Xenotransplantation assays
Jurkat cells (1×106) transduced with
vectors expressing a shRNA directed against SIRT1 or scrambled
shRNA were transplanted via tail vein injection into sub-lethally
irradiated (2.5 Gy) NOD-SCID mice (Shanghai SLAC Laboratory Animal,
Co., Ltd., Shanghai, China). The survival time was subsequently
recorded. Twenty days after transplantation, three mice from each
group were euthanized, and bone marrow and spleen were collected
for subsequent experiments.
Statistical analyses
Unless otherwise specified, data are presented as
mean ± SD. For data analysis, unpaired two-tailed Student's t-test
or Mann-Whitney U test were performed. For the animal studies,
Kaplan-Meier survival analysis was performed and survival was
calculated using the log-rank test. Statistical computations were
performed using Prism 6.0 (GraphPad Software).
Results
SIRT1 is overexpressed in Jurkat
cells
We assessed the relative SIRT1 mRNA levels in
several cell lines and 4 PBMC samples of the healthy human
controls. As shown in Fig. 1A,
Karpas 299, K562, Namalwa, Su-DHL-4 and Jurkat demonstrated a
significantly higher expression of SIRT1 than in the normal
controls. The Jurkat cell line showed the highest SIRT1 mRNA level.
Consistent with the mRNA expression levels, the immunoblot analysis
revealed that Jurkat cells manifested markedly higher SIRT1 protein
levels than the remaining four cell lines (Fig. 1B and C). Subsequently, Jurkat cells
were selected for subsequent experiments.
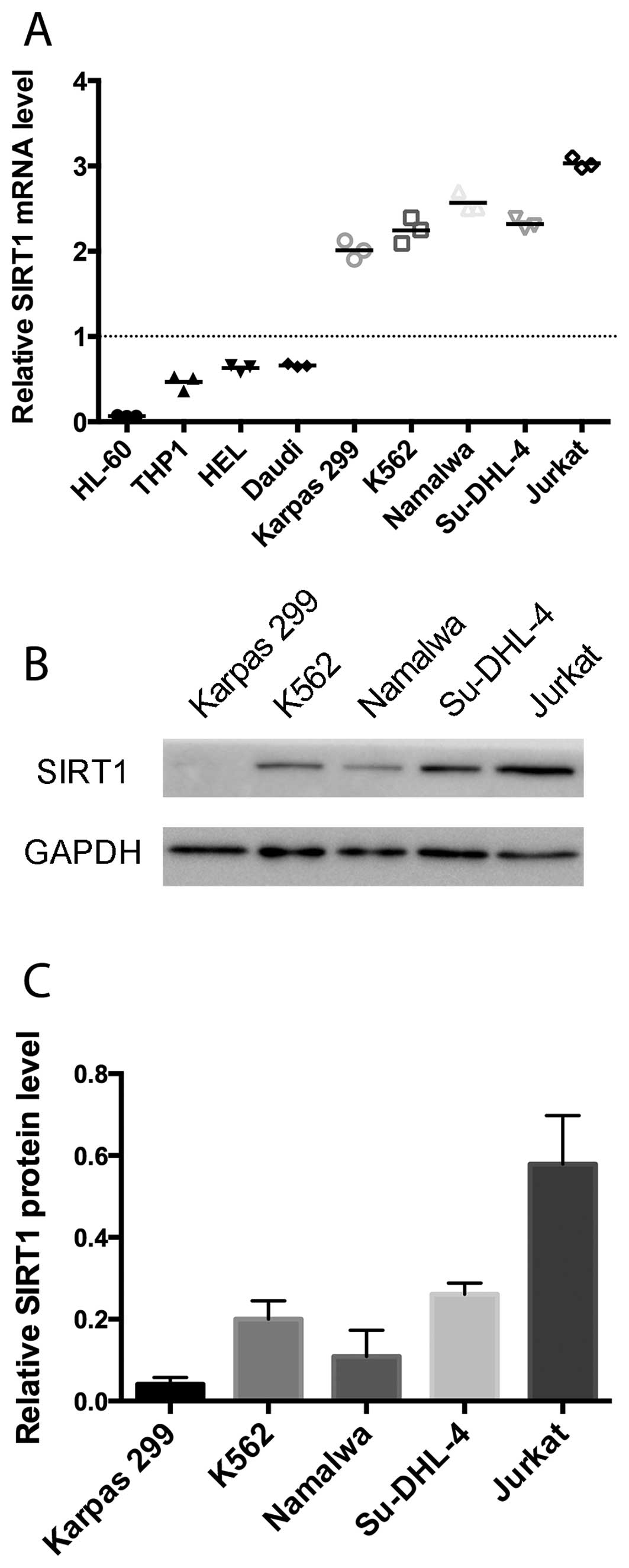 | Figure 1SIRT1 expression levels in the cell
lines and normal control PBMCs. (A) SIRT1 mRNA levels of HL-60,
THP-1, HEL, Daudi, Karpas 299, K562, Namalwa, Su-DHL-4 and Jurkat
were analyzed by RT-qPCR relative to β-actin as the internal
control. Horizontal bars indicate the mean percentages of mRNA
expression. Dotted line is the mean of SIRT1 mRNA levels of 4
normal control PBMCs. (B) Protein expression levels of SIRT1 and
GAPDH in the cell lysates of Karpas 299, K562, Namalwa, Su-DHL-4
and Jurkat were determined by immunoblotting. (C) SIRT1 protein
expression was quantified by densitometric analysis and normalized
to GAPDH expression. The normalized SIRT1 expression levels of
Karpas 299, K562, Namalwa, Su-DHL-4 and Jurkat are shown. |
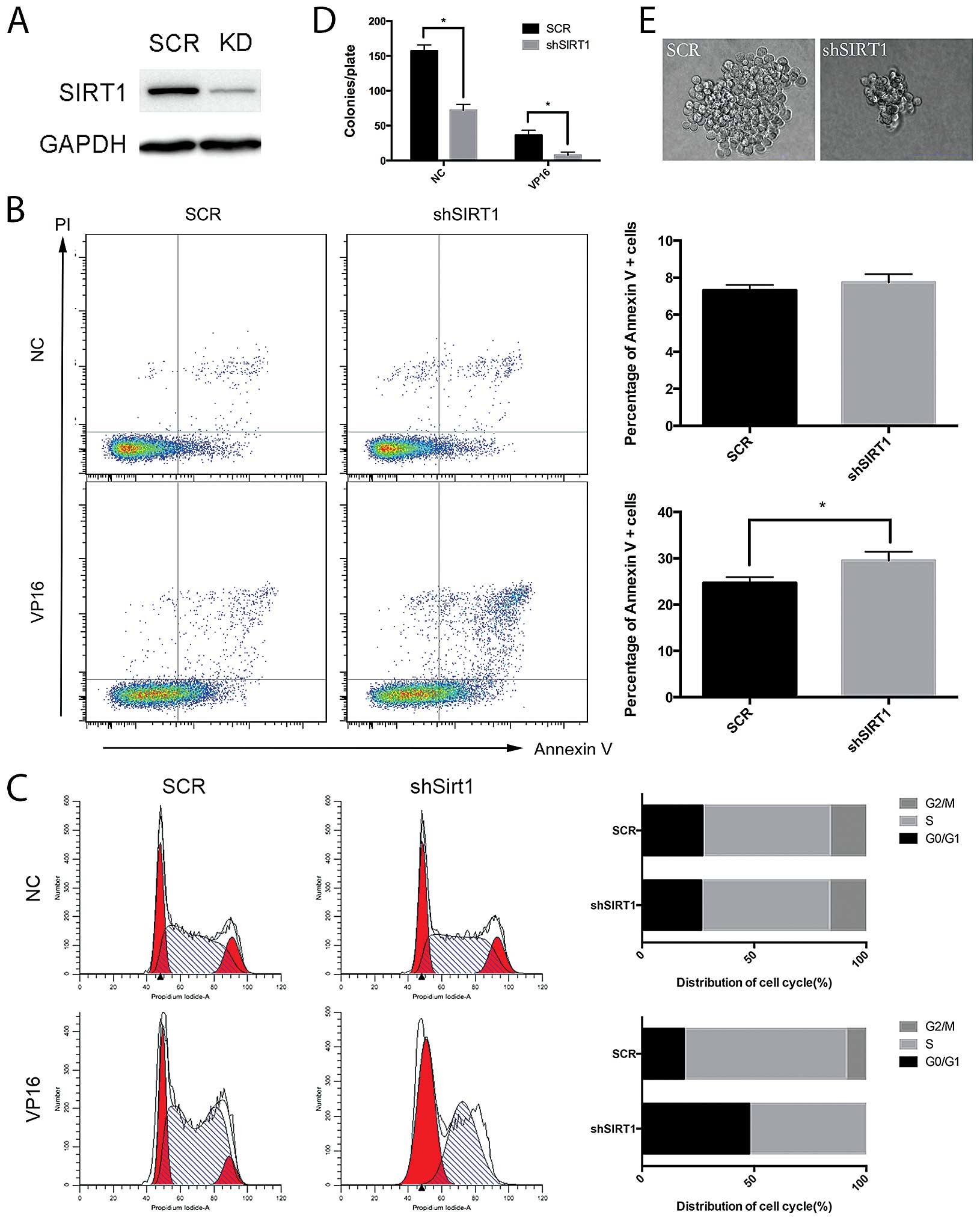
shRNA-mediated knockdown of SIRT1
sensitizes Jurkat cells to etoposide
To investigate the role of SIRT1 in response to DNA
damage of Jurkat cells, we downregulated SIRT1 by shRNA and
analyzed the apoptosis, cell cycle distribution and colony
formation ability of Jurkat cells following etoposide treatment.
The shRNA against SIRT1 specifically decreased SIRT1 protein level
in Jurkat cells (Fig. 2A). Compared
with the control shRNA, SIRT1 shRNA did not apparently alter cell
apoptosis under normal growth conditions, but significantly
enhanced the induction of apoptosis by etoposide in Jurkat cells
(Fig. 2B). Similarly, the cell
cycle analysis revealed that SIRT1 shRNA did not alter apoptosis
under normal growth conditions. However, SIRT1 knockdown increased
the cell population at G0/G1 phase with a marked reduction of cells
in the G2/M phase following treatment with etoposide (Fig. 2C). Furthermore, the shRNA-mediated
knockdown of SIRT1 decreased the colony-forming potential of Jurkat
cells by 2-fold under normal growth conditions, and further
suppressed the colony-forming potential of Jurkat cells by 4-fold
following etoposide treatment (Fig.
2D). In addition, the colony size of Jurkat cells with SIRT1
shRNA was much smaller than that of cells with control shRNA
(Fig. 2E). Taken together, these
results demonstrated that the shRNA-mediated knockdown of SIRT1
reduced the cell viability, and increased the induction of
apoptosis and G0/G1 arrest by etoposide in Jurkat cells.
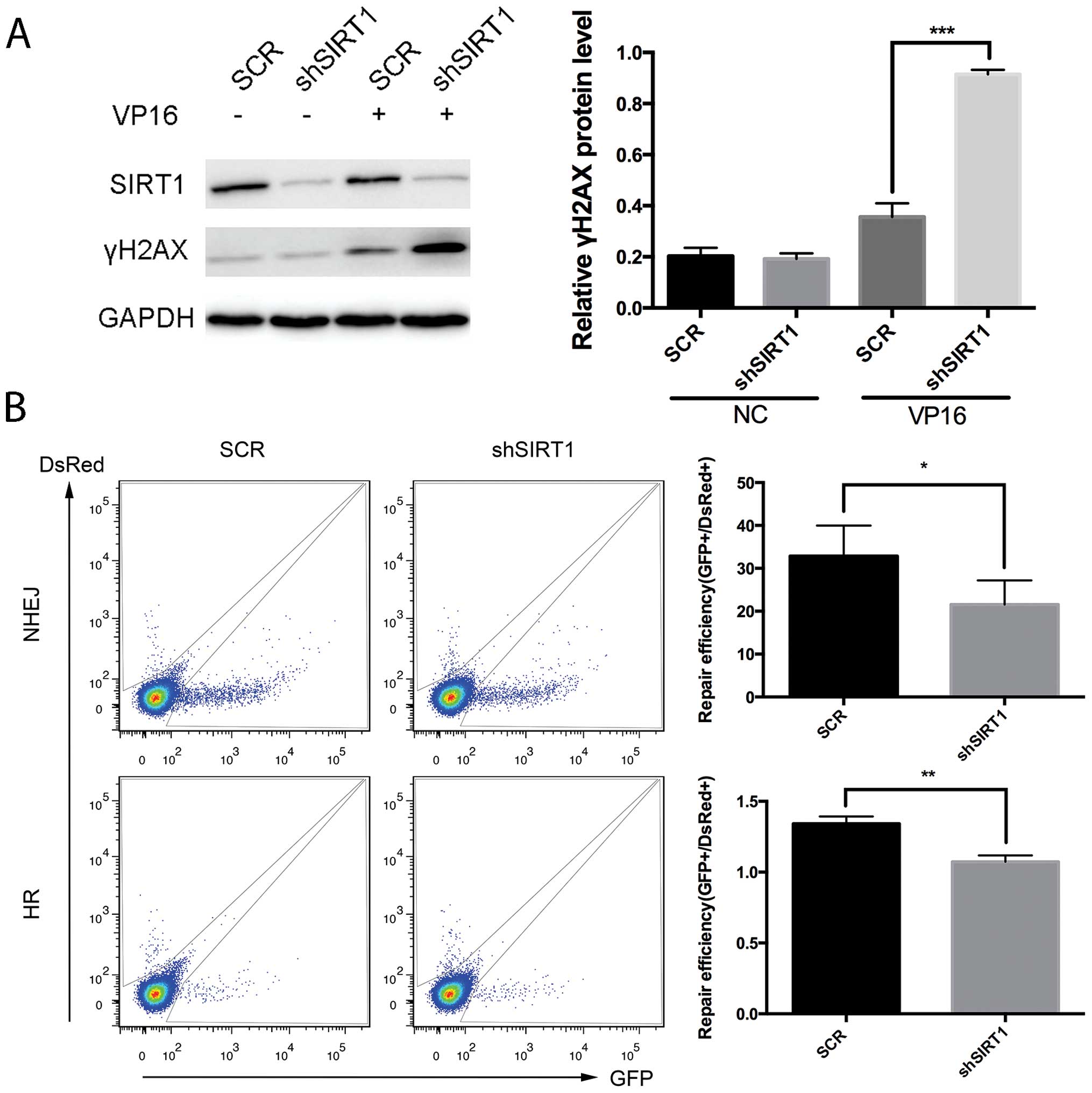
Silencing of SIRT1 results in impairs DNA
repair by HR and NHEJ in Jurkat cells
Previous results suggested that SIRT1 increases cell
survival under stressful conditions including hydrogen peroxide,
anticancer drugs (24), and
ionizing radiation (25). The
impaired cell viability and increased sensitivity to etoposide
following SIRT1 knockdown suggests that SIRT1 may promote DNA
damage repair. To test this hypothesis, we analyzed the levels of
DSBs in Jurkat cells after SIRT1 knockdown with etoposide
treatment. The phosphorylation of H2AX (γH2AX), a marker of DSBs,
increased in response to the etoposide treatment in Jurkat cells;
whereas, the level of γH2AX was significantly higher in Jurkat
cells transduced with SIRT1 shRNA than that with control shRNA
(Fig. 3A), indicating that SIRT1
was required for the repair of DSBs in Jurkat cells. DSBs are
repaired by NHEJ and HR. To monitor the efficiency of NHEJ and HR
in a quantitative manner, we used DNA repair reporter plasmids
containing fluorescent reporter constructs in which a functional
GFP gene was reconstituted following the activity of HR or NHEJ
event. The results showed that inhibition of SIRT1 by shRNA reduced
the efficiency of NHEJ by 25% and HR by 50% (Fig. 3B), suggesting that SIRT1 was
required for both HR and NHEJ.
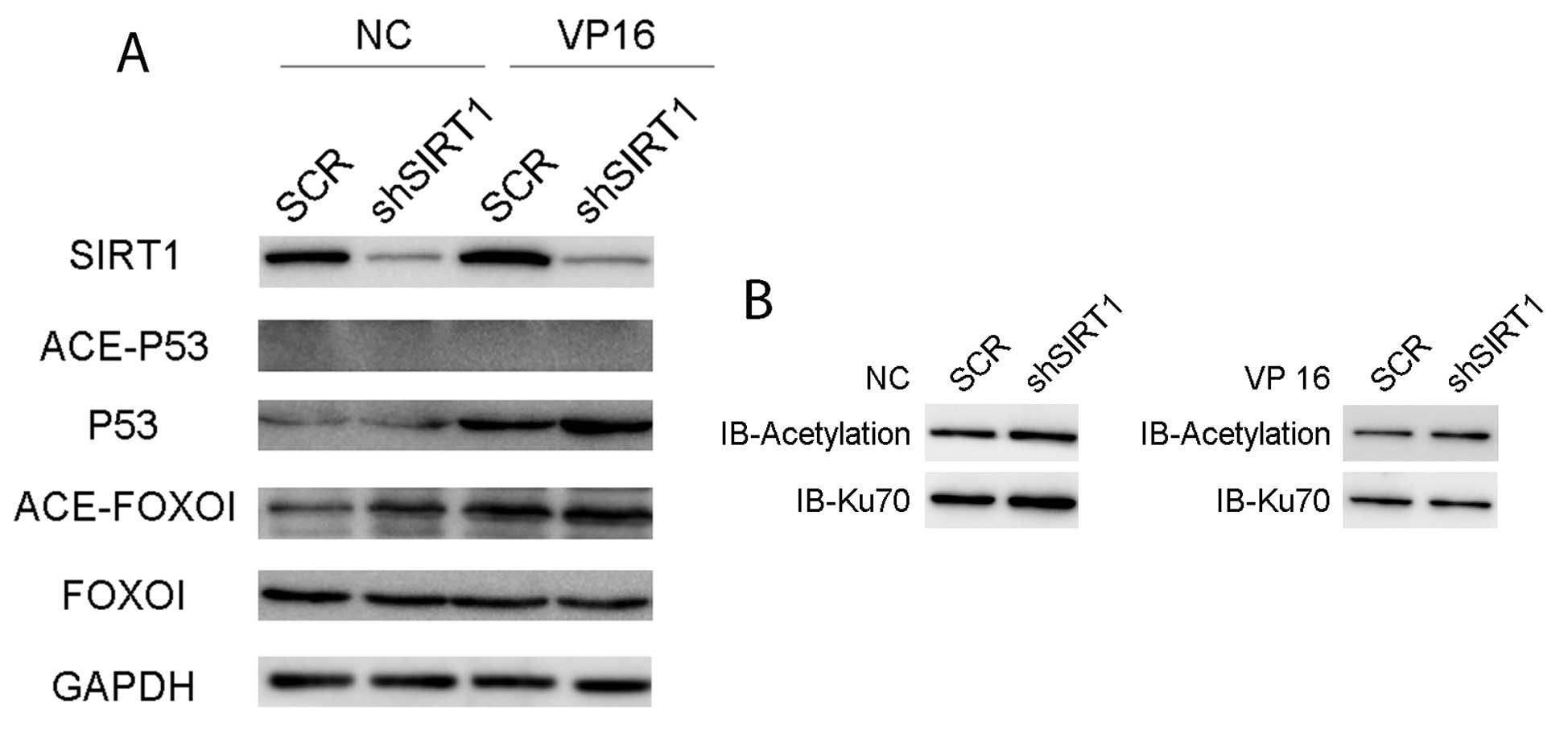
SIRT1 knockdown by shRNA leads to the
acetylation of p53, FOXO1 and Ku70 in Jurkat cells
SIRT1 has been shown to deacetylate various proteins
involved in DNA damage response, including p53, FOXO1 and Ku70
(26). We therefore determined the
acetylation levels of p53, FOXO1 and Ku70 in Jurkat cells after
SIRT1 knockdown with or without etoposide treatment. Etoposide
treatment resulted in the increased acetylation level of FOXO1.
SIRT1 knockdown led to the elevated acetylation of FOXO1 under
normal growth conditions and slightly enhanced FOXO1 acetylation by
etoposide (Fig. 4A). Total p53
protein level was significantly elevated by SIRT1 knockdown after
the etoposide treatment, but the acetylation level of p53 was
undetectable in Jurkat cells. Similarly, SIRT1 shRNA increased the
acetylation level of Ku70 under both normal growth conditions and
etoposide treatment (Fig. 4B). Ku70
is a core component of the NHEJ repair pathway and FOXO
transcription factors regulate the expression of genes related to
cell cycle, DNA repair and apoptosis in response to DNA damage and
oxidative stress. The findings suggested that SIRT1 inhibition
impairs the viability of Jurkat cells by reducing DNA repair
efficiency via elevation of the acetylation levels of Ku70 and
FOXO1.
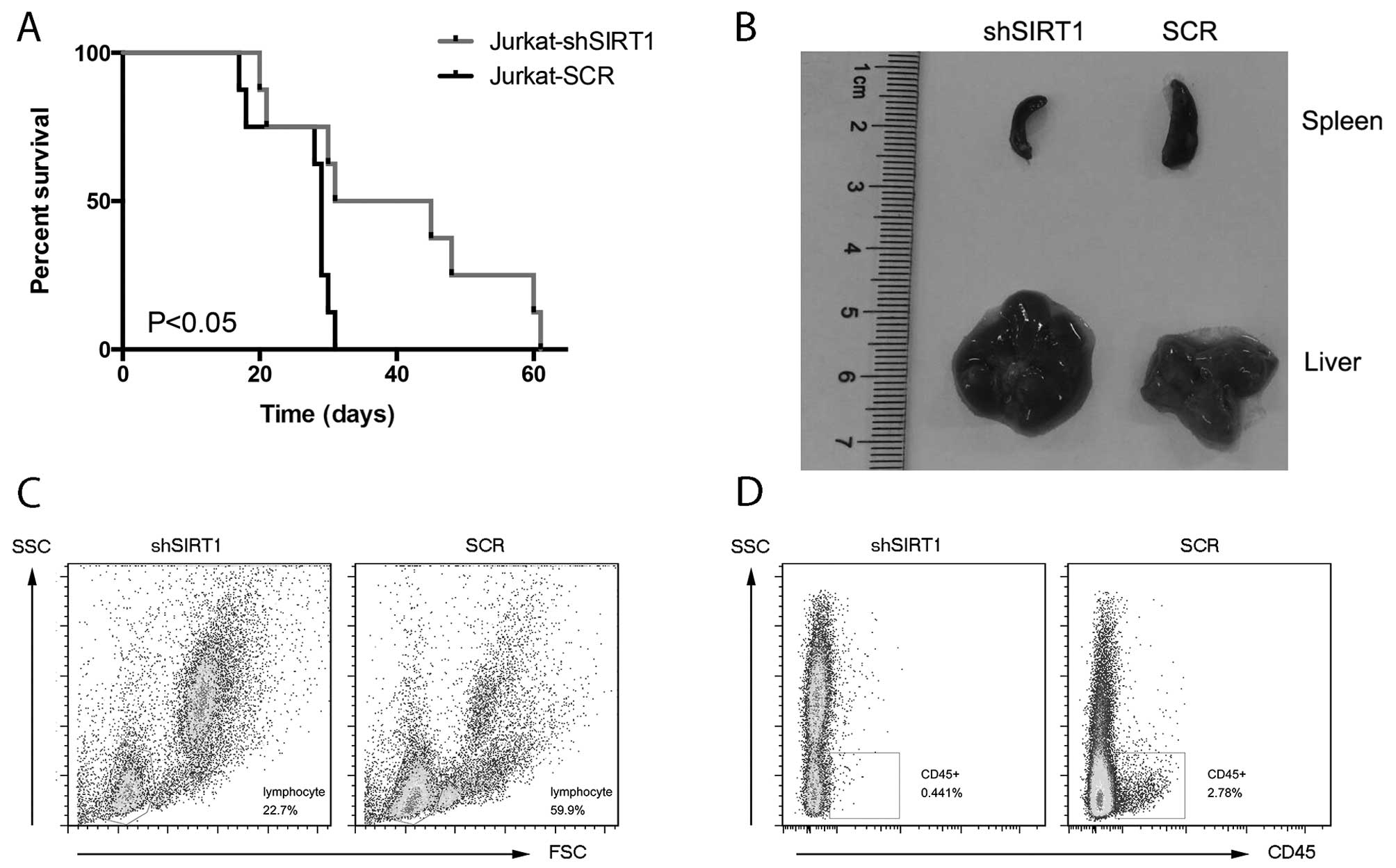
SIRT1 silencing prolongs the survival
time of Jurkatxenografted mice
We determined whether inhibition of SIRT1 induces
the regression of leukemic blasts in vivo. The results
showed that the lifespan of the shSIRT1 group was 38 days, while
that of the SCR group was ~29 days (Fig. 5A). The difference in survival time
was statistically significant (P<0.05). On day 20 after the
Jurkat cell injection, three mice from each group were sacrificed
for histopathological analyses. Compared with the mice injected
with Jurkat cells transfected with SIRT1 shRNA, the mice injected
with Jurkat cells transfected with control shRNA presented larger
splenic size and pallor of liver (Fig.
5B). Furthermore, the bone marrow FACS assay showed that the
SIRT1 shRNA group had a lower percentage of lymphocytes (Fig. 5C) and human CD45+ cells
(Fig. 5D) compared with that of the
control siRNA group. These results indicated that SIRT1 inhibition
delayed the leukemic blasts of Jurkat-xenografted mice.
Discussion
In the present study, we have demonstrated that
SIRT1 was overexpressed in Jurkat cells and SIRT1 silencing by
shRNA reducing the cell viability, and increasing the induction of
apoptosis and G0/G1 arrest by etoposide in Jurkat cells.
Additionally, SIRT1 downregulation resulted in impaired DNA repair
capacity, accompanied by reduced activity of the HR and NHEJ in
Jurkat cells. Further analyses showed that SIRT1 knockdown by shRNA
elevated the acetylation of FOXO1 and Ku70 in Jurkat cells.
Notably, we found that SIRT1 silencing prolonged the survival time
and delayed leukemic blasts in Jurkat-xenografted mice. Our
findings suggest that targeting SIRT1 is a promising strategy for
the sensitization of ATL to DNA damage-based chemotherapies.
SIRT1, a well-known longevity factor, is a
NAD(+)-dependent protein deacetylase that is involved in a wide
variety of cell processes from cancer to aging (11). SIRT1 is also consistently
upregulated in malignant cells or tissues from patients with
glioblastoma, prostate, colorectal or skin cancers (27). In the present study, SIRT1
demonstrated the highest expression level in Jurkat cells, a
typical ATL cell line, suggesting that the deregulation of SIRT1
may contribute to the anticancer drug resistance of Jurkat
cells.
Etoposide, targeting DNA topoisomerase II (TOP2), is
widely used as an anticancer drug, which increases the TOP2
cleavage complex and thus TOP2-mediated chromosome DNA breakage
(28,29). In the present study, etoposide
treatment was administered at a concentration of 20 µM for 4
h (30), and further experiments
were performed 48 h later, to induce DNA damage and provide
adequate time for DNA repair in Jurkat cells. Our results showed
that SIRT1 knockdown enhanced etoposide-induced γH2AX (31), suggesting that SIRT1 was required
for the repair of etoposide-induced DNA damage. NHEJ for direct
DSBs (32) and HR for
replication-associated DSBs (33,34),
are two core DNA repair mechanisms for DSB lesions. The results
showed that SIRT1 knockdown leads to impaired capacity of the NHEJ
and HR pathways.
The homologous recombination repair pathway is
essential during the proliferative stages of development and during
somatic cell renewal in adults to protect against cell death and
mutagenic outcomes from DNA damage. HR is a crucial DNA repair
pathway in mammalian cells. RAD51, as a core protein of HR,
catalyses the defining biochemical step of HR (35). SIRT1 deacetylates the HR repair
factor Nijmegen breakage syndrome (NBS) (16), a component of MRE11-RAD50-NBS1 (MRN)
complex, and regulates the recruitment of NBS1 and RAD51 to DNA
damage foci for repair (36). In
the present study, we have shown that SIRT1 silencing reduced the
HR by 50% in Jurkat cells. These findings indicate that SIRT1 was
required for efficient HR repair pathway.
Ku70 and Ku80 heterodimer is essential for NHEJ
(10). Both biochemical and in
vivo systems have shown that incompatible end joining can occur
without Ku, but the joining is much less efficient (32). SIRT1 protein physically interacts
with Ku70 (13), and this
interaction may modulate Ku70 protein activity by controlling its
acetylation status. In this study, acetylated Ku70 levels, which
represent inactive Ku70 status, increased and the repair efficiency
of the NHEJ pathway decreased in Jurkat cells following SIRT1
knockdown. Notably, it was reported that SIRT1 regulated NBS1 and
RAD51, and inhibition of either NBS1 or Rad51 resulted in impaired
NHEJ repair (37). Therefore, SIRT1
may be involved in the NHEJ repair pathway by modulating various
proteins, including acetylation levels of Ku70 protein in ATL.
In the present study, we found that inhibition of
SIRT1 rendered Jurkat cells more sensitive to etoposide treatment,
as demonstrated by the higher level of apoptosis, G0/G1 phase
arrest and impaired colony-formation capacity. Our results suggest
that impaired DNA repair by SIRT1 inhibition may lead to
accumulation of lesions in cells following treatment with DNA
damage-inducing drugs, which in turn induces apoptosis and cell
cycle arrest. SIRT1 has been shown to deacetylate various proteins
involved in the DNA damage response, including p53 (24,25),
FOXO (15), NF-κB (19) and Ku70. Of these, acetylated p53 and
active status were reported to induce apoptosis and cell cycle
arrest. Our results showed a significant elevation of total p53,
however, the expression level of p53 was too low for us to detect
the acetylation level. The FOXO1 acetylation level was elevated
after SIRT1 knockdown in Jurkat cells. FOXO1 regulates cell
proliferation and differentiation (38). Acetylated FOXO1, the inactive form,
may therefore contribute to reduced colony formation of cultured
Jurkat cells and longer survival time of Jurkat-xenografted mice by
affecting cell viability and proliferation.
In conclusion, the present study provides evidence
that SIRT1 inhibition induces the acetylation of various substrate
proteins, affects DNA damage repair efficiency in ATL cells,
renders ATL cells more sensitive to DNA damage drugs and prolongs
the survival time of xenografted mice. The findings suggest that
SIRT1 is a novel target for the development of ATL treatment.
Acknowledgments
The present study was in part supported by a grant
from the National Nature Science Foundation of China (nos. 81270615
and 81400111), leading talent project (no. 2014054) and the
Shanghai Municipal Health Bureau (nos. XBR2013077 and
20134445).
References
|
1
|
Uchiyama T: Human T cell leukemia virus
type I (HTLV-I) and human diseases. Annu Rev Immunol. 15:15–37.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yoshida M: Multiple viral strategies of
HTLV-1 for dysregulation of cell growth control. Annu Rev Immunol.
19:475–496. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Linker C, Damon L, Ries C and Navarro W:
Intensified and shortened cyclical chemotherapy for adult acute
lymphoblastic leukemia. J Clin Oncol. 20:2464–2471. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hijiya N, Thomson B, Isakoff MS, Silverman
LB, Steinherz PG, Borowitz MJ, Kadota R, Cooper T, Shen V, Dahl G,
et al: Phase 2 trial of clofarabine in combination with etoposide
and cyclophosphamide in pediatric patients with refractory or
relapsed acute lymphoblastic leukemia. Blood. 118:6043–6049. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chabner BA and Roberts TG Jr: Timeline:
Chemotherapy and the war on cancer. Nat Rev Cancer. 5:65–72. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Helleday T, Petermann E, Lundin C, Hodgson
B and Sharma RA: DNA repair pathways as targets for cancer therapy.
Nat Rev Cancer. 8:193–204. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Grady S, Finn SP, Cuffe S, Richard DJ,
O'Byrne KJ and Barr MP: The role of DNA repair pathways in
cisplatin resistant lung cancer. Cancer Treat Rev. 40:1161–1170.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rich T, Allen RL and Wyllie AH: Defying
death after DNA damage. Nature. 407:777–783. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khanna KK and Jackson SP: DNA
double-strand breaks: Signaling, repair and the cancer connection.
Nat Genet. 27:247–254. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lombard DB, Chua KF, Mostoslavsky R,
Franco S, Gostissa M and Alt FW: DNA repair, genome stability, and
aging. Cell. 120:497–512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brooks CL and Gu W: How does SIRT1 affect
metabolism, senescence and cancer? Nat Rev Cancer. 9:123–128. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bordone L and Guarente L: Calorie
restriction, SIRT1 and metabolism: Understanding longevity. Nat Rev
Mol Cell Biol. 6:298–305. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeong J, Juhn K, Lee H, Kim SH, Min BH,
Lee KM, Cho MH, Park GH and Lee KH: SIRT1 promotes DNA repair
activity and deacetylation of Ku70. Exp Mol Med. 39:8–13. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Furukawa-Hibi Y, Kobayashi Y, Chen C and
Motoyama N: FOXO transcription factors in cell-cycle regulation and
the response to oxidative stress. Antioxid Redox Signal. 7:752–760.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brunet A, Sweeney LB, Sturgill JF, Chua
KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et
al: Stress-dependent regulation of FOXO transcription factors by
the SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan Z, Zhang X, Sengupta N, Lane WS and
Seto E: SIRT1 regulates the function of the Nijmegen breakage
syndrome protein. Mol Cell. 27:149–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li K, Casta A, Wang R, Lozada E, Fan W,
Kane S, Ge Q, Gu W, Orren D and Luo J: Regulation of WRN protein
cellular localization and enzymatic activities by SIRT1-mediated
deacetylation. J Biol Chem. 283:7590–7598. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ming M, Shea CR, Guo X, Li X, Soltani K,
Han W and He YY: Regulation of global genome nucleotide excision
repair by SIRT1 through xeroderma pigmentosum C. Proc Natl Acad Sci
USA. 107:22623–22628. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD,
Jones DR, Frye RA and Mayo MW: Modulation of NF-kappaB-dependent
transcription and cell survival by the SIRT1 deacetylase. EMBO J.
23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yuan H, Wang Z, Li L, Zhang H, Modi H,
Horne D, Bhatia R and Chen W: Activation of stress response gene
SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 119:1904–1914.
2012. View Article : Google Scholar :
|
|
21
|
Lopez-Royuela N, Rathore MG, Allende-Vega
N, Annicotte JS, Fajas L, Ramachandran B, Gulick T and Villalba M:
Extracellular-signal-regulated kinase 5 modulates the antioxidant
response by transcriptionally controlling Sirtuin 1 expression in
leukemic cells. Int J Biochem Cell Biol. 53:253–261. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sasca D, Hähnel PS, Szybinski J, Khawaja
K, Kriege O, Pante SV, Bullinger L, Strand S, Strand D, Theobald M,
et al: SIRT1 prevents genotoxic stress-induced p53 activation in
acute myeloid leukemia. Blood. 124:121–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kozako T, Aikawa A, Shoji T, Fujimoto T,
Yoshimitsu M, Shirasawa S, Tanaka H, Honda S, Shimeno H, Arima N,
et al: High expression of the longevity gene product SIRT1 and
apoptosis induction by sirtinol in adult T-cell leukemia cells.
International journal of cancer. Int J Cancer. 131:2044–2055. 2012.
View Article : Google Scholar
|
|
24
|
Luo J, Nikolaev AY, Imai S, Chen D, Su F,
Shiloh A, Guarente L and Gu W: Negative control of p53 by Sir2alpha
promotes cell survival under stress. Cell. 107:137–148. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vaziri H, Dessain SK, Ng Eaton E, Imai SI,
Frye RA, Pandita TK, Guarente L and Weinberg RA: hSIR2(SIRT1)
functions as an NAD-dependent p53 deacetylase. Cell. 107:149–159.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saunders LR and Verdin E: Sirtuins:
Critical regulators at the crossroads between cancer and aging.
Oncogene. 26:5489–5504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu T, Liu PY and Marshall GM: The
critical role of the class III histone deacetylase SIRT1 in cancer.
Cancer Res. 69:1702–1705. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nitiss JL: Targeting DNA topoisomerase II
in cancer chemotherapy. Nat Rev Cancer. 9:338–350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu
YJ, Yen TJ, Chiang CW and Chan NL: Structural basis of type II
topoisomerase inhibition by the anticancer drug etoposide. Science.
333:459–462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Z, Sun B, Clewell RA, Adeleye Y,
Andersen ME and Zhang Q: Dose-response modeling of
etoposide-induced DNA damage response. Toxicol Sci. 137:371–384.
2014. View Article : Google Scholar
|
|
31
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lieber MR: NHEJ and its backup pathways in
chromosomal translocations. Nat Struct Mol Biol. 17:393–395. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sargent RG, Brenneman MA and Wilson JH:
Repair of site-specific double-strand breaks in a mammalian
chromosome by homologous and illegitimate recombination. Mol Cell
Biol. 17:267–277. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Arnaudeau C, Lundin C and Helleday T: DNA
double-strand breaks associated with replication forks are
predominantly repaired by homologous recombination involving an
exchange mechanism in mammalian cells. J Mol Biol. 307:1235–1245.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moynahan ME and Jasin M: Mitotic
homologous recombination maintains genomic stability and suppresses
tumorigenesis. Nat Rev Mol Cell Biol. 11:196–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oberdoerffer P, Michan S, McVay M,
Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner
A, Loerch P, et al: SIRT1 redistribution on chromatin promotes
genomic stability but alters gene expression during aging. Cell.
135:907–918. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Z, Yuan H, Roth M, Stark JM, Bhatia R
and Chen WY: SIRT1 deacetylase promotes acquisition of genetic
mutations for drug resistance in CML cells. Oncogene. 32:589–598.
2013. View Article : Google Scholar
|
|
38
|
Arden KC: Multiple roles of FOXO
transcription factors in mammalian cells point to multiple roles in
cancer. Exp Gerontol. 41:709–717. 2006. View Article : Google Scholar : PubMed/NCBI
|