Introduction
For decades, advances in the breast cancer field
have dramatically promoted the overall 5-year survival rate for
women in the United States from 75% between 1975 to 1977, to 90%
between 2002 to 2008 (1). Despite
these advances, breast cancer is still the second cause of death
from cancer. Most of the patients died of metastases, rather than
the primary tumors, and the 5-year survival rate is a mere 23% for
the women diagnosed with distant metastatic disease (1). It was reported that metastasis to
distant sites accounts for >90% of breast cancer-related
mortality (2). Despite its clinical
importance, metastasis remains the most insidious aspect of breast
cancer. Consequently, there are few successful treatments that
directly target this stage of carcinogenesis. Therefore, unraveling
the complexities of the genetic and biochemical determinants of
metastasis and developing effective therapies are urgently needed
to improve treatment regimens and ultimately prognostic
outcomes.
A recent focus in breast cancer metastasis research
is the epithelial-mesenchymal transition (EMT). Originally, EMT is
reported to be a critical developmental program that entails
epithelial cells to mesenchymal cells, mobiling cells and giving
rise to bone, muscle, connective tissue and blood vessels (3). Increasing data have shown that EMT
processes have been implicated in cancer progression and metastasis
including carcinogenesis, increased motility, invasion, and
angiogenesis in various solid tumor types (4). A wide spectrum of alterations occurs
during EMT processes, promoting loss of cell-cell adhesion, leading
to a shift in cytoskeletal dynamics and a change from epithelial
morphology and physiology to the mesenchymal phenotype (5). Several oncogenic pathways that respond
to extracellular cues have been shown to contribute to EMT of
carcinoma cells, such as transforming growth factor-β (TGF-β) and
bone morphogenetic protein (BMP), Wnt/β-catenin, Notch and Hedgehog
signaling pathway. These pathways have activated transcription
factors including Snail, Slug, and the Twist to generate many
intermediate phenotypes between epithelial and mesenchymal states
(6).
CLT, also named atractylenolide III, is the major
bioactive component of Atractylodes lancea and also found in
a range of medical herbs, such as Codonopsis pilosula,
Atractylodes macrocephala Koidz and Chloranthus henryi
Hemsl (7). This sesquiterpene
lactone has been demonstrated to exhibit a range of activities,
including anti-allergic activity (8,9),
anti-inflammatory (10),
gastroprotective and neuroprotective activity (11,12).
Furthermore, CLT was shown recently to induce apoptosis of A549
cells via mitochondria-mediated death pathway (13). Previously, we focused our interest
on antitumor activities of CLT, and revealed that this natural
compound exhibited anti-metastatic effects in metastatic breast
cancer cells, and these effects may be mediated by inhibition of
matrix metalloproteinases (MMPs) via downregulation of the
transcriptional activity of Runx2 (14). Runx2 is a key transcription factor
associated with osteoblast differentiation and breast cancer
metastasis. Τhere is close association between abnormal activation
of Runx2 and EMT, matrix degradation, angiogenesis and colonizing
in bone marrow (15). Moreover,
this transcription factor is also the common target of TGF-β/BMP
signaling (16,17). Based on their critical roles in
TGF-β signaling and Runx2 during EMT, we hypothesized that
EMT-involved mechanisms may contribute to the anti-metastatic
effects of CLT. Therefore, we evaluated the effects of CLT and its
mechanisms on EMT processes in breast cancer.
Materials and methods
Materials
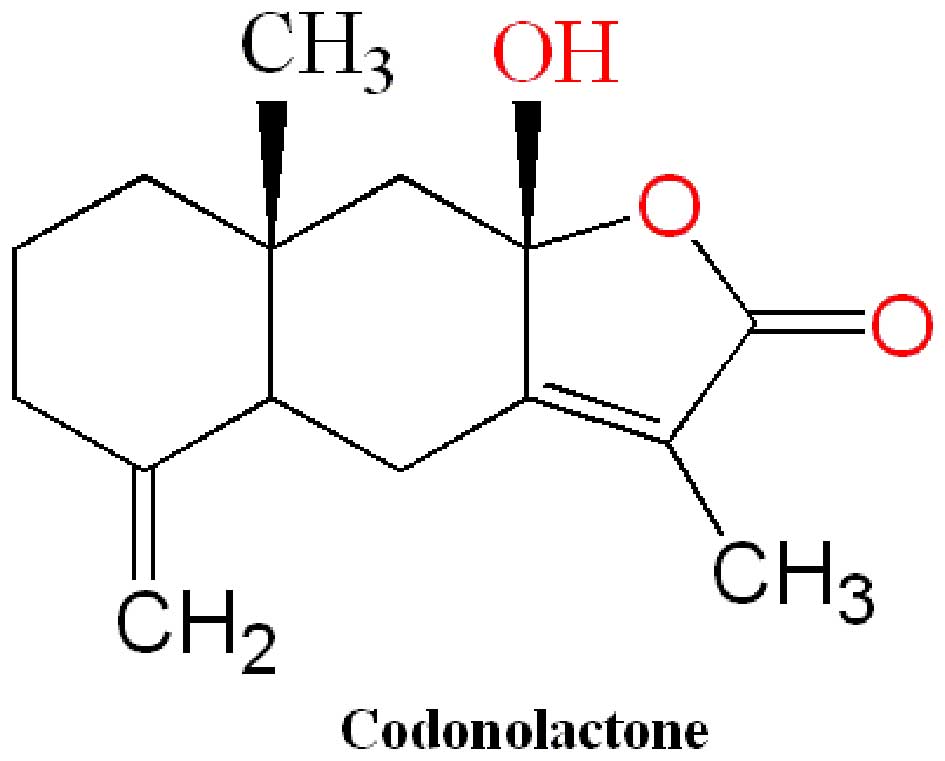
Codonolactone (CLT) with a purity of up to 98.0% was
from Shanghai PureOne Biotechnology (P0110; Shanghai, China). A
stock solution was used throughout the study by dissolving CLT in
100% dimethyl sulfoxide (DMSO). The final DMSO concentration did
not exceed 0.1%. The molecular structure of CLT is presented in
Fig. 1. TGF-β1 recombinant human
protein (PHG9211) is the product of Life Technologies, and
5-fluorouracil (5-FU, cat no. PHR1227; Sigma-Aldrich, Beijing,
China) was used as positive controls in in vivo study.
Batimastat (Bat, cat no. SC-203833; Santa Cruz Biotechnology Inc.,
Santa Cruz, CA, USA) was used in in vitro invasion and
migration assay and served as a positive control. This compound is
a potent, broad spectrum MMP inhibitor, and exhibits anti-invasive
and anti-metastatic activity.
Cell culture
Human breast cancer MDA-MB-468 and MDA-MB-231 cells,
purchased from Cell Resource Center, Institute of Basic Medical
Sciences, Chinese Academy of Medical Sciences, were maintained in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS; Gibco, Inc.), 100 IU/ml penicillin, and
100 µg/ml streptomycin (Invitrogen, Inc.) in a humidified
incubator containing 5% CO2 at 37°C.
Animals and ethics statement
Five-to 6-week-old female NOD/SCID mice were
purchased from Vital River Laboratories (Beijing, China) to
establish an orthotopic xenograft tumor model. All animal studies
were conducted in strict accordance with the requirements of animal
experimental protocol, which was approved by the Animal Ethics
Committees of Jiangxi University of Traditional Chinese Medicines.
All surgery was performed under sodium pentobarbital anesthesia,
and all efforts were made to minimize suffering.
In vivo growth assay
For the growth assay, MDA-MB-468 cell xenografts
were established by injection of 1×107 cells at the
mammary fat pad in NOD/SCID mice. After 3 weeks of growth, the
tumors were removed and chopped into 1×1×1-mm tumor pieces, and
these pieces were then implanted into the mammary fat pad of mice.
The mice bearing tumor chunks were randomly divided into 4 groups:
control, 5-FU (100 mg/kg/week) treatment group, CLT (25 mg/kg/day)
treatment group and CLT (75 mg/kg/day) treatment group. Forty-eight
hours later, CLT and was administered by gavage on a regimen of
6-day dosing per week for 5 weeks. 5-FU was treated by
intraperitoneal injection on a regimen of 3-day dosing per week for
5 weeks. Tumor growth was assessed by measuring the length and
width of tumors with electronic calipers every 3–4 days
continuously. Volumes were calculated using the formula: (length) ×
(width)2/2.
Immunohistochemistry for tumor
tissues
Cytokeratin 19 (CK19) and vimentin in tumor tissues
were determined by immunostaining. Formalin-fixed,
paraffin-embedded MDA-MB-468 tumor tissues were cut into
4-µm-thick sections. After dewaxing and hydration, the
slides were incubated with proteinase K at 37°C for 15 min to
retrieve antigen. Then the sections were treated with 3%
H2O2 in methanol for 10 min. Followed by
blocking with 10% normal goat serum (Cell Signaling Technology,
Danvers, MA, USA), the slides were incubated with CK19 (1:50,
ab15463; Abcam) or vimentin antibody (1:50, 3932; Cell Signaling
Technology) or PBS (0.01 mol/l) at 4°C overnight. The slices were
incubated with second antibody (HRP-linked secondary antibody) at
room temperature for 60 min, and then peroxidase activity was
detected by SignalStain® DAB Substrate kit (Cell
Signaling Technology) or AEC Substrate system (ab64252; Abcam). All
the slides were checked under light microscopy (BX-63; Olympus),
and images were analyzed by Image Pro Plus software 5.0 (Media
Cybernetics Inc., Silver Spring, MD, USA).
In vitro invasion assay
Effects of CLT on TGF-β1-induced invasion of breast
cancer cells were measured by a 48-well microchemotaxis system (AP
48; Neuro Probe, Gaithersburg, MD, USA). Briefly, individual
filters were coated with 5 µg Matrigel and fibronectin
(served as a chemoattractant) on the smooth (upper) and rough
(lower) surface, respectively. Thirty microliters Medium (30
µl) containing 0.1% BSA was added into each lower chamber,
and then the chambers were fixed. Cells (5×105/well) in
100 µl FBS-free medium containing vehicle, Bat or CLT were
added separately to the top of this Matrigel layer in the presence
of 10 ng/ml TGF-β1 (final concentration). After 14-h incubation,
the filters were fixed with methanol and stained with 0.5% crystal
violet for 60 min. The cells on the upper surface of the filters
were removed by wiping with cotton swabs. The cells invading to the
lower surface of the filter through Matrigel and filter were
quantified with Image Pro Plus software 5.0 (Media Cybernetics
Inc.) and the most representative results are illustrated in the
figures. Each assay was performed in triplicate.
In vitro migratory assay
In vitro migration of breast cancer cells
induced by TGF-β1 was measured using Oris™ Cell Migration Assay
(Platypus Technologies, Madison, WI, USA) following the
manufacturer's instructions. Log-phase cells were harvested, washed
three times with serum-free medium, and resuspended into a final
concentration of 4×106/ml in culture medium. Suspended
cells (100 µl) were pipetted into each test well through one
of the side ports of the Oris™ Cell Seeding Stopper. The seeded
plate containing the Oris™ Cell Seeding Stoppers was incubated in a
humidified chamber (37°C, 5% CO2) for 24 h to permit
cell attachment. Using the Stopper Tool, all the stoppers were
removed. The media was removed gently and the wells washed with PBS
to remove the unattached cells. After that, 100 µl of
FBS-free medium containing Calcein AM (final concentration 0.5
µg/ml) was added, and the cells were incubated in a
humidified chamber (37°C, 0.5% CO2) for 40 min. The
images were captured under a fluorescence microscope (DM 3000B;
Leica), and these data served as initial control. After
fluorescence intensity examination, media were removed gently, and
washed twice with PBS, and 100 µl of medium containing
vehicle, Bat or CLT was added, and the cells were incubated in a
humidified chamber (37°C, 0.5% CO2) for 24 h. At the end
of treatment, data were obtained as indicated above.
Cell immunofluorescence
E-cadherin and vimentin immunofluorescence in
MDA-MB-468 and MDA-MB-231 cells were analyzed. Briefly, log-phase
cells were seeded into wells of 8-well-Chamber Slide system
(Nunc®). Cells were grown at 37°C in a humidified
CO2 incubator until 50–70% confluence, and exposed to
CLT and/or vehicle for 24 h. After exposure, the chambers were
gently removed, and slides were rinsed twice in PBS. Cells fixed by
4% paraformaldehyde solutions were blocked against non-specific
antibody interactions with 5% (w/v) BSA in TBST for 1 h at room
temperature and incubated overnight at 4°C with anti-E-cadherin
(1:100, 3195) or vimentin antibody (1:100, 3932) (both from Cell
Signaling Technology). After washing in TBST, slides were incubated
with a secondary antibody conjugated with Alexa Fluor 555 (4413,
1:500; Cell Signaling Technology) for 1 h. Cells were then washed
and incubated with ProLong Gold antifade reagent with DAPI
(Invitrogen). Samples were examined under a fluorescent microscope
(BX63; Olympus).
Protein extraction and western blot
analysis
Cells were rinsed twice with PBS, and total proteins
were extracted in 500 µl lysis buffer. Aliquots of whole
cell lysates were separated by 10% SDS-PAGE and then transferred to
Hybond nitro blotting membranes. The membranes were blocked with 3%
BSA in Tris-buffered saline containing 0.5 ml/l Tween-20 and then
incubated with primary antibodies against E-cadherin (3195; Cell
Signaling Technology), N-cadherin (ab18203; Abcam), vimentin (3932;
Cell Signaling Technology), cytokeratin 19 (ab15463), Snail
(ab82846) and Slug (ab27568) (all from Abcam), Twist (sc-6070;
Santa Cruz Biotechnology), Smad2 (ab55478; Abcam), phosphor-Smad2/3
(sc-11769) and Runx2 (SC-10758) (both from Santa Cruz
Biotechnology), phospho-Runx2 (AP3559a, Abgent), JNK1/2/3 (ab59227)
and phospo-JNK1/2/3 (ab192200) (both from Abcam), ERK1/2
(sc-292838) and phosphor-ERK1/2 (sc-16982) (both from Santa Cruz
Biotechnology), p38MAPK (9212) and phospho-p38MAPK (9211) (both
from Cell Signaling Technology) followed by incubation with
horseradish peroxidase (HRP)-conjugated secondary antibodies.
Immunoreactive proteins were detected using an enhanced
chemiluminescence kit (Millipore). β-actin (SC-130301; Santa Cruz
Biotechnology) served as an internal control.
Data analysis
The data are presented as mean ± SD and were
analyzed with SPSS for Windows (13.0) software program (Chicago,
IL, USA). Comparison among different groups was carried out by
one-way analysis of variance (the one-way ANOVA). Differences
between means were considered statistically significant at
P<0.05.
Results
CLT shows significant inhibition on
TGF-β1-mediated EMT in breast cancer cells
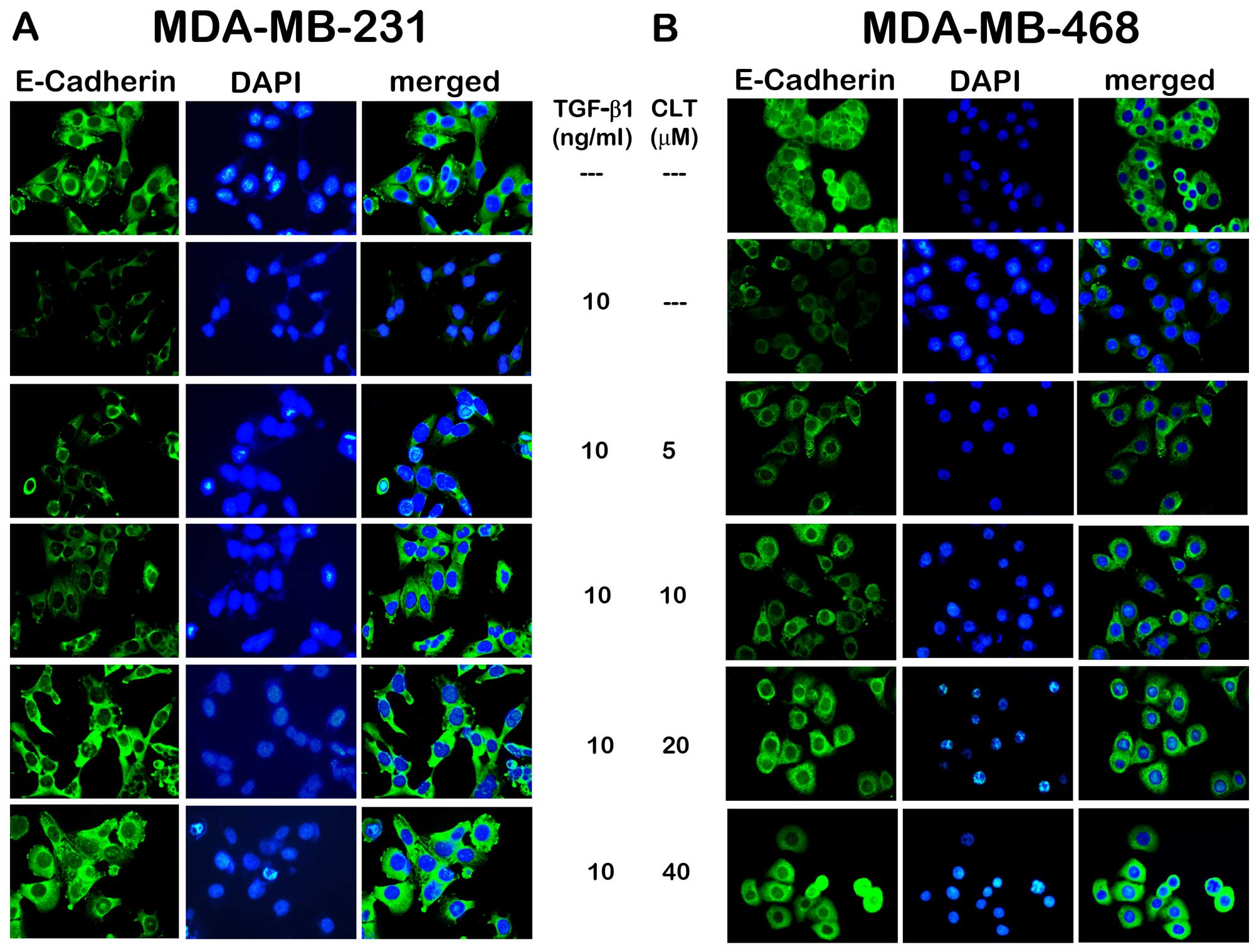
To understand the effects of CLT on EMT, the initial
study was to check expression of E-cadherin in CLT-treated
MDA-MB-231 and MDA-MB-468 cells. As shown in Fig. 2A, when MDA-MB-231 cells were
incubated with TGF-β1, the epithelial cell marker E-cadherin was
significantly inhibited. After exposure to CLT solution at various
concentrations for 24 h, expression of this epithelial cell marker
was increased in a dose-dependent manner. In addition, to verify
the anti-EMT effects of CLT, another breast cancer cell line was
studied, which has been demonstrated to be susceptible to undergo
EMT changes under appropriate stimuli (18). Reduced E-cadherin was observed when
MDA-MB-468 cells were incubated with TGF-β1, and CLT increased the
expression of this marker significantly (Fig. 2B).
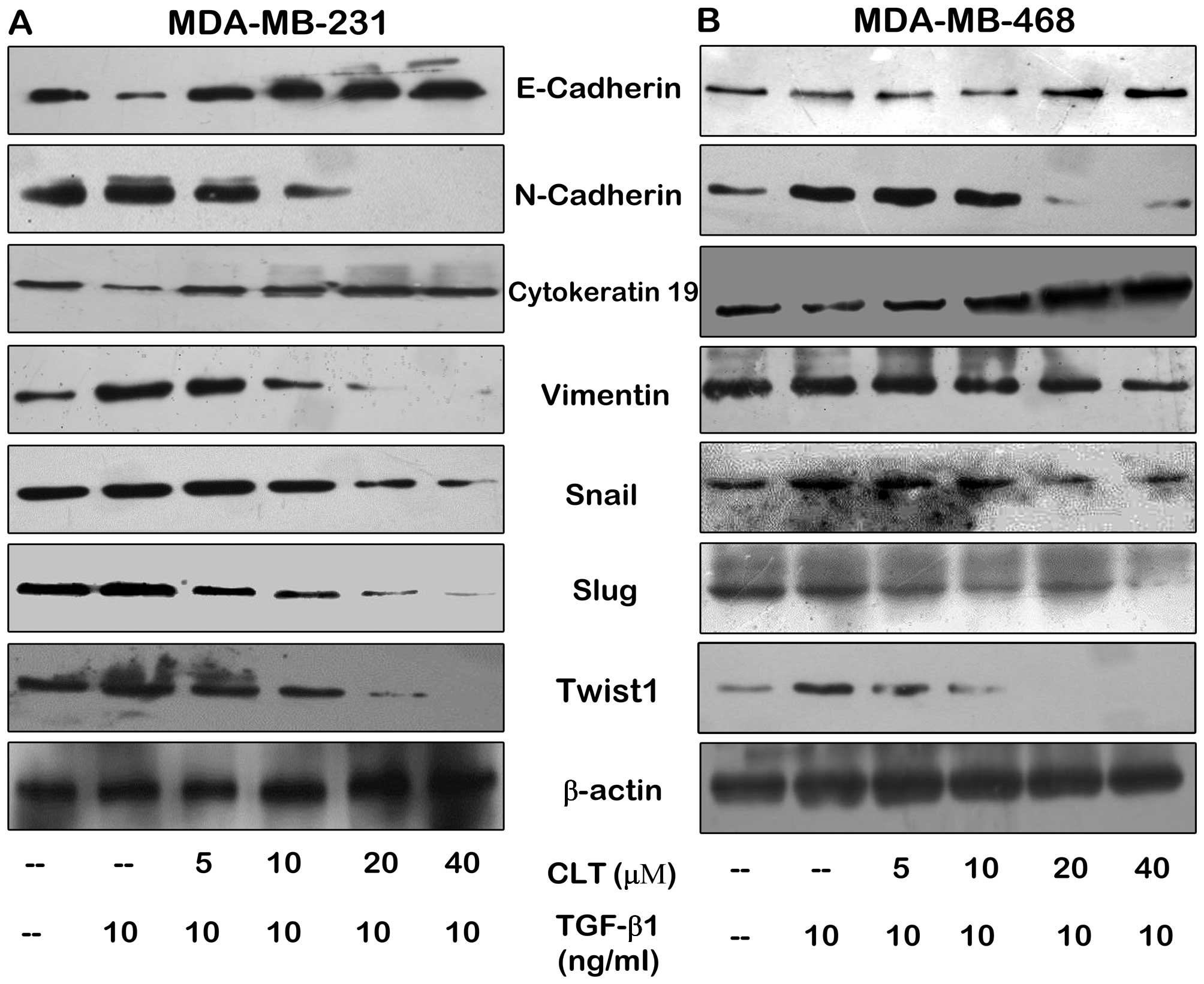
EMT programs are associated classically with a set
of change of cell-surface and cytoskeleton markers, which are
orchestrated by a set of pleiotropically acting transcription
factors. Thus, we next determined the effects of CLT on several
prototypical epithelial and mesenchymal markers by western blotting
assay, including cell surface molecular, cytoskeleton proteins and
transcription factors. As shown in Fig.
3, CLT enhanced E-cadherin and CK19 expression in both
MDA-MB-231 and MDA-MB-468 cells. In addition, CLT significantly
inhibited expression of N-cadherin and vimentin, which are
mesenchymal markers and acquired during EMT. Furthermore, we also
found that expression of several transcription factors, Snail, Slug
and Twist 1, was significantly blocked by CLT (Fig. 3) in a dose-dependent manner. These
data demonstrated the effects of CLT on TGF-β1-induced EMT in
vitro.
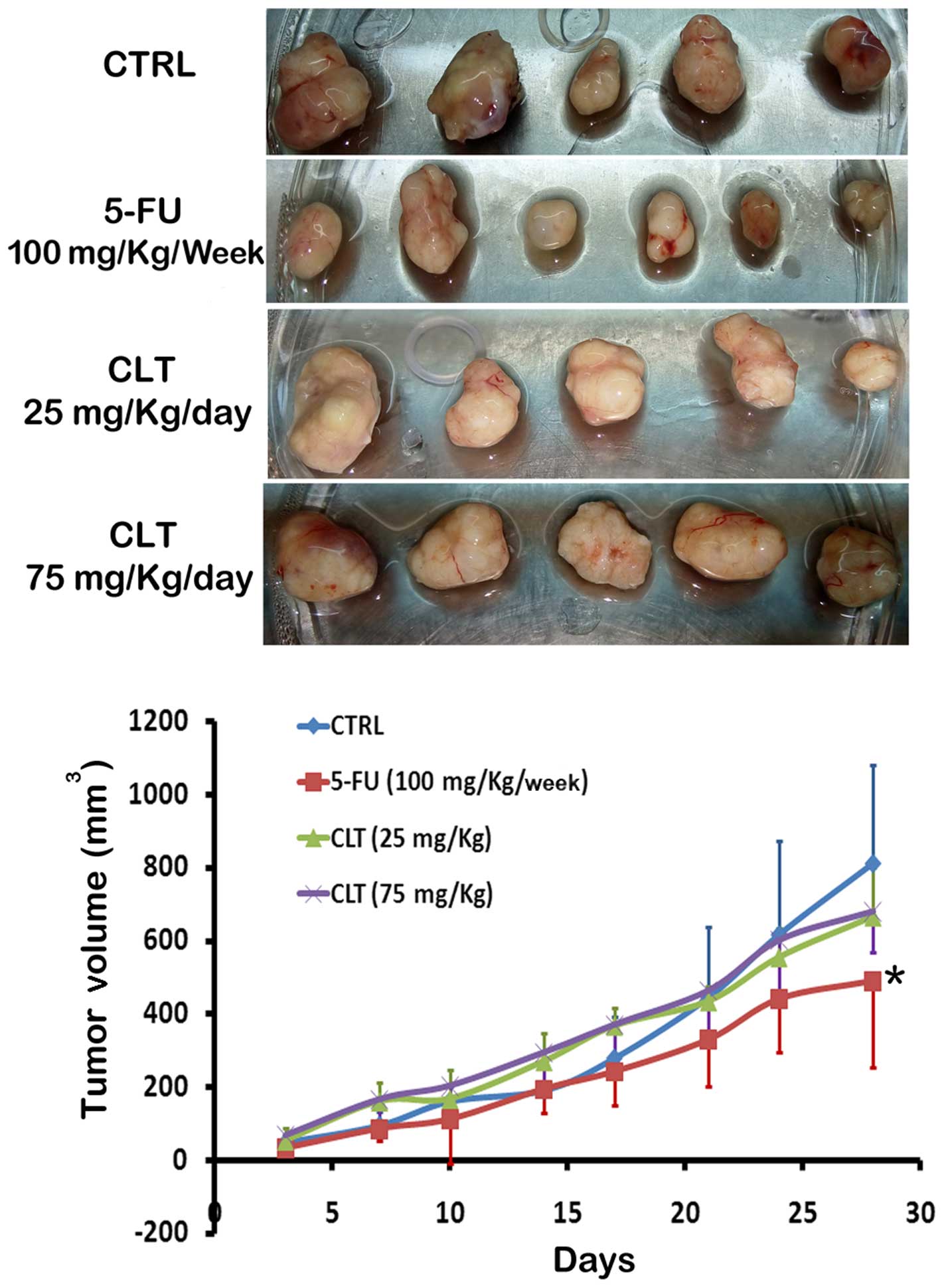
CLT inhibits EMT of MDA-MB-468 tumors in
vivo
According to the study by the Gilles group (18), MDA-MB-468 is an inducible model of
EMT in vivo. To determine whether CLT impairs EMT in
vivo, we established an orthotopic xenograft model by injection
of MDA-MB-468 cells into the mammary fat pad in NOD/SCID mice
following the method of Gilles et al. It was found that
compared with vehicle-treated animals, CLT administered at 75
mg/kg/day caused very slight inhibition on primary tumor growth
(Fig. 4), which is in agreement
with our previous results. Next, the anti-EMT effects of CLT were
evaluated by examining expression of two kinds of cytoskeleton
proteins in tumor tissues by IHC. The most representative results
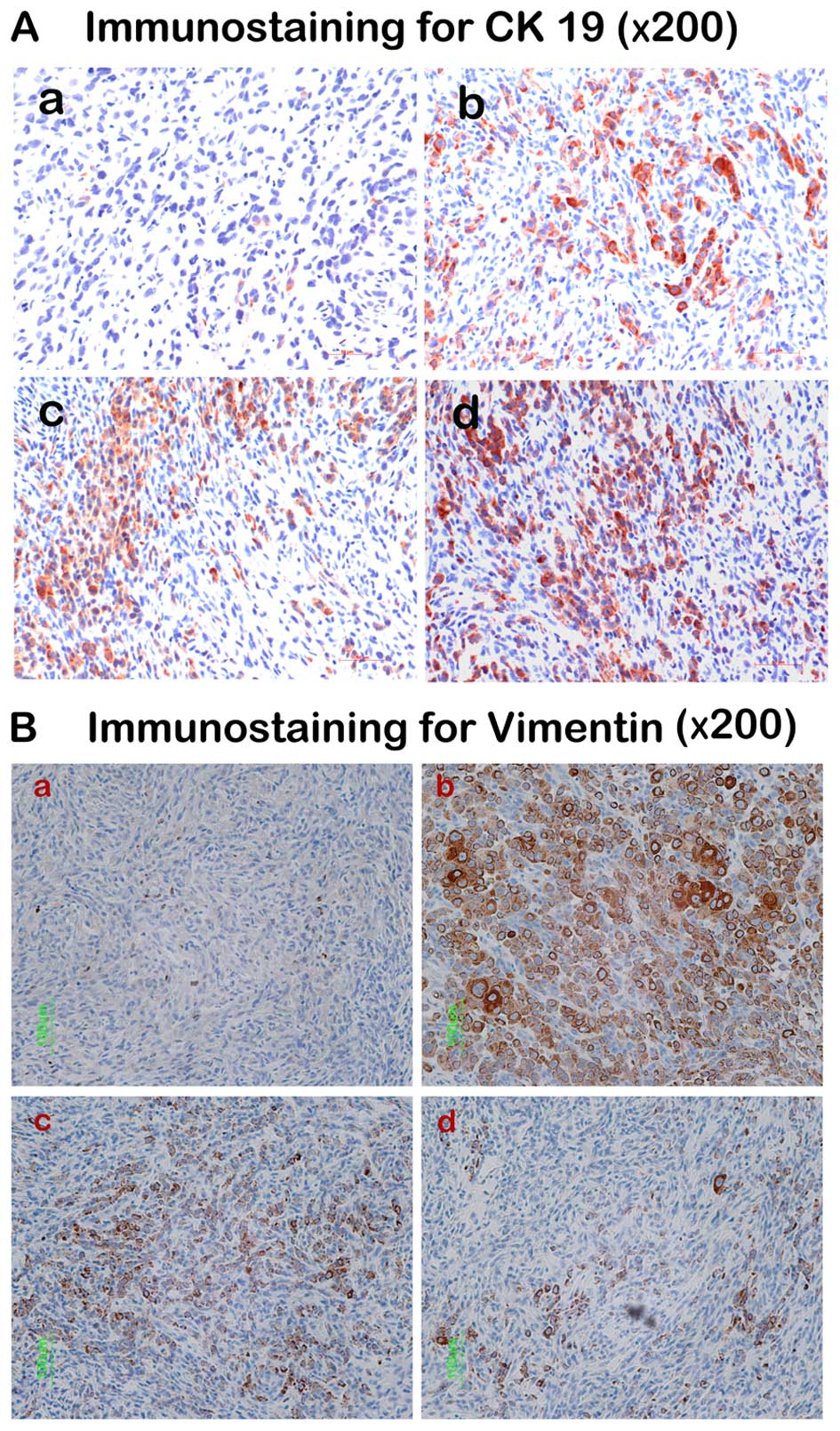
are illustrated in Fig. 5 and
semi-quantitative analysis in the tumor blocks is shown in Table I. It was found that CLT
significantly enhanced CK19 expression, and the increased rate of
CK19 is 126.62% compared with control (CTRL) when CLT was
administered at 75 mg/kg/day. In addition, a 72.03% (P<0.01
compared with CTRL) and 88.73% (P<0.01 compared with CTRL)
reduction of positive area of vimentin staining were observed when
the nude mice were treated with 25 and 75 mg/kg/day CLT,
respectively. These data suggested that CLT attenuated EMT in
metastatic breast cancer, which may contribute to its
anti-metastatic property.
 | Table ISemi-quantitative analysis of CK19
and vimentin in MDA-MB-468 tumor tissues. |
Table I
Semi-quantitative analysis of CK19
and vimentin in MDA-MB-468 tumor tissues.
| Group | CK19 immunostaining
| Vimentin
immunostaining
|
|---|
| Relative area
(%)a | Increased rate
(%) | Relative area
(%) | Decreased rate
(%) |
|---|
| Blank | 1.12±0.30b | – | 0.47±0.15 | – |
| CTRL | 21.30±2.56c | – | 73.37±8.04c | – |
| CLT 25 mg/kg | 26.93±3.74 | 26.43 | 20.52±1.92d | 72.03 |
| CLT 75 mg/kg | 48.27±7.91d | 126.62 | 8.27±1.23d | 88.73 |
Repression of TGF-β signaling contributes
to CLT-mediated EMT inhibition
Known for their ability to stimulate bone formation,
TGF-β signaling forms a vicious cycle in tumor progression and
metastasis (19) and TGF-β1 is
regarded as the key growth factor involved in driving EMT (20). To determine whether CLT-mediated
inhibition of EMT programming was through TGF-β signaling pathway
suppression, the effects of CLT on activity of TGF-β signaling
transducers were examined. The initial assay was to check the
expression of Smad2 and phosph-Smad2/3, the TGF-β selective Smad
complex, by western blotting assay in MDA-MB-468 cells. Our data in
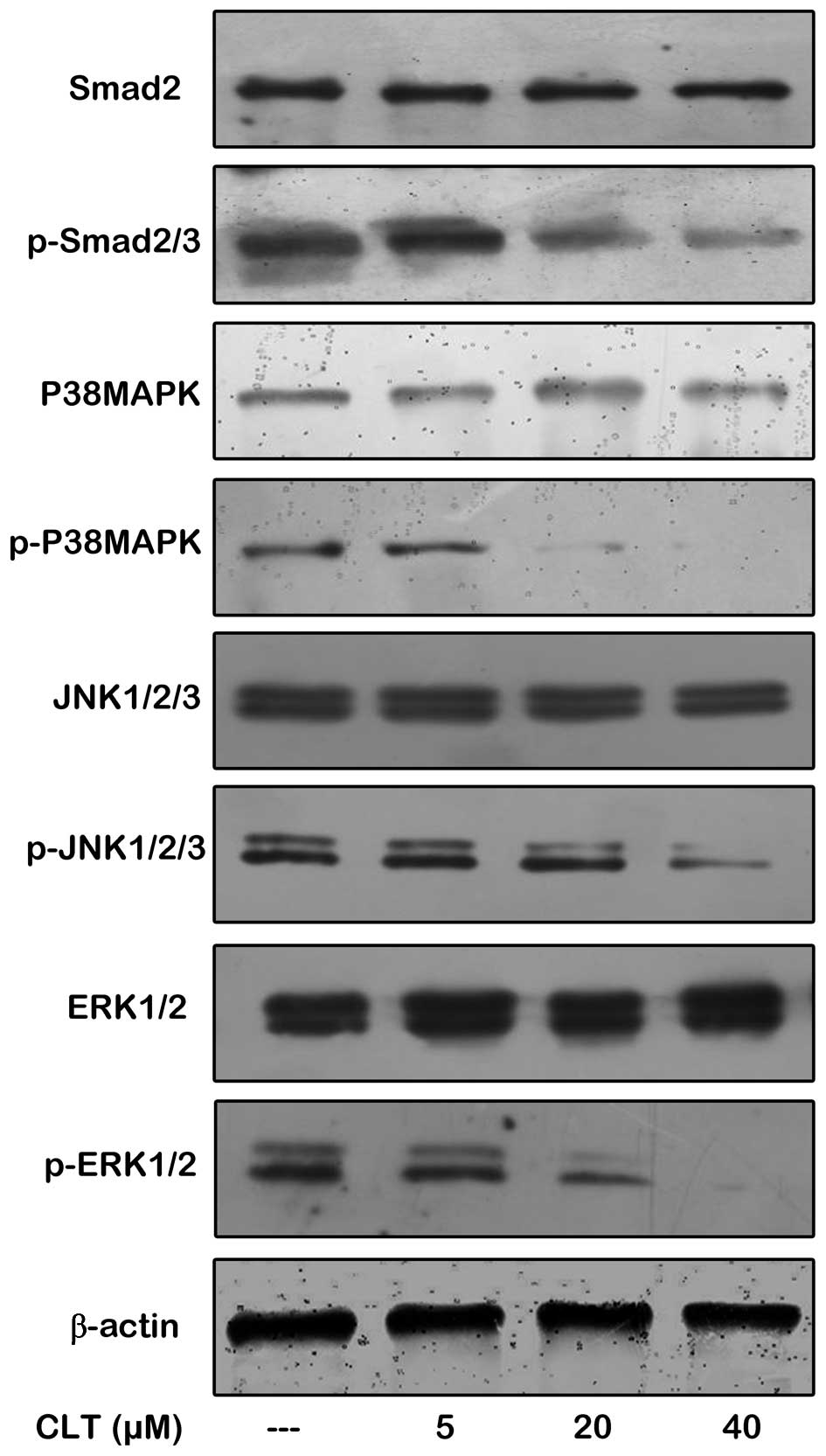
Fig. 6 show that CLT significantly
reduced phosph-Smad 2/3 expression in the presence of 10 ng/ml
TGF-β1, but there was no obvious change on expression of Smad2.
Next, the effects of CLT on Smad-independent TGF-β signaling and
crosstalk with other pathway were also checked. It was found that
phosphorylation of p38MAPK, ERK1/2 and JNK1/2/3 were decreased by
CLT in a dose-dependent manner when MDA-MB-468 cells were
co-incubated with 10 ng/ml TGF-β1. Collectively, these findings
indicated that CLT inhibited TGF-β1-driven EMT programming by
interfering with TGF-β signaling in breast cancer cells.
CLT inhibits TGF-β1 induced Runx2
phosphorylation in both MDA-MB-231 and MDA-MB-468 cells
In a previous study, we demonstrated that the
activation of Runx2, a key transcription factor of skeletal
formation, was abolished by CLT in metastatic breast cancer cells.
To determine whether TGF-β1 induces Runx2 activation, and whether
Runx2 activation induced by TGF-β1 is suppressed by CLT in
metastatic breast cancer cells, we examined the effects of CLT on
Runx2 and phosph-Runx2 expression when 10 ng/ml of TGF-β1 was added
in the culture system. The initial study was to evaluate expression
of Runx2 and phosph-Runx2 in MDA-MB-231 and MDA-MB-468 cells in the
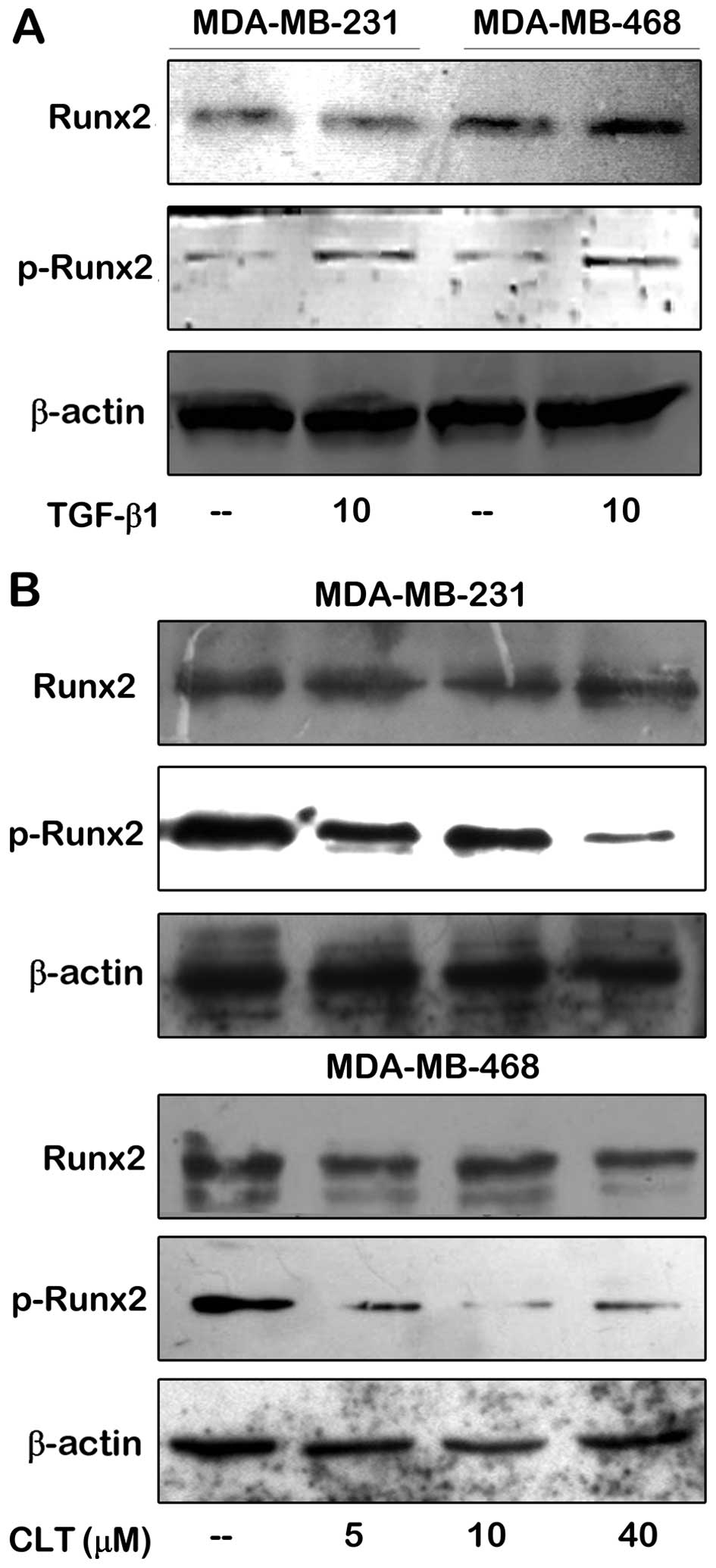
presence and/or the absence of TGF-β1. As shown in Fig. 7A, Runx2 was expressed in both
MDA-MB-231 and MDA-MB-468 cells in the absence of TGF-β1. When
TGF-β1 was added in the culture system, there was no significant
change of expression of Runx2 in either of the breast cancer lines.
However, the situation of p-Runx2 expression was different. When
there was TGF-β1 in the culture system, phosphorylation of Runx2
was stimulated drastically in both cell lines. Next, effects of CLT
on Runx2 and p-Runx2 expression were evaluated in the presence of
10 ng/ml of TGF-β1. As shown in Fig.
7B, expression of p-Runx2 induced by TGF-β1 in MDA-MB-231 and
MDA-MB-468 cells was decreased, respectively, by CLT in a
dose-dependent manner, but there was no effect on Runx2 expression
compared to untreated control. These data indicated that in breast
cancer cells, activation of Runx2 stimulated by TGF-β1 may be
abolished by CLT.
CLT inhibits TGF-β1-induced motility of
metastatic breast cancer cells in vitro
Through EMT, epithelial-derived tumor cells acquire
migratory and invasive capacity. The migration and invasion of
epithelial-derived carcinoma cells are the essential steps to form
metastatic foci. Thus, we next determined the effects of CLT on
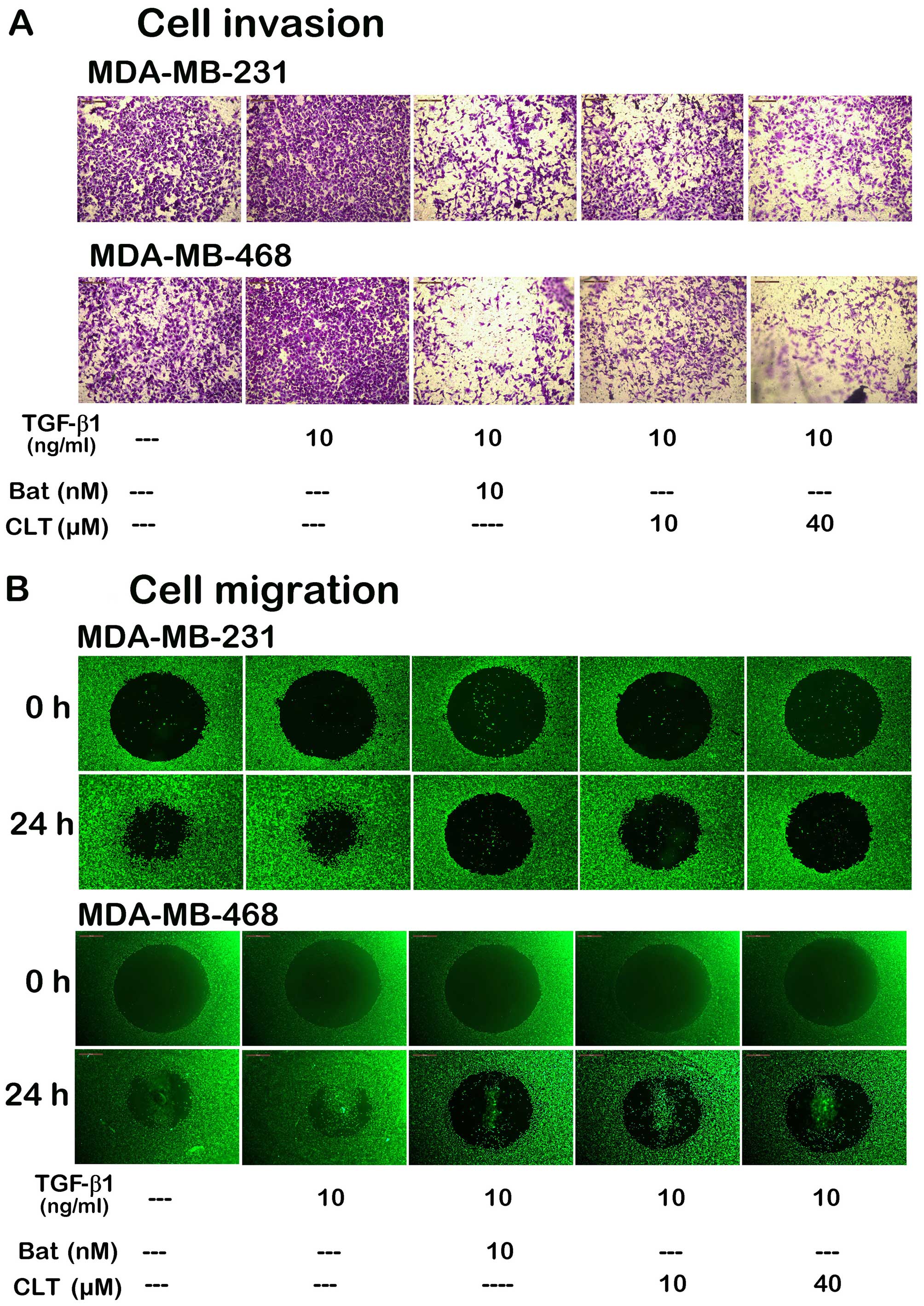
invasion and migration induced by TGF-β1. As shown in Fig. 8A, CLT blocked TGF-β1-induced
invasion of breast cancer cells through re-constitutive basement
membrane significantly, when the cells were incubated with 10 and
40 µM CLT for 14 h in the presence of 10 ng/ml TGF-β1. We
next evaluated the effects of CLT on cell migration with the Oris™
cell migration system. Here, we also found that migration of both
breast cancer cell types was significantly blocked by CLT (Fig. 8B) in a dose-dependent manner.
Discussion
The EMT is a biologic process that allows a
polarized epithelial cell, which normally interacts with basement
membrane via its basal surface, to undergo multiple biochemical
changes that enable it to assume a mesenchymal cell phenotype,
which includes enhanced migratory capacity, invasiveness, elevated
resistance to apoptosis, and greatly increased production of
extracellular matrix (ECM) components (21). For the progression of human
carcinoma, EMT is also seen in a set of carcinoma cells undergoing
phenotypic conversion for invasion and metastasis. Through EMT,
epithelial-derived tumor cells promote genetic alterations and
acquire a mesenchymal cell phenotype, including down-modulating
cell-cell adhesion structures, altering their polarity,
reorganizing their cytoskeleton, and becoming isolated, motile, and
resistant to anoikis (21).
Previously, we revealed that CLT exhibited anti-metastatic effects
in metastatic breast cancer cells, and these effects may be
mediated by inhibition of MMPs via downregulation of the
transcriptional activity of Runx2 (14). Runx2 is a key transcription factor
associated with osteoblast differentiation and breast cancer
metastasis (15). Extensive
evidence shows that there is a close association between abnormal
activation of Runx2 and matrix degradation, colonizing in bone
marrow, angiogenesis and EMT. In addition, it is reported that
Runx2 is a common target of TGF-β signaling (16). By activating Smad proteins and other
signal transduction pathways to stimulate expression of Runx2,
TGF-β1 stimulate osteoblast-specific gene expression (17). Led by these concepts, we sought to
find out whether the EMT-involved mechanisms and related signaling
contribute to the anti-metastatic effects of CLT in breast
cancer.
EMT describes a series of rapid changes in cellular
phenotype, including epithelial cells loosening cell-cell adhesion
structures, modulating their polarity and rearranging their
cytoskeleton (22). A set of
molecules is involved in transition, some of which are acquired,
and some are attenuated. Recently, most of them have been used as
biomarkers of EMT to monitor EMT. These molecules include
expression of specific cell-surface proteins, reorganization and
expression of cytoskeletal proteins, production of ECM-degrading
enzymes, and changes in the expression of specific microRNAs
(23). cadherins (named for
'calcium-dependent adhesion') are a class of type-1 transmembrane
proteins, which constitute a large family of cell surface proteins
including cadherins, protocadherins, desmogleins, and desmocollins.
They play important roles in cell adhesion, forming adherens
junctions to bind cells within tissues (24). E-cadherin, one of cadherins
expressed on the epithelial cell surface, is decreased during EMT
in cancer progression, but N-cadherin, a mesenchymal cadherin, is
increased during EMT. This kind of changes in the level of
expression of E-cadherins and N-cadherin, so-called cadherin
switches, has been increasingly used to monitor progress of EMT
during cancer progression (25).
Vimentin, which is a type III intermediate filament protein
expressed in mesenchymal cells, is acquired during EMT. This
cytoskeleton protein is commonly used as a biomarker to identify
cells undergoing EMT in cancers (26). In the present study, these typical
biomarkers were studied to check the effects of CLT on EMT. We
demonstrated using cell immunofluorescence assay that CLT
significantly inhibited TGF-β1-driven cadherin switch in both
MDA-MB-231 and MDA-MB-468 cells. By western blot assay, we verified
that CLT enhanced expression of epithelial markers in both breast
cancer cells, such as epithelial cell surface marker E-cadherin and
cytoskeleton marker CK19, and that CLT also attenuated expression
of mesenchymal markers, such as mesenchymal cell surface marker
N-cadherin and cytoskeleton marker vimentin. Moreover, in
vivo study demonstrated that CLT increased CK19 expression and
decreased vimentin expression in MDA-MB-468 tumors significantly,
which means CLT blocked EMT programming of MDA-MB-468 tumors. Taken
together, our data indicated that CLT arrests programming of EMT,
which may contribute to its anti-metastatic properties in breast
cancer.
In order to initiate an EMT and enable it to reach
completion, a number of distinct transcription factors are engaged
in this process. Snail transcription factors are one prominent
example of a common trans-acting regulatory factor binding
to the promoter of various EMT-associated genes. The Snail family
of zinc-finger transcription factors consist of Snail1 (Snail),
Snail2 (Slug) and Snail3 (Smuc), and the members are the most
widely recognized as key regulators of various aspects of the EMT
phenotype, such as increased expression of mesenchymal cells
(fibronectin and vitronectin), decreased expression of various
epithelial markers (E-cadherin and cytokeratins) (27). Twist proteins are highly conserved
basic helix-loop-helix (bHLH) transcription factors that have
important regulatory functions during lineage determination and
cell differentiation. Numerous studies have demonstrated that this
master regulator of embryonic morphogenesis plays a crucial role in
metastasis (28). According to Yang
et al, Twist can act independently of Snail to repress
E-cadherin and to upregulate fibronectin and N-cadherin in the
development of metastatic cancer cells by EMT (29). Thus, we sought to determine whether
CLT inhibits expression of EMT-associated transcription factors.
Our data showed that CLT reduced TGF-β1-driven expression of Snail,
Slug and Twist1 in both breast cancer cell types. Collectively,
these data indicated that CLT-induced decrease of the
EMT-associated transcription factors may result in change of
expression of cell surface and cytoskeleton proteins driven by
TGF-β1, leading to EMT blockade in breast cancer.
Several signaling pathways are involved in
transcription program switching of EMT, such as TGF-β, BMP,
Wnt/β-catenin, Notch, Hedgehog signaling, and TGF-β signaling
pathway is the most well-characterized pathway that is known to
induce EMT (6,30-32).
By activating Smad-dependent signaling, TGF-β promotes the
transcription of key genes associated with EMT. Complexes of
R-Smads are able to bind directly to the promoter of snai1
to induce its transcription, and can form complexes with Snail to
suppress the expression of genes encoding E-cadherin and occludin
(33,34). Other factors directly influenced by
the binding of R-Smads include the ZEB transcription factors and
the high mobility group factor HGMA2, which also regulates the
expression of snai1, snai2, and twist genes
(33,35). Besides acting through Smad pathway,
TGF-β also activates Smad-independent pathway during EMT. These
pathways have non-transcriptional roles in EMT, including
dissolution of epithelial junctions, cytoskeletal reorganization
and motility, and translational control. They also target Smads and
thus help define their functions, while controling the expression
and activation of transcription factors, with which Smad complexes
cooperate in the control of gene expression (6). The JNK and p38MAPK cascades can be
activated by TGF-β-activated kinase 1 (TAK1) which is involved in
the induction of snai1 transcription in mammary epithelial
cells in a Smad-independent manner during EMT (36). Early evidence indicated that MAPK
kinase (MEK)-ERK signaling is activated by TGF-β, which is
consistent with the observation of induction of EMT transcription
factors, such as Twist1 (37). As
TGF-β and the TGF-β-related proteins have been shown as major
inducers of EMT, we hypothesized that CLT-induced EMT inhibition
was through suppression of TGF-β signal pathway. This hypothesis
was supported by our data from western blotting. Our data showed
that CLT significantly inhibited TGF-β1 induced activation of
Smad-dependent and -independent signaling in MDA-MB-468 cells.
These findings indicated that CLT inhibited TGF-β1-driven EMT
programming by interfering with TGF-β signaling in breast cancer
cells.
Runx2 (also named PEBP2αA/AML3/Cbfa1) is a
transcription factor of the runx gene family, encoding proteins
homologous to Drosophila Runt (38). Originally, this transcription factor
was thought to contribute to skeletal formation (39), but extensive research indicates the
role of Runx2 in metastatic growth of breast cancer cells. In
breast cancer cells, several genes correlated with the occurrence
of skeletal metastasis regulated by Runx2, such as bsp,
opn, mmp-9, mmp-13, and vegf (40). Therefore, Runx2 is reported to be a
'master' transcription factor of metastatic growth of breast cancer
cells, including homing to bone, induction of high bone turnover,
angiogenesis and tissue invasion (41). Studies from the signal pathway in
osteoblast differentiation and cancer metastasis reveal that this
transcription factor is a common target of the TGF-β/BMP signal
pathway. By activating Smad proteins and other signal transduction
pathways to stimulate expression of Runx2, TGF-β stimulated
osteoblast-specific gene expression (16,17).
More recently, studies have demonstrated that Runx2 is a crucial
regulator of EMT. Chimge et al demonstrated that Slug is a
Runx2 target required for mediating its pro-metastatic property
(42). By transient interference
RNA assay, Niu et al demonstrated that EMT-associated
transcription factors, such as Snail, Slug and Twist1, are target
of Runx2 in thyroid carcinoma cells (43). Thus, Runx2 is a crucial transducer
of EMT and metastasis, which is regulated by TGF-β/BMP signal and
regulates the EMT phenotype. Previously, we showed that
downregulation of Runx2 transcription activity may be involved in
inhibition of MMP expression induced by CLT. Thus, we investigated
whether Runx2 is activated by TGF-β and CLT blocks activation of
Runx2 in breast cancer cells. Our data from western blot assay
showed that TGF-β1 significantly stimulated Runx2 phosphorylation
in both MDA-MB-231 and MDA-MB-468 cells, and CLT inhibited
TGF-β1-induced p-Runx2 expression, indicating that the
transcriptional ability of Runx2 is impaired by CLT in breast
cancer cells.
Collectively, our present study demonstrated that
CLT inhibited programming of EMT in vitro and in
vivo, resulting in inhibition of motility of metastatic breast
cancer cells. The inhibitory effect of CLT was due to its ability
to inhibit TGF-β signaling and Runx2 phosphorylation.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81560639, 81160530,
and 81260656), the Key Research Project from the Ministry of
Education of China (grant no. 211091), and the Natural Science
Foundation of Jiangxi Province (grant no. 2010GQY0147).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu S, Goldstein RH, Scepansky EM and
Rosenblatt M: Inhibition of rho-associated kinase signaling
prevents breast cancer metastasis to human bone. Cancer Res.
69:8742–8751. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Son H and Moon A: Epithelial-mesenchymal
transition and cell invasion. Toxicol Res. 26:245–252. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Q, He H, Li P, Zhu J and Xiong M:
Identification and quantification of atractylenolide I and
atractylenolide III in Rhizoma Atractylodes macrocephala by liquid
chromatographyion trap mass spectrometry. Biomed Chromatogr.
27:699–707. 2013. View
Article : Google Scholar
|
|
8
|
Kang TH, Bang JY, Kim MH, Kang IC, Kim HM
and Jeong HJ: Atractylenolide III, a sesquiterpenoid, induces
apoptosis in human lung carcinoma A549 cells via
mitochondria-mediated death pathway. Food Chem Toxicol. 49:514–519.
2011. View Article : Google Scholar
|
|
9
|
Zhang NN, Park DK and Park HJ: The
inhibitory activity of atractylenolide III, a sesquiterpenoid, on
IgE-mediated mast cell activation and passive cutaneous anaphylaxis
(PCA). J Ethnopharmacol. 145:278–285. 2013. View Article : Google Scholar
|
|
10
|
Li CQ, He LC and Jin JQ: Atractylenolide I
and atractylenolide III inhibit lipopolysaccharide-induced
TNF-alpha and NO production in macrophages. Phytother Res.
21:347–353. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu C, Zhao H, Ji ZH and Yu XY:
Neuroprotection of atractylenolide III from Atractylodis
macrocephalae against glutamate-induced neuronal apoptosis via
inhibiting caspase signaling pathway. Neurochem Res. 39:1753–1758.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang KT, Chen LG, Wu CH, Chang CC and Wang
CC: Gastroprotective activity of atractylenolide III from
Atractylodes ovata on ethanol-induced gastric ulcer in vitro and in
vivo. J Pharm Pharmacol. 62:381–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kang TH, Han NR, Kim HM and Jeong HJ:
Blockade of IL-6 secretion pathway by the sesquiterpenoid
atractylenolide III. J Nat Prod. 74:223–227. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, Chen B, Zou R, Tu X, Tan S, Lu H,
Liu Z and Fu J: Codonolactone, a sesquiterpene lactone isolated
from Chloranthus henryi Hemsl, inhibits breast cancer cell
invasion, migration and metastasis by downregulating the
transcriptional activity of Runx2. Int J Oncol. 45:1891–1900.
2014.PubMed/NCBI
|
|
15
|
Ferrari N, McDonald L, Morris JS, Cameron
ER and Blyth K: RUNX2 in mammary gland development and breast
cancer. J Cell Physiol. 228:1137–1142. 2013. View Article : Google Scholar
|
|
16
|
Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C,
Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM, et al: Runx2 is a
common target of transforming growth factor beta1 and bone
morphogenetic protein 2, and cooperation between Runx2 and Smad5
induces osteoblast-specific gene expression in the pluripotent
mesenchymal precursor cell line C2C12. Mol Cell Biol. 20:8783–8792.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Phimphilai M, Zhao Z, Boules H, Roca H and
Franceschi RT: BMP signaling is required for RUNX2-dependent
induction of the osteoblast phenotype. J Bone Miner Res.
21:637–646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonnomet A, Syne L, Brysse A, Feyereisen
E, Thompson EW, Noël A, Foidart JM, Birembaut P, Polette M and
Gilles C: A dynamic in vivo model of epithelial-to-mesenchymal
transitions in circulating tumor cells and metastases of breast
cancer. Oncogene. 31:3741–3753. 2012. View Article : Google Scholar
|
|
19
|
Zhang Q, Yu N and Lee C: Vicious cycle of
TGF-β signaling in tumor progression and metastasis. Am J Clin Exp
Urol. 2:149–155. 2014.
|
|
20
|
Borthwick LA, Gardner A, De Soyza A, Mann
DA and Fisher AJ: Transforming growth factor-β1 (TGF-β1) driven
epithelial to mesenchymal transition (EMT) is accentuated by tumour
necrosis factor α (TNFα) via crosstalk between the SMAD and NF-κB
pathways. Cancer Microenviron. 5:45–57. 2012. View Article : Google Scholar
|
|
21
|
Savagner P: The epithelial-mesenchymal
transition (EMT) phenomenon. Ann Oncol. 21(Suppl 7): vii89–vii92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Angst BD, Marcozzi C and Magee AI: The
cadherin superfamily: Diversity in form and function. J Cell Sci.
114:629–641. 2001.PubMed/NCBI
|
|
25
|
Gheldof A and Berx G: Cadherins and
epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci.
116:317–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eriksson JE, Dechat T, Grin B, Helfand B,
Mendez M, Pallari HM and Goldman RD: Introducing intermediate
filaments: From discovery to disease. J Clin Invest. 119:1763–1771.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Shi J, Chai K, Ying X and Zhou BP:
The role of Snail in EMT and tumorigenesis. Curr Cancer Drug
Targets. 13:963–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khan MA, Chen HC, Zhang D and Fu J: Twist:
A molecular target in cancer therapeutics. Tumour Biol.
34:2497–2506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang Z, Zhang X, Gang H, Li X, Li Z, Wang
T, Han J, Luo T, Wen F and Wu X: Up-regulation of gastric cancer
cell invasion by Twist is accompanied by N-cadherin and fibronectin
expression. Biochem Biophys Res Commun. 358:925–930. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Derynck R, Muthusamy BP and Saeteurn KY:
Signaling pathway cooperation in TGF-β-induced
epithelial-mesenchymal transition. Curr Opin Cell Biol. 31:56–66.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thomson S, Petti F, Sujka-Kwok I, Mercado
P, Bean J, Monaghan M, Seymour SL, Argast GM, Epstein DM and Haley
JD: A systems view of epithelial-mesenchymal transition signaling
states. Clin Exp Metastasis. 28:137–155. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Peinado H, Quintanilla M and Cano A:
Transforming growth factor beta-1 induces snail transcription
factor in epithelial cell lines: Mechanisms for epithelial
mesenchymal transitions. J Biol Chem. 278:21113–21123. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vincent T, Neve EP, Johnson JR, Kukalev A,
Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL,
et al: A SNAIL1-SMAD3/4 transcriptional repressor complex promotes
TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol.
11:943–950. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thuault S, Tan EJ, Peinado H, Cano A,
Heldin CH and Moustakas A: HMGA2 and Smads co-regulate SNAIL1
expression during induction of epithelial-to-mesenchymal
transition. J Biol Chem. 283:33437–33446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamashita M, Fatyol K, Jin C, Wang X, Liu
Z and Zhang YE: TRAF6 mediates Smad-independent activation of JNK
and p38 by TGF-beta. Mol Cell. 31:918–924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stein GS, Lian JB, van Wijnen AJ, Stein
JL, Montecino M, Javed A, Zaidi SK, Young DW, Choi JY and Pockwinse
SM: Runx2 control of organization, assembly and activity of the
regulatory machinery for skeletal gene expression. Oncogene.
23:4315–4329. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Komori T: Runx2, a multifunctional
transcription factor in skeletal development. J Cell Biochem.
87:1–8. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Inman CK and Shore P: The osteoblast
transcription factor Runx2 is expressed in mammary epithelial cells
and mediates osteopontin expression. J Biol Chem. 278:48684–48689.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gupta GP and Massagué J: Cancer
metastasis: Building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chimge NO, Baniwal SK, Little GH, Chen YB,
Kahn M, Tripathy D, Borok Z and Frenkel B: Regulation of breast
cancer metastasis by Runx2 and estrogen signaling: The role of
SNAI2. Breast Cancer Res. 13:R1272011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Niu DF, Kondo T, Nakazawa T, Oishi N,
Kawasaki T, Mochizuki K, Yamane T and Katoh R: Transcription factor
Runx2 is a regulator of epithelial-mesenchymal transition and
invasion in thyroid carcinomas. Lab Invest. 92:1181–1190. 2012.
View Article : Google Scholar : PubMed/NCBI
|