Introduction
Glioblastoma (GBM) is the most frequent primary
brain tumor in adults and presents a very aggressive course with
few therapeutic options. Recent studies have shown that GBM is
composed of cell populations that are heterogeneous in terms of
morphology and differentiation status (1). It has been proposed that a small
population of tumor cells, named glioblastoma-initiating cells
(GICs), plays a crucial role in the origin, growth, recurrence and
drug resistance of GBM (2). Studies
suggest that the limited therapeutic efficacy of conventional
approaches to GBM is due to the resistant nature of GICs to
chemotherapy and radiotherapy, allowing the survival of GICs to
regenerate the tumor (3). GICs are
thus considered the major barrier to GBM therapy, and elimination
of GICs is considered to be a key strategy for achieving the
long-term survival of GBM patients (1,4). At
present, temozolomide (TMZ) is the standard chemotherapy drug for
GBM, yet significant GICs resistance toward TMZ has been widely
reported (5,6). Recent findings have even noted the
significant expansion of a newly converted GIC population from
differentiated GBM cells in vitro and in vivo after
long-term exposure to TMZ (7).
Therefore, it is vital to develop strategies to enhance the
efficacy of TMZ in treating GICs (8).
Resveratrol (RES) is a natural polyphenolic compound
that is widely present in plants and is enriched in red wine,
peanuts, and grapes. It has exhibited a broad range of
chemo-preventive and therapeutic properties in a variety of animal
models (9,10). As a potential candidate for treating
cancer, RES presents low toxicity and few adverse effects upon
administration at relatively high doses (11). It has been shown that RES alters
multiple signaling pathways, such as the JAK/STAT, NF-κB/p50/p65
and p53 pathways, to induce cell cycle arrest, apoptosis and
autophagy in various types of tumor cells (12–14).
Specifically, RES potentiates the toxicity of TMZ in GBM cell lines
such as SHG44 and T98 (15,16). However, to the best of our
knowledge, little is known regarding the effect of TMZ combined
with RES on treating GICs which have distinct properties when
compared to GBM cell lines.
The aim of the present study was to investigate
whether RES could enhance the antitumor effect of TMZ on GICs both
in vitro and in vivo, and the involved mechanisms in
response to the enhanced effects.
Materials and methods
GIC culture and cell
immunofluorescence
GICs were derived from neurosurgical samples of two
GBM patients at the Department of Neurosurgery, Beijing Tiantan
Hospital, and informed consent was obtained from the patients. The
use of human tissue specimens had been approved by the ethics board
in our hospital. The tumor tissues were dissociated into single
cells according to a previous study (6). To induce differentiation, the GICs
were cultured in DMEM/F12 medium containing 10% fetal bovine serum
(FBS) for two weeks. GICs and differentiated cells were
immunofluorescence-stained with CD133, nestin, GFAP, NF, and CNP.
The cell nuclei were stained with DAPI (Sigma-Aldrich, USA).
Limiting dilution assay
A limiting dilution assay was used to indicate the
number of cells from a primary neurosphere (NS) that was needed to
form a secondary NS. The specific method was described in a
previous study (17).
Determination of GICs in NOD/SCID
mice
The GICs (2×104/mouse, 6 mice) were
injected stereotactically into the right corpus striatum (2.5 mm
anterior and 2.5 mm lateral to the bregma and 3.0-mm deep) of
6-week-old female NOD/SCID mice (VitalStar, China). Once the mice
were incapable, the brains were harvested. The brains were stained
with hematoxylin and eosin (H&E), nestin (GTX39578), and glial
acidic fibrillary protein (GFAP, GTX84438) (both from GeneTex). All
animal procedures were performed in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals.
Cell viability, apoptosis and sphere
counting assay
MTT (Sigma-Aldrich, USA) assay was used to evaluate
cell viability. The calculation of the combination index value (CI)
was based on a previous study (18). Synergism, addition, and antagonism
were defined as CI<1, CI=1, and CI>1, respectively. To
analyze apoptosis, the Annexin V/FITC and PI apoptosis detection
kit was used according to the instructions provided by the
manufacturer (Becton-Dickinson, USA). Quantification of apoptotic
cells was performed using a FACScan flow cytometer. For the sphere
counting assay which evaluates the self-renewal ability of GICs,
colonies (>50 µm in diameter) were counted after GICs
were exposed to drugs for 2 weeks, and the number of
colonies/number of cells in each well was calculated according to a
previous study (19) with minor
revisions. Each assay was repeated in three independent
experiments.
Measurement of caspase-3 activity
Caspase-3 activity was assayed
spectrophotometrically via the detection of pNA cleavage from
caspase-3-specific substrates (Ac-LEVD-pNA) according to a
commercially available kit (Beyotime, China).
DNA double-strand break (DSB) assay
DSBs were confirmed according to the OxiSelect™ DSB
staining kit protocol. GICs exposed to etoposide at 100 µM
were regarded as the positive control. Cell images were acquired
and analyzed under a Zeiss fluorescence microscope.
Western blot analysis
The cells were lysed in buffer (Cell Signaling
Technology) containing 100 mM NaF, and 1:500 protease inhibitor
mixture (Roche, USA). Equal amounts of proteins were separated by
7–15% SDS-PAGE for electrophoresis. The protein was hybridized by
overnight incubation with the primary antibodies. ChemiDoc XRS+
image analyzer (Bio-Rad, USA) and ImageJ software (http://rsb.info.nih.gov/ij) were used to quantify the
protein band density.
In vivo xenograft study and
immunohistochemical study
GICs (2×105) were subcutaneously injected
into the left hind flank of the 6-week-old female NOD/SCID mice.
When the tumors grew to the 10th day, TMZ administration was began
by oral gavage at doses of 68 mg/kg, which corresponded to the
murine equivalent of the standard clinical dose of 200
mg/m2 and schedule as used by Yuan et al
(15). TMZ was used for 5 days and
halted for 10 days, and then repeated again. Meanwhile, 12.5 mg/kg
RES was injected intraperitoneally once a day. Animal body weight,
hair color and appearance were assessed every 5 days. The tumor
sizes were measured every 5 days with a caliper and were calculated
as 1/2 × length × width2 in mm3. At day 40 of
the inoculation, the mice were sacrificed and tumor tissues were
excised for GFAP and nestin immunohistochemical study.
Nestin-positive cells and GFAP-positive cells were counted in 5
staining fields chosen randomly.
Statistical analysis
The data are presented as the means ± SD, and the
Student's t-test was used for comparing paired sample sets. A
P-value <0.05 was considered to indicate a statistically
significant result.
Results
Determination of GICs in vitro and in
vivo
Using the methods described above, we successfully
isolated two patient-derived GIC cell lines and named them GIC400
and GIC411, respectively. The representative results of GIC400
determination are shown in Fig. 1
(similar results were also determined for GIC411; data not shown).
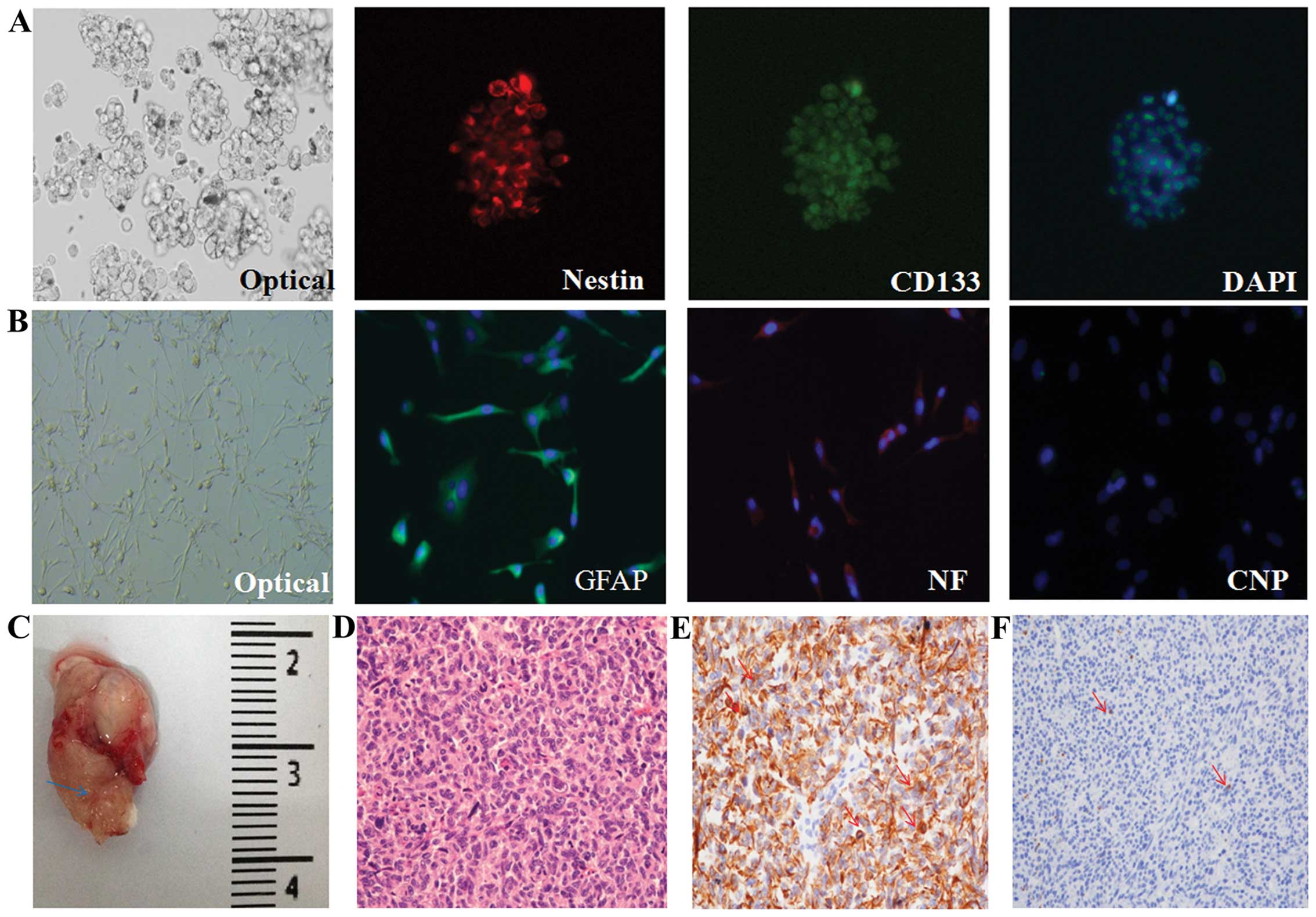
As shown in Fig. 1A, these cells
formed characteristic renewable neurospheres and could proliferate
indefinitely, and all the GICs exhibited a high expression of
nestin which is a neural progenitor cell marker while a large
portion of the cells were stained positive for CD133 by
immunofluorescence. Furthermore, upon serum exposure, GICs acquired
the glial- and neurite-like cell features with protrusions and
adherence to the flask under optical microscope, and the GICs
showed a GFAP (astrocyte)-, NF (neuron)-, and CNP
(oligodendrocyte)-directed differentiation morphology (Fig. 1B). The results were consistent with
previous literature (17), showing
that GICs could be efficiently induced into astrocytic, neural and
partly induced into oligo-dendrocytic lineages after incubation
with serum-containing DMEM/F12 media for 2 weeks. We then performed
limiting dilution assays to confirm the enrichment of GICs in the
primary cultures. GIC400 and GIC411 cells required a small number
of cells to generate a secondary neurosphere (7.6 cells for GIC400,
and 10.6 for GIC411) (data not shown). Furthermore, the potential
for tumorigenesis was determined through intra-cranial tumor
formation in the NOD/SCID mice for 2 months after inoculation with
only 2×104 GICs (Fig.
1C), while GBM tumorigenesis generally requires 106
non-GICs such as U87 cells. The tumors histologically resembled the
GBM in patients through H&E staining with the aid of a
qualified expert pathologist in the brain tumor field, suggesting
the successful establishment of the xenograft model of GICs
according to the literature (20)
(Fig. 1D). The xenograft samples
also presented high expression levels of nestin (Fig. 1E) and acicular expression of GFAP
(Fig. 1F), implying that the
xenograft samples contained a high abundance of GICs.
RES has a synergistic effect with TMZ on
GIC viability
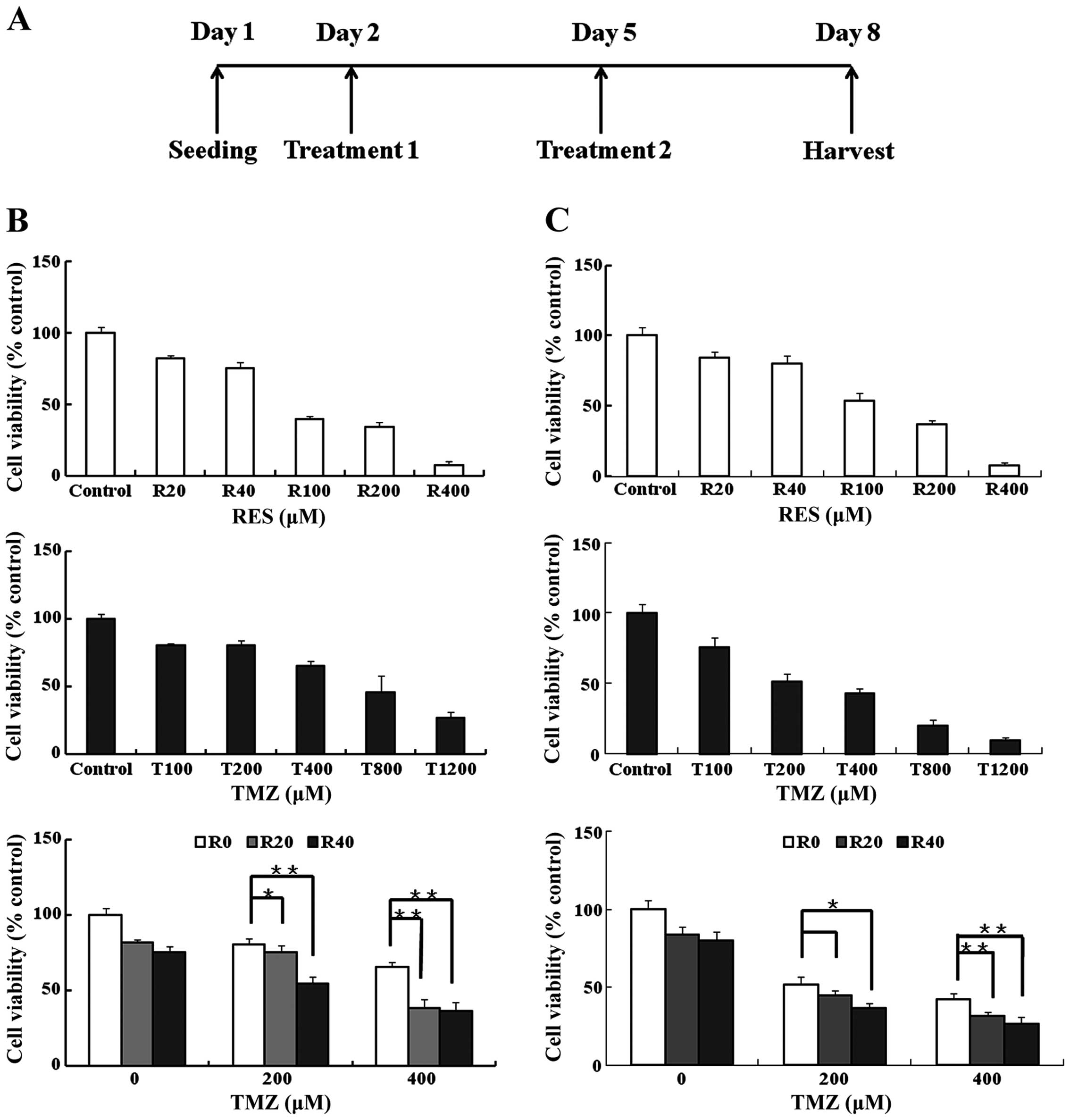
The cell viability of GIC400 and GIC411 cells to
TMZ, RES and the drug combination was evaluated after 6 days of
exposure (Fig. 2A). The GIC400 cell
line exhibited high resistance to TMZ while the half maximal
inhibitory concentration (IC50) of TMZ was 578 µM
and the IC50 of RES was 83 µM. Compared with the
GIC400 cell line, the GIC411 cell line showed moderate resistant to
TMZ; the IC50 of TMZ was 331 µM. To evaluate
whether RES could enhance the TMZ cytotoxicity on cell viability,
we chose RES concentrations of 20 and 40 µM (denoted R20 and
R40 in short) and TMZ concentrations of 200 and 400 µM
(denoted T200 and T400 in short) for the combination treatment due
to their moderate toxicity toward GICs when used alone (Fig. 2B and C). Synergistic effects on
GIC400 and GIC411 cell viability were observed with the combined
usage of R20 and T200 (CI=0.88 for GIC400), R20 and T400 (CI=0.72
for GIC400; CI=0.94 for GIC411), R40 and T200 (CI=0.9 for GIC400;
CI=0.93 for GIC411), and R40 and T400 (CI=0.74 for GIC400; CI=0.9
for GIC411). In addition, an additive effect was observed in the
combination of R20 and T200 for GIC411 cells (CI=1.03). Thus, we
chose GIC400 cells for the following studies due to their highly
resistant property to TMZ and a more obvious synergism of drug
combination observed.
RES enhances TMZ-induced apoptosis of
GICs via activation of the DSBs/pATM/pATR/p53 pathway
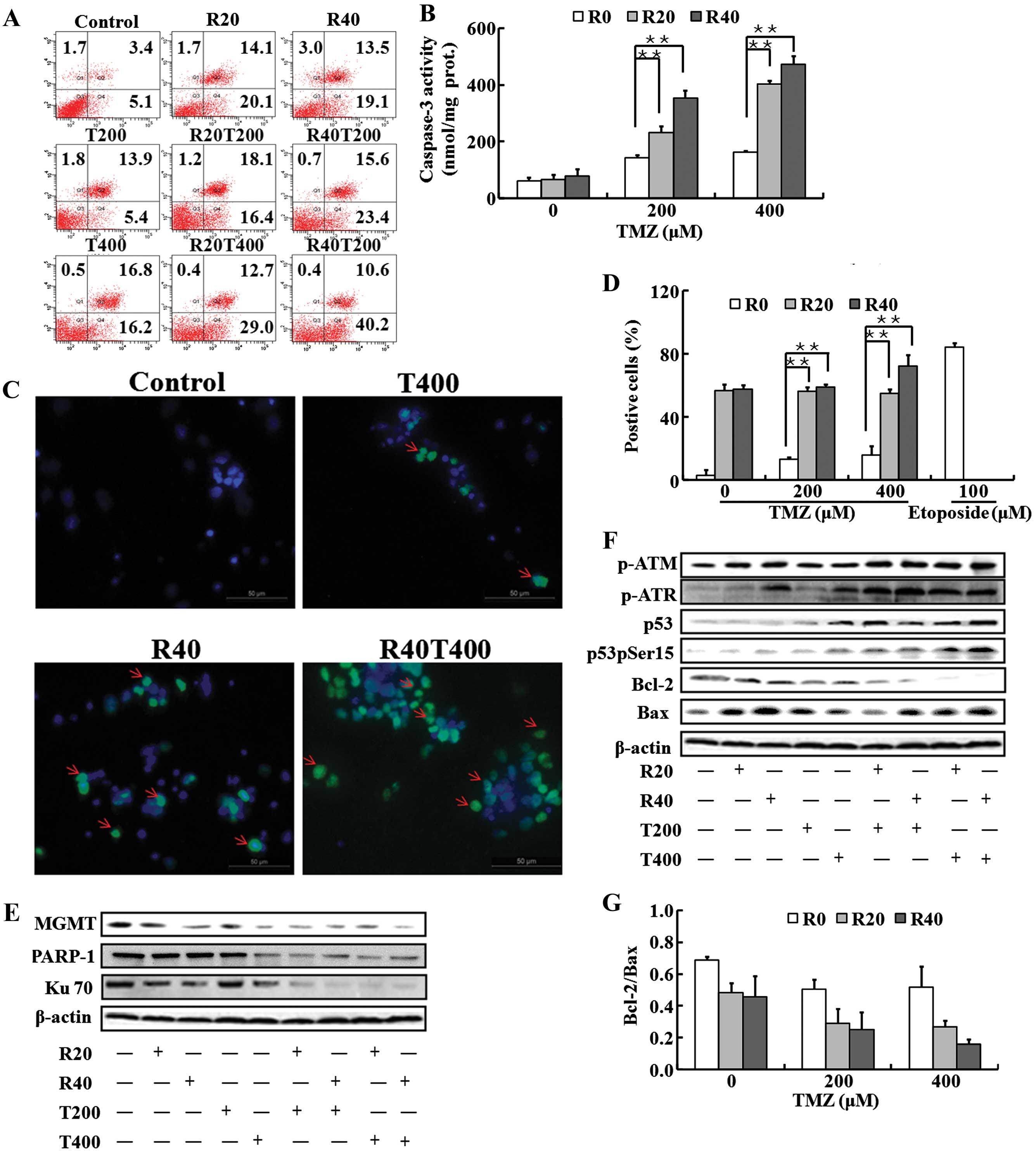
The treatment is shown in Fig. 2A, and the representative results of
apoptosis are demonstrated in Fig.
3A. The percentage of total apoptosis of GICs was ~20% when
exposed to T200 and increased to ~30% when exposed to T400.
Combined with RES, TMZ induced the apoptosis significantly (the
percentage of apoptosis was 34.5% when T200 was combined with R20,
39% when T200 was combined with R40, 41.7% when T400 was combined
with R20, 50.8% when T400 combined with R40).
The enhancement of apoptosis was evidenced by the
elevation of caspase-3 activity. TMZ-treated GICs presented ~2- to
3-fold higher caspase-3 activity than the control (P<0.05). A 4-
to 9-fold increase in caspase-3 activity compared with the control
was observed in the groups treated with the TMZ and RES
combinations (Fig. 3B).
The presence of DSBs is determined by the elevation
of phosphorylated histone H2AX (γH2AX), which are DNA-damage
downstream effectors (21). A
marked increase in γH2AX expression was observed in the GICs
exposed to R20 and R40, whereas a moderate increase was detected in
the GICs exposed to T200 and T400. Furthermore, a significant
increase in γH2AX expression was observed in the GICs exposed to
the combination treatment with TMZ and RES (especially for R40 and
T400) compared with that obtained with each agent alone (Fig. 3C and D).
Ku70 and MGMT which can repair DSBs and protect
cells from death (22,23), were moderately reduced in the GICs
exposed to R20 and R40 in a dose-dependent manner, while the
expression remained unchanged when GICs were exposed to TMZ alone
except T400 (T400 inhibited MGMT expression). In addition, PARP1,
another DSB repair protein, was decreased following treatment with
T400 rather than with R20, R40 and T200. However, Ku70, MGMT and
PARP1 were markedly reduced in the GICs exposed to the drug
combinations compared with the levels obtained with each agent
alone (Fig. 3E).
Important sensors of DSBs are ATM (ataxia
telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3
related) proteins, which signal downstream to p53 (24). The level of pATM was moderately
upregulated while pATR, p53 and pp53 were significantly increased
when TMZ was combined with RES. The representative graphics are
shown in Fig. 3F. Moreover, the
ratio of Bcl-2 to Bax, which is regulated by p53 expression, was
significantly reduced in the GICs exposed to the combination
treatment with TMZ and RES than the control (P<0.01) (Fig. 3F and G).
Inhibition of self-renewal capacity and
induction of cell differentiation via STAT3 inactivation in GICs
exposed to the combinations with TMZ and RES
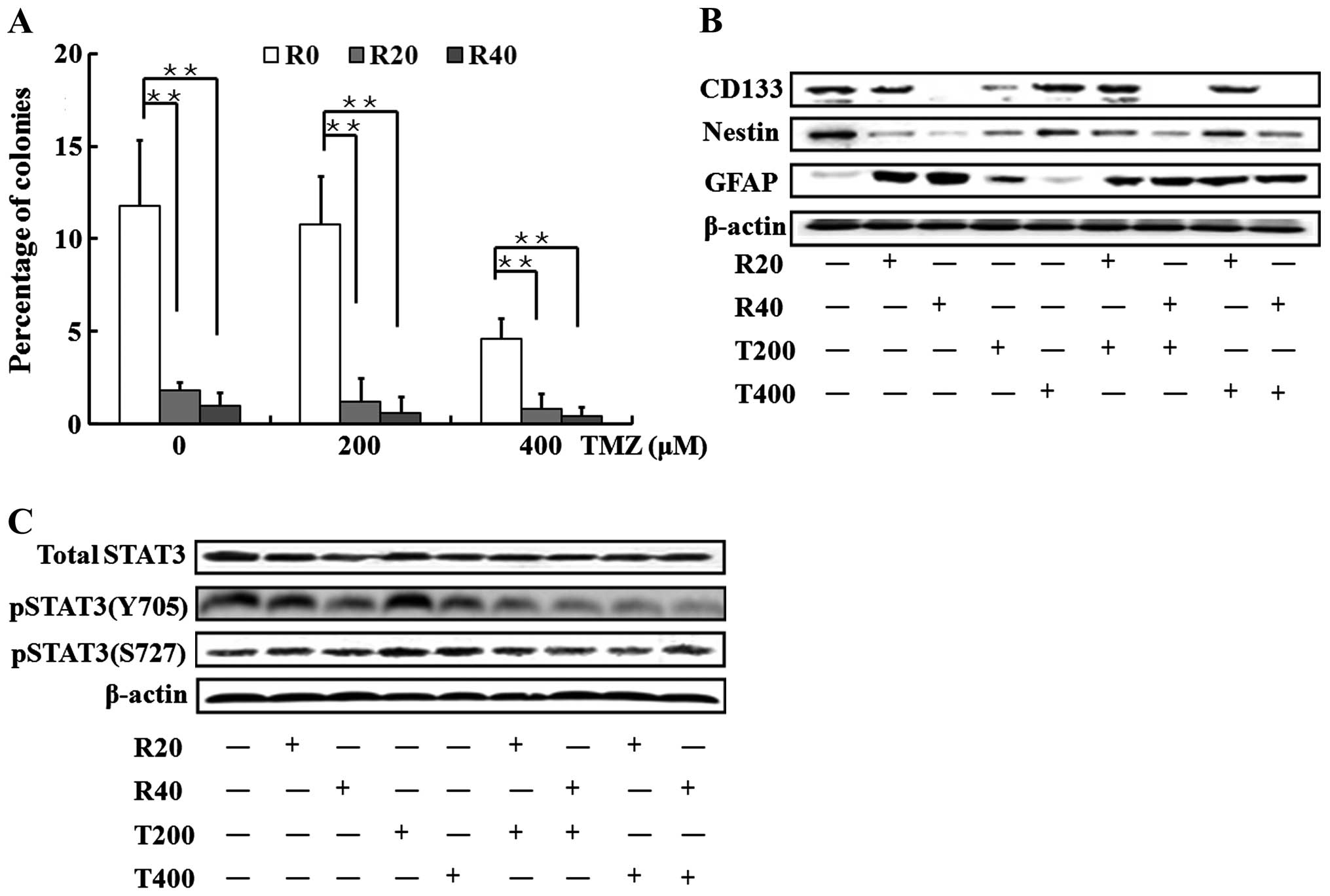
As demonstrated in Fig.
4A, the sphere-formation ability of GICs was slightly affected
by T200, but a ~50% reduction in this ability was induced by T400
(P<0.01, T400 vs. control). Combined with RES, the TMZ
treatments significantly impaired the capacity of GICs to form
spheres, similar to the effects observed in the GICs exposed to RES
rather than TMZ (Fig. 4A).
Moreover, slight changes in the expression levels of
CD133, nestin and GFAP were observed in the GICs exposed to T200
and T400. When involved by RES, the drug combinations significantly
increased GFAP expression levels and decreased CD133 and nestin
levels.
pSTAT3 (Y705) rather than pSTAT3 (S727) and the
total STAT3 was found to be significantly suppressed when the
self-renewal capacity was lost and differentiation was promoted in
the GICs (Fig. 4C).
RES enhances TMZ-induced inhibition of
the tumor growth in a xenograft model of GICs and promotes
differentiation
To translate our findings into a clinically relevant
approach, the drug combination effects were determined in
vivo. Compared with an intracranial xenograft model, a
subcutaneous xenograft model of GICs makes it easier to observe the
tumor growth rate and measure tumor volume. Therefore, we adopted a
subcutaneous xenograft model as described in a previous study
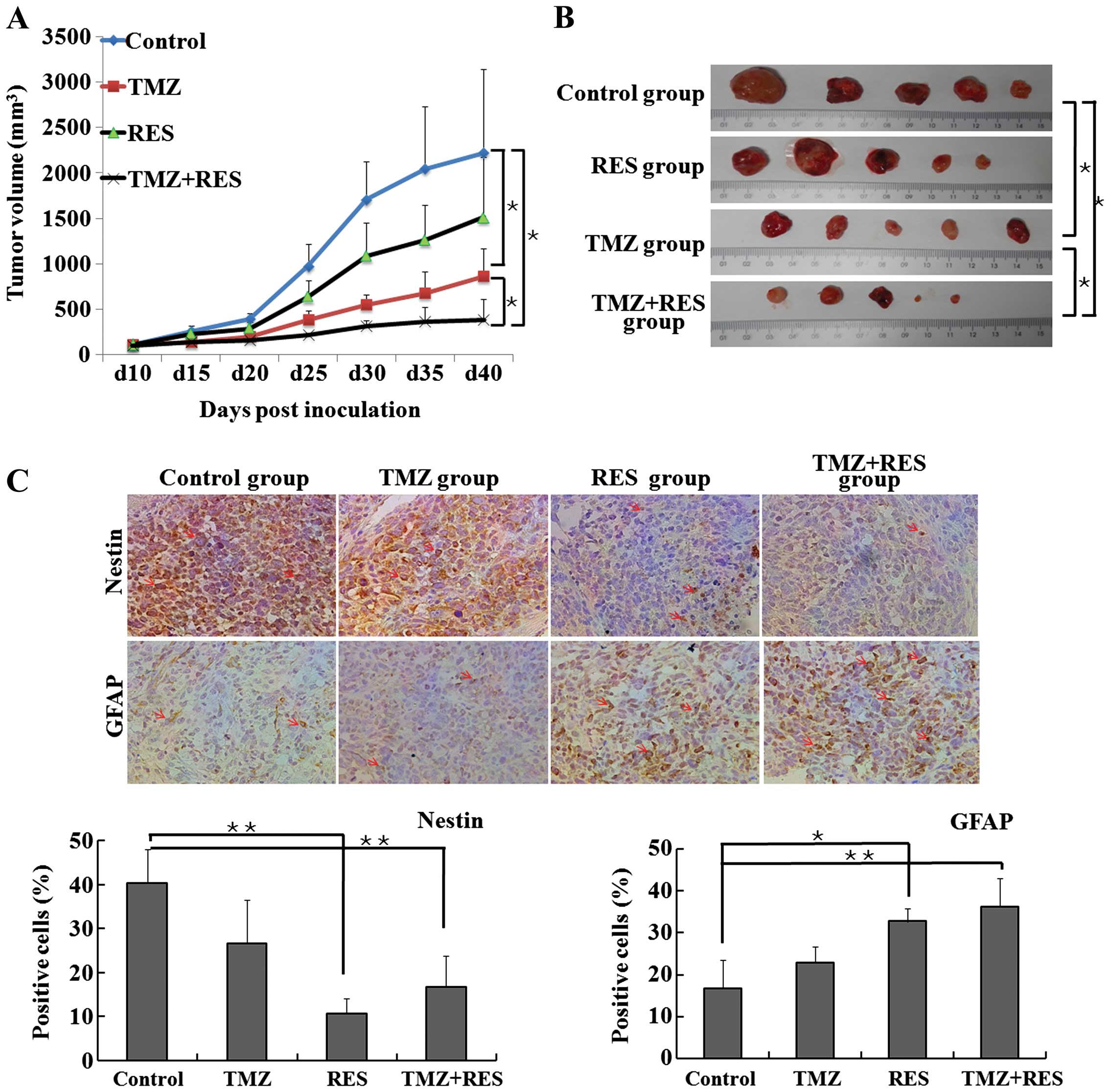
(25). The results (Fig. 5A) showed that 68 mg/kg TMZ treatment
alone exhibited an antitumor effect in regards to tumor volume
growth inhibition (P=0.013, TMZ vs. control at day 40). RES
treatment alone did not significantly inhibit the growth of the
tumors at the dose of 12.5 mg/kg (P=0.113 vs. control at day 40).
RES significantly potentiated TMZ in inhibiting tumor volume growth
(P=0.035, TMZ+RES vs. TMZ). As shown in Fig. 5B, the combination of 68 mg/kg TMZ
and 12.5 mg/kg RES significantly reduced the excised tumor volume
by 77.1% compared with that observed in the control group (P=0.034
vs. control). Animal body weight was monitored serially to assess
the tolerability of the regimens tested in all mice (data not
shown). In the course of treatment, there was no significant
difference in body weight between the control group and RES group.
Moreover, there was no significant increase or decrease in the
average mouse weight in the TMZ alone and TMZ+RES groups between
days 0 and 40, with the exception of day 15, where a decrease in
body weight was observed. However, the body weight was 15% and 17%
lower compared with the control group in the TMZ and TMZ+RES
groups, respectively. Of note, there were no deaths during the
treatment course. The results indicated that the combination
therapy did not have increased toxicity compared to TMZ alone.
Moreover, as shown in Fig. 5C, the density of the
nestin-immunoreactive 'stemness' cell surface in the TMZ group was
similar to that in the control group (P=0.15). TMZ combined with
RES robustly downregulated nestin expression by 58.5% compared with
the control group (P=0.005 TMZ+RES vs. control). GFAP
immunostaining was rare in the control group and the TMZ group;
treatment with RES combined with TMZ greatly increased the
GFAP-immunoreactive astrocytic surface density by 116.1% (P=0.0009,
TMZ+RES vs. control) and 57.9% (P=0.002, TMZ+RES vs. TMZ).
Discussion
Using highly resistant GICs isolated from patient
samples, the present study demonstrated that RES enhanced the
sensitivity of TMZ by inducing the apoptosis of GICs via activation
of the DSBs/pATM/pATR/p53 pathway. Moreover, the involvement of RES
in TMZ therapy also reduced the self-renewal ability and promoted
the differentiation of GICs by the inactivation of pSTAT3.
RES enhances TMZ-induced GIC apoptosis
via activation of the DSBs/pATM/pATR/p53 pathway
A recent study demonstrated that RES potentiated the
efficacy of TMZ by suppressing TMZ-induced autophagy and
subsequently increasing apoptotic cell death in glioma cell lines
(26). However, whether RES
enhances the TMZ-induced apoptosis in GICs from glioma cell lines
due to their distinct properties and the mechanism underlying the
enhanced apoptosis induced by TMZ and RES combination has not yet
been reported.
In the present study, we found that RES
significantly enhanced TMZ-induced apoptosis of GICs. As an
alkylating agent, TMZ induces the O6-methylguanine
lesion which leads to DSBs via collapse of a replication fork at
the site of a blocking DNA lesion and results in cell death via
apoptosis and/or autophagy (27).
In addition, several studies have shown that RES poisons TOPOIIa
thus inducing DSBs and activating the DSB signaling pathway to
induce apoptosis (28). Given that
TMZ or RES alone leads to tumor cell apoptosis through DSB
formation capacity, we found that their combinations significantly
induced DSBs underlying apoptosis, evidenced by γH2AX focus
formation. Furthermore, it has been suggested that DNA damage
repair proteins may prevent DSBs and protect tumor cells from death
and they are considered as important determinants of
chemoresistance in many tumors including GBM (29,30).
Thus, we assessed the expression levels of three DNA damage repair
proteins MGMT, PARP1 and Ku70 due to their important roles in
removing DNA lesions, alternative end-joining (A-EJ) repair and the
non-homologous end joining (NHEJ) repair mechanism, respectively
(22,31,32).
Our results showed that combined usage of TMZ and RES markedly
repressed MGMT, PARP1 and Ku70 compared with that obtained with
either chemical alone, suggesting that the DSB repair system was
broadly inhibited.
The signaling pathway orchestrated by the ATM and
ATR kinases is the central regulator in bridging DSBs to final
apoptosis (24,33). Although ATM and ATR often work
together to signal DSBs, ATM is primarily activated through DSBs
caused by extrinsic stress such as irradiation and chemical
toxicity while ATR is considered mainly activated in response to
replication stress (34). Compared
with slight increase in pATM expression, there was a significant
upregulation of pATR when TMZ was combined with RES in our study.
This implies that p-ATR activation principally functions as the
bridge to pass down the DSB signal to downstream effectors, and
replication stress might be mainly responsible for DSB formation
induced by the current concentrations of TMZ, RES and their
combinations.
p53 contributes significantly to the maintenance of
genomic stability and is the core component of the network in
regulating apoptosis (35). ATM and
ATR could both directly phosphorylate p53 to activate and stabilize
the protein. Thus, the DSBs/pATM/pATR/p53 signaling pathway has
been widely reported to play key roles in regulating apoptosis
induced by DNA damage in previous studies (36,37).
In accordance with these results, we found that p53 and its
phosphorylated form were highly elevated accompanying the
activation of pATM/pATR in the use of drug combinations.
Furthermore, due to the role of p53 activation in the regulation of
Bcl-2/Bax both transcriptionally and at the protein level (38), we also demonstrated that the
Bcl-2/Bax ratio was significantly decreased when TMZ was combined
with RES.
The involvement of RES in TMZ treatment
reduces the self-renewal ability and promotes differentiation of
GICs with the inactivation of pSTAT3
The 'stemness' of GICs is commonly believed to be
the significant property accounting for therapeutic resistance and
the capability of repopulation for GBM (39). In our study, GICs exposed to TMZ
exhibited no change in their 'stemness' phenotype as shown in
Fig. 4B, which is in line with
previous studies (6,7). The suppression of sphere-formation
ability of GICs by TMZ may be due to increases in apoptosis and
cell cycle arrest (data not shown) induced by DNA damage.
Resveratrol was previously found to promote the
acquisition of a long-lasting differentiated phenotype in human GBM
cells and to inhibit the tumorigenicity of GICs (40). In our study, a marked decreased
tendency of GIC expansion was observed when TMZ was combined with
RES as these cells lost their self-renewal capacity and underwent a
conversion from 'stemness' to a differentiation phenotype,
evidenced by decreases in the expression levels of CD133 and nestin
and an increase in the expression of GFAP.
STAT3 is important in maintaining the self-renewal
of GICs and the inactivation of STAT3 in GICs is identified as the
onset signal for the conversion from a 'stemness' phenotype to a
differentiation phenotype (19,41–43).
For instance, suppression of STAT3 with siRNA significantly induced
GIC differentiation with a decrease in CD133, increase in GFAP and
a decrease in capacity for GICs to initiated a tumor. STAT3 is
activated mainly by phosphorylation at Tyr705, which induces
dimerization, nuclear translocation, and DNA binding. In addition,
phosphorylation of STAT3 at Ser727 is usually considered to be
necessary for the maximal transcriptional activity (44,45).
Moreover, constitutive activation of STAT3 at both Tyr705 and
Ser727 has been observed in many human malignancies including GBM
(46). In the present study, we
found that STAT3-Tyr705 was significantly suppressed when GICs were
converting from a 'stemness' phenotype to a differentiation status
while STAT3-Ser727 and STAT3 remained almost the same, suggesting
that STAT3-Tyr705 suppression by RES and drug combinations plays a
key role in inhibition of self-renewal ability and promotion of
differentiation.
In summary, the present study introduces evidence
that RES may act in concert with TMZ to eradicate GICs, thereby
providing a foundation for the combined usage of RES and TMZ on GBM
patients, particularly those with abundant GICs in their
histopathological sections. This new strategy targeting GICs may
benefit patients who show slight responsiveness to TMZ therapy and
provide a promising long-term survival effect compared with TMZ
therapy alone.
Acknowledgments
The authors would like to thank Dr Pingyang Liu at
the University of California, San Francisco for critically reading
and discussing this manuscript. This study was supported by a grant
from the National Natural Science Foundation of China (81102463),
funds from the China National Clinical Research Center for
Neurological Diseases, the Training Plan for Beijing High-Level
Healthcare Personnel (2011-3-28), and funds of the Capital Medical
University Clinical-Basic Cooperation Research (11JL16).
References
|
1
|
Cheng L, Bao S and Rich JN: Potential
therapeutic implications of cancer stem cells in glioblastoma.
Biochem Pharmacol. 80:654–665. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Orza A, Soriţău O, Tomuleasa C, Olenic L,
Florea A, Pana O, Bratu I, Pall E, Florian S, Casciano D, et al:
Reversing chemore-sistance of malignant glioma stem cells using
gold nanoparticles. Int J Nanomedicine. 8:689–702. 2013. View Article : Google Scholar :
|
|
3
|
Higgins DM, Wang R, Milligan B, Schroeder
M, Carlson B, Pokorny J, Cheshier SH, Meyer FB, Weissman IL,
Sarkaria JN, et al: Brain tumor stem cell multipotency correlates
with nanog expression and extent of passaging in human glioblastoma
xenografts. Oncotarget. 4:792–801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Neman J and Jandial R: Decreasing glioma
recurrence through adjuvant cancer stem cell inhibition. Biologics.
4:157–162. 2010.PubMed/NCBI
|
|
5
|
Johannessen TC, Bjerkvig R and Tysnes BB:
DNA repair and cancer stem-like cells - potential partners in
glioma drug resistance? Cancer Treat Rev. 34:558–567. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beier D, Schriefer B, Brawanski K, Hau P,
Weis J, Schulz JB and Beier CP: Efficacy of clinically relevant
temozolomide dosing schemes in glioblastoma cancer stem cell lines.
J Neurooncol. 109:45–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Auffinger B, Tobias AL, Han Y, Lee G, Guo
D, Dey M, Lesniak MS and Ahmed AU: Conversion of differentiated
cancer cells into cancer stem-like cells in a glioblastoma model
after primary chemotherapy. Cell Death Differ. 21:1119–1131. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Persano L, Rampazzo E, Basso G and Viola
G: Glioblastoma cancer stem cells: Role of the microenvironment and
therapeutic targeting. Biochem Pharmacol. 85:612–622. 2013.
View Article : Google Scholar
|
|
9
|
Sales JM and Resurreccion AV: Resveratrol
in peanuts. Crit Rev Food Sci Nutr. 54:734–770. 2014. View Article : Google Scholar
|
|
10
|
Borriello A, Bencivenga D, Caldarelli I,
Tramontano A, Borgia A, Zappia V and Della Ragione F: Resveratrol:
From basic studies to bedside. Cancer Treat Res. 159:167–184. 2014.
View Article : Google Scholar
|
|
11
|
Pallàs M, Ortuño-Sahagún D, Benito-Andrés
P, Ponce-Regalado MD and Rojas-Mayorquín AE: Resveratrol in
epilepsy: Preventive or treatment opportunities? Front Biosci
(Landmark Ed). 19:1057–1064. 2014. View
Article : Google Scholar
|
|
12
|
Aggarwal BB, Bhardwaj A, Aggarwal RS,
Seeram NP, Shishodia S and Takada Y: Role of resveratrol in
prevention and therapy of cancer: Preclinical and clinical studies.
Anticancer Res. 24:2783–2840. 2004.PubMed/NCBI
|
|
13
|
Harikumar KB and Aggarwal BB: Resveratrol:
A multitargeted agent for age-associated chronic diseases. Cell
Cycle. 7:1020–1035. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delmas D, Solary E and Latruffe N:
Resveratrol, a phytochemical inducer of multiple cell death
pathways: Apoptosis, autophagy and mitotic catastrophe. Curr Med
Chem. 18:1100–1121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan Y, Xue X, Guo RB, Sun XL and Hu G:
Resveratrol enhances the antitumor effects of temozolomide in
glioblastoma via ROS-dependent AMPK-TSC-mTOR signaling pathway. CNS
Neurosci Ther. 18:536–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang H, Lin H, Zhang X and Li J:
Resveratrol reverses temo-zolomide resistance by downregulation of
MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway.
Oncol Rep. 27:2050–2056. 2012.PubMed/NCBI
|
|
17
|
Aldaz B, Sagardoy A, Nogueira L, Guruceaga
E, Grande L, Huse JT, Aznar MA, Díez-Valle R, Tejada-Solís S,
Alonso MM, et al: Involvement of miRNAs in the differentiation of
human glioblastoma multiforme stem-like cells. PLoS One.
8:e770982013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Romanelli S, Perego P, Pratesi G, Carenini
N, Tortoreto M and Zunino F: In vitro and in vivo interaction
between cisplatin and topotecan in ovarian carcinoma systems.
Cancer Chemother Pharmacol. 41:385–390. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang YP, Chang YL, Huang PI, Chiou GY,
Tseng LM, Chiou SH, Chen MH, Chen MT, Shih YH, Chang CH, et al:
Resveratrol suppresses tumorigenicity and enhances radiosensitivity
in primary glioblastoma tumor initiating cells by inhibiting the
STAT3 axis. J Cell Physiol. 227:976–993. 2012. View Article : Google Scholar
|
|
20
|
Gedye C and Ailles L: Isolation and
characterization of cancer stem cells in vitro. Methods Mol Biol.
946:181–204. 2013. View Article : Google Scholar
|
|
21
|
Roos WP and Kaina B: DNA damage-induced
cell death: From specific DNA lesions to the DNA damage response
and apoptosis. Cancer Lett. 332:237–248. 2013. View Article : Google Scholar
|
|
22
|
Ponnala S, Veeravalli KK, Chetty C, Dinh
DH and Rao JS: Regulation of DNA repair mechanism in human glioma
xenograft cells both in vitro and in vivo in nude mice. PLoS One.
6:e261912011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Silber JR, Bobola MS, Blank A and
Chamberlain MC: O(6)-methylguanine-DNA methyltransferase in glioma
therapy: Promise and problems. Biochim Biophys Acta. 1826:71–82.
2012.PubMed/NCBI
|
|
24
|
Park I and Avraham HK: Cell
cycle-dependent DNA damage signaling induced by ICRF-193 involves
ATM, ATR, CHK2, and BRCA1. Exp Cell Res. 312:1996–2008. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eyler CE, Wu Q, Yan K, MacSwords JM,
Chandler-Militello D, Misuraca KL, Lathia JD, Forrester MT, Lee J,
Stamler JS, et al: Glioma stem cell proliferation and tumor growth
are promoted by nitric oxide synthase-2. Cell. 146:53–66. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin CJ, Lee CC, Shih YL, Lin TY, Wang SH,
Lin YF and Shih CM: Resveratrol enhances the therapeutic effect of
temozolomide against malignant glioma in vitro and in vivo by
inhibiting autophagy. Free Radic Biol Med. 52:377–391. 2012.
View Article : Google Scholar
|
|
27
|
Johannessen TC and Bjerkvig R: Molecular
mechanisms of temozolomide resistance in glioblastoma multiforme.
Expert Rev Anticancer Ther. 12:635–642. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leone S, Basso E, Polticelli F and Cozzi
R: Resveratrol acts as a topoisomerase II poison in human glioma
cells. Int J Cancer. 131:E173–E178. 2012. View Article : Google Scholar
|
|
29
|
Srivastava M and Raghavan SC: DNA
double-strand break repair inhibitors as cancer therapeutics. Chem
Biol. 22:17–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aparicio T, Baer R and Gautier J: DNA
double-strand break repair pathway choice and cancer. DNA Repair
(Amst). 19:169–175. 2014. View Article : Google Scholar
|
|
31
|
Villalva C, Cortes U, Wager M, Tourani JM,
Rivet P, Marquant C, Martin S, Turhan AG and Karayan-Tapon L:
O6-Methyl-guanine-methyltransferase (MGMT) promoter
methylation status in glioma stem-like cells is correlated to
temozolomide sensitivity under differentiation-promoting
conditions. Int J Mol Sci. 13:6983–6994. 2012. View Article : Google Scholar
|
|
32
|
Haince JF, McDonald D, Rodrigue A, Déry U,
Masson JY, Hendzel MJ and Poirier GG: PARP1-dependent kinetics of
recruitment of MRE11 and NBS1 proteins to multiple DNA damage
sites. J Biol Chem. 283:1197–1208. 2008. View Article : Google Scholar
|
|
33
|
Gobbini E, Cesena D, Galbiati A, Lockhart
A and Longhese MP: Interplays between ATM/Tel1 and ATR/Mec1 in
sensing and signaling DNA double-strand breaks. DNA Repair (Amst).
12:791–799. 2013. View Article : Google Scholar
|
|
34
|
Cooper TJ, Wardell K, Garcia V and Neale
MJ: Homeostatic regulation of meiotic DSB formation by ATM/ATR. Exp
Cell Res. 329:124–131. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Speidel D: The role of DNA damage
responses in p53 biology. Arch Toxicol. 89:501–517. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shimada M and Nakanishi M: Response to DNA
damage: Why do we need to focus on protein phosphatases? Front
Oncol. 3:82013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Loewer A, Karanam K, Mock C and Lahav G:
The p53 response in single cells is linearly correlated to the
number of DNA breaks without a distinct threshold. BMC Biol.
11:1142013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kolb JP: Mechanisms involved in the pro-
and anti-apoptotic role of NO in human leukemia. Leukemia.
14:1685–1694. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nakano I: Stem cell signature in
glioblastoma: Therapeutic development for a moving target. J
Neurosurg. 122:324–330. 2015. View Article : Google Scholar
|
|
40
|
Sato A, Okada M, Shibuya K, Watanabe E,
Seino S, Suzuki K, Narita Y, Shibui S, Kayama T and Kitanaka C:
Resveratrol promotes proteasome-dependent degradation of Nanog via
p53 activation and induces differentiation of glioma stem cells.
Stem Cell Res (Amst). 11:601–610. 2013. View Article : Google Scholar
|
|
41
|
Li GH, Wei H, Lv SQ, Ji H and Wang DL:
Knockdown of STAT3 expression by RNAi suppresses growth and induces
apoptosis and differentiation in glioblastoma stem cells. Int J
Oncol. 37:103–110. 2010.PubMed/NCBI
|
|
42
|
Yang L, Guo H, Dong L, Wang L, Liu C and
Wang X: Tanshinone IIA inhibits the growth, attenuates the stemness
and induces the apoptosis of human glioma stem cells. Oncol Rep.
32:1303–1311. 2014.PubMed/NCBI
|
|
43
|
Liu M, Inoue K, Leng T, Guo S and Xiong
ZG: TRPM7 channels regulate glioma stem cell through STAT3 and
Notch signaling pathways. Cell Signal. 26:2773–2781. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lin J, Jin X, Rothman K, Lin HJ, Tang H
and Burke W: Modulation of signal transducer and activator of
transcription 3 activities by p53 tumor suppressor in breast cancer
cells. Cancer Res. 62:376–380. 2002.PubMed/NCBI
|
|
45
|
Yang F, Zhang W, Li D and Zhan Q: Gadd45a
suppresses tumor angiogenesis via inhibition of the mTOR/STAT3
protein pathway. J Biol Chem. 288:6552–6560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gray GK, McFarland BC, Nozell SE and
Benveniste EN: NF-κB and STAT3 in glioblastoma: Therapeutic targets
coming of age. Expert Rev Neurother. 14:1293–1306. 2014. View Article : Google Scholar : PubMed/NCBI
|