Introduction
Liver kinase B1 (LKB1) also known as
serine/threonine kinase 11 (STK11), is a potent tumor-suppressor
gene mutationally inactivated in the autosomal dominant
Peutz-Jeghers syndrome (PJS), which is characterized by the
development of multiple gastrointestinal hamartomatous polyps and
distinct pigmentation of the skin and mucous membranes (1–3). PJS
is a cancer predisposition syndrome, with an 18-fold increased
incidence of malignancy in a wide variety of tissues, including the
colon, breast, stomach, uterine cervix, lung and liver (4). In addition, multiple LKB1 mutations
have been identified in various sporadic cancers, particularly
those of the lung and also those of the genitourinary tract and
breast (5–7), further highlighting the bona fide
tumor-suppressor role of the LKB1 gene.
Encoding of the serine/threonine protein kinase,
LKB1, is highly conserved throughout evolution from worms to
mammals and is ubiquitously expressed in both embryonic and adult
tissues with a notable higher expression in the pancreas, liver and
skeletal muscle (8). The universal
expression of LKB1 transcripts in all mammalian tissues is
consistent with the elevated risk of multiple cancer types in PJS
patients, and suggests that LKB1 is important to general cell
function. Genetic inactivation of LKB1 in mice leads to embryonic
lethality at midgestation and a variety of developmental
abnormalities including the yolk sac and placenta (9,10),
indicating that LKB1 is essential for normal development. Over the
past 16 years, the LKB1 tumor-suppressor kinase has been
extensively studied and implicated in a broad range of cellular
processes, including cell cycle arrest, energy metabolism,
apoptosis, senescence, differentiation and angiogenesis (11,12).
Currently, there is no doubt that LKB1 is at the center of an
important signaling node affecting numerous cellular processes
whose deregulation contributes to tumorigenesis, resulting in
pathologies such as PJS and various sporadic cancers (4,13).
The human hepatic cell line L02 was derived from
primary normal human hepatocytes and immortalized in 1980 (14). Over the past 35 years, the L02 cell
line has been widely used in studies of human hepatocellular
functions, particularly those related to hepatic steatosis, drug
hepatotoxicity and chemical carcinogenesis (15–18),
but the genetic background of LKB1 signaling in L02 cells remains
unknown. In the present study, we examined endogenous LKB1
expression in the L02 cell line and investigated the effect of LKB1
expression on proliferation, anchorage-independent growth and
tumorigenesis of L02 cells. The present study provides strong
evidence that endogenous LKB1 expression is deficient in the L02
cell line, which may confer observable tumorigenicity both in
vitro and in vivo and suggests that the immortalized L02
cell line is not a normal cell line, but cancerous.
Materials and methods
Materials
Cell culture media, fetal bovine serum (FBS), G418
and Lipofectamine 2000 reagent were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). Primary antibodies against Rb and
phospho-Rb (Ser807/811) were purchased from Cell Signaling
Technology (Beverly, MA, USA). Primary antibodies against LKB1 and
GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies
were obtained from Jackson ImmunoResearch Laboratories (West Grove,
PA, USA). Unless otherwise noted, all chemicals and reagents were
purchased from Sigma-Aldrich (Taufkirchen, Germany) and were of the
highest grade.
Cell culture and generation of stable L02
cell lines
Human hepatic cell line L02 was purchased from the
Type Culture Collection of the Chinese Academy of Sciences
(Shanghai) and cultured in RPMI-1640 medium containing 20% FBS.
HEK-293T and HeLa cells, and HUVECs, were maintained in minimum
essential medium (MEM) supplemented with 10% FBS. Cells were
cultured in a humidified incubator at 37°C containing 5%
CO2. Cultured cells were used between passage 3 and 10.
Transfection was performed using Lipofectamine 2000 according to
the manufacturer's protocols. L02 cells were transfected with
either an empty mock plasmid or a plasmid expressing wild-type
LKB1, as previously described (19). After selection for 16 days in G418
(600 µg/ml)-containing medium, the resistant isolated single
clones were selected, expanded and propagated. LKB1 expression was
verified by northern blot and western blot analyses, respectively.
One maximally expressing clone from the LKB1-transfected L02 cells
was obtained and maintained in the same manner as the parental
cells.
Cell proliferation assay
Cell proliferation was examined using the CellTiter
96 AQueous One Solution Cell Proliferation Assay (Promega, Madison,
WI, USA) according to the manufacturer's protocol. Cells
(5,000/well) were plated into 96-well plates in triplicates for 96
h and incubated in culture medium containing MTS every day for 4
days. SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA) was
used to detect the absorbance at 490 nm. Results are presented as
the average absorbance of 3-wells/experiment.
Soft agar colony formation assay
Cells were seeded in complete media at a density of
1×103 cells in 60-mm dishes containing a top layer of
0.35% low melting point (LMP) agar and a bottom layer of 0.6% LMP
agar. The plates were incubated at 37°C in 5% CO2 for 4
weeks. Every 7 days, fresh medium was added to each plate. Crystal
violet 0.2% was used to stain the plates. Visible colonies were
photographed and counted manually.
Western blot analysis
Total proteins were analyzed by SDS-PAGE
electrophoresis and blotted as previously described (19,20).
Specific primary anti bodies recognizing LKB1 (1:500), Rb
(1:1,000), phospho-Rb (1:1,000) and GAPDH (1:1,000) were used at
the indicated dilutions. HRP-conjugated secondary antibodies were
used at 1:10,000 dilutions. The signals of HRP were detected by
Substrate SuperSignal West Pico Chemiluminescent Substrate kit
(Pierce Biotechnology, Rockford, IL, USA). The protein signal was
quantified using Quantity One software (Bio-Rad, Hercules, CA,
USA).
Northern blot analysis
Total RNA was isolated using TRIzol (Invitrogen Life
Technologies), resolved in 1% formaldehyde-agarose gels,
transferred to nylon membranes and cross-linked. Blots were probed
with digoxigenin (DIG)-labeled cDNAs. The LKB1 probe was prepared
using the forward primer (5′-TCGGTGGGTATGGACACGTTC-3′) and the
reverse primer (5′-GTTCGTACTCAAGCATCCCTTTCA-3′), and labeled with
DIG using End Tailing kit (Roche Applied Science, Indianapolis, IN,
USA). β-actin gene was used as a loading control. Hybridization
signals were detected with chemiluminescence and recorded on x-ray
film.
Reverse transcription PCR (RT-PCR) and
sequencing
Total RNA (2 µg) was transcribed with M-MLV
reverse transcriptase and used for subsequent PCR amplification
with Taq polymerase (both from Promega). PCR products were
analyzed on 2% agarose gels and verified by DNA sequencing.
Housekeeping gene β-actin was amplified as an internal control.
LKB1-specific primers designed to detect LKB1 transcriptional
variants are described in Table
I.
 | Table IPrimer pair sequences of the LKB1
gene and related information. |
Table I
Primer pair sequences of the LKB1
gene and related information.
| Loci | Sequence | Annealing temp.
(°C) | Size (bp) |
|---|
| P1 | F
5′-CTCCACCGAGGTCATCTACC-3′ | 57 | 341 |
| R
5′-AAATGCTGGACAGCGTGC-3′ | | |
| P2 | F
5′-TACGGCAAGGTGAAGGAGG-3′ | 59 | 413 |
| R 5′-ATCTCCGACCTG
GGCGT-3′ | | |
| P3 | F
5′-CCTGCTGAAAGGGATGCT-3′ | 56 | 366 |
| R
5′-AGCACCAAATCCAGGGC-3′ | | |
| P4 | F
5′-CCTGCTGAAAGGGATGCT-3′ | 59 | 688 |
| R
5′-GCTTGTTGACTTCGCAGCCC-3′ | | |
| P5 | F
5′-AACCTGCTGCTCACCACCG-3′ | 58 | 338 |
| R
5′-TGAAAGGGATGCTTGAGTACG-3′ | | |
| P6 | F
5′-CCGGGACTGACGTGTAGAAC-3′ | 56 | 393 |
| R
5′-GGTGAAGGAGGTGCTGGA-3′ | | |
Tumorigenicity assay
Aliquots of the particular cell populations were
counted and injected into the axillary region (2×106
cells/injection site) of 6-week-old male BALB/c nude mice (Animal
Center of Chongqing Medical University, Chongqing, China). The mice
were housed in sterile cages under laminar flow hoods in a
temperature-controlled room and were fed autoclaved chow and water
ad libitum. All mice (n=4/group) were weekly weighed and
examined every other day for morbidity and for tumor growth
(measured using a digital caliper). After 6 weeks, all of the mice
were euthanized with excess CO2 and necropsied to
determine possible gross metastases.
All experiments using mice were in accordance with
the Guide for the Care and Use of Laboratory Animals and was
approved by the Institutional Animal Care and Use Committee (IACUC)
of Chongqing Medical University (Chongqing, China).
Histological analysis and
immunohistochemistry
Tumor tissues were fixed in 4% paraformaldehyde and
embedded in paraffin wax before sectioning at 5-µm
thickness. For histopathological examinations, the sections were
stained with hematoxylin and eosin (H&E). For
immunohistochemistry, serial sections were deparaffinized, treated
with 0.3% H2O2 in methanol to inactivate
endogenous peroxidase, and incubated with anti-LKB1 monoclonal
antibody at a 100-fold dilution for 60 min. Immunodetection of LKB1
was performed using Vector Stain Elite kit (Vector Research,
Burlingame, CA, USA). A positive control was included with each
batch of staining to ensure consistency between consecutive runs.
Immunostaining was evaluated by a pathologist.
Statistical analysis
All cell culture experiments were repeated at least
3 times, unless otherwise indicated, and paired t-tests were used
to determine statistical significance.
Results
LKB1 expression is deficient in the L02
cell line
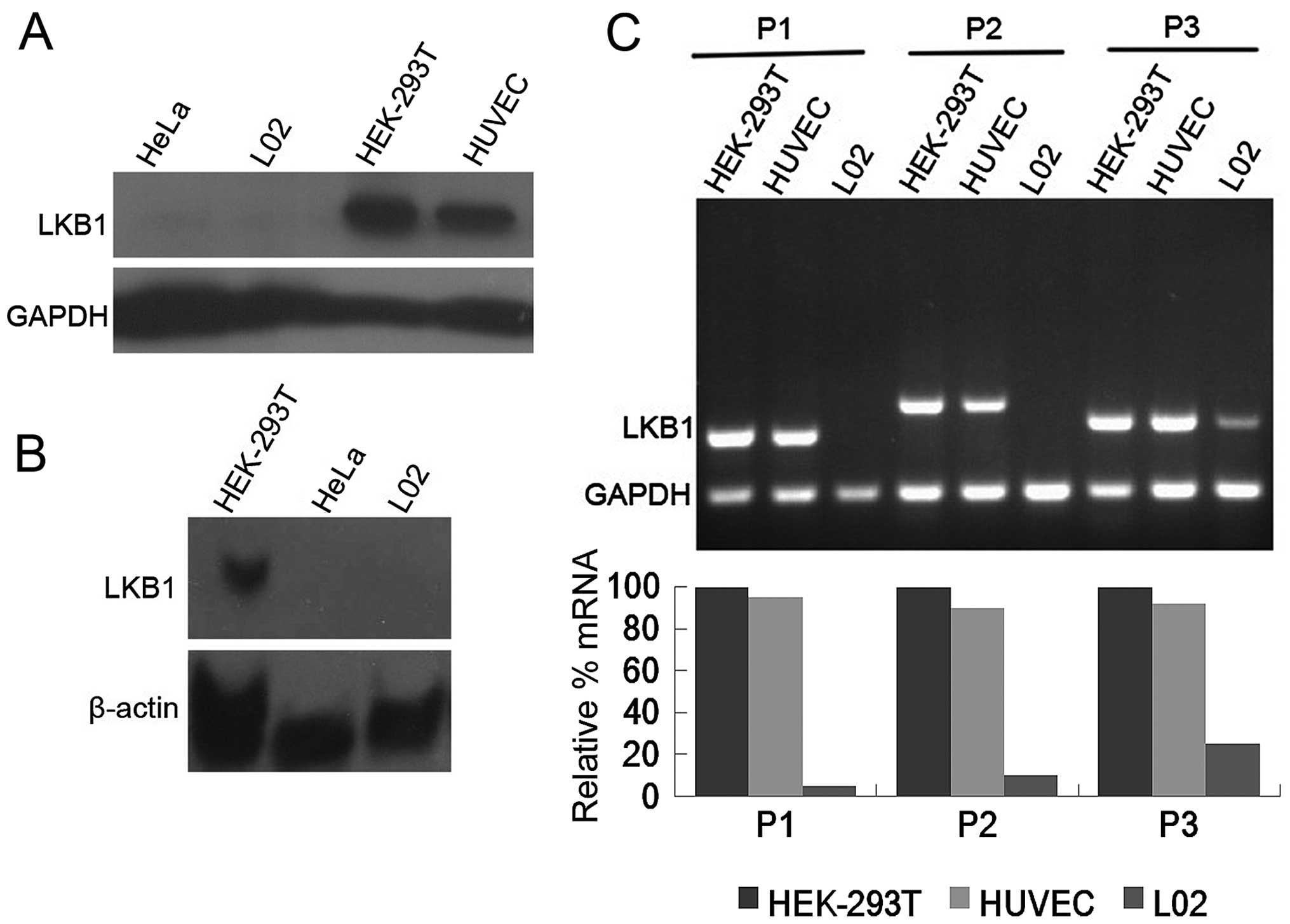
We first examined the protein expression of LKB1 in
the L02 cell line by western blot analysis. The HeLa cell line,
which has been extensively characterized as endogenous
LKB1-deficient (21), was included
as a negative control. Human embryonic kidney 293T cells (HEK-293T
cells) and human umbilical vein endothelial cells (HUVECs) were
used as positive controls. As depicted in Fig. 1A, a specific band for LKB1 was
detected in 25 µg total protein of HEK-293T cells and
HUVECs, whereas the LKB1 signal was not detectable in 180 µg
total protein of L02 cells and 140 µg total protein of HeLa
cells, suggesting that LKB1 protein expression is severely
compromised in L02 cells. In concert with the deficiency of LKB1
protein, northern blot analysis indicated that LKB1 mRNA levels
were barely detectable in the L02 and HeLa cells, in contrast to
the HEK-293T cells which expressed abundant LKB1 transcript
(Fig. 1B), supporting the lack of
LKB1 expression in the L02 cell line.
RT-PCR and sequencing
To better evaluate the genetic defects underlying
LKB1 deficiency in the L02 cell line, we performed LKB1 mutation
analysis. The transcript was screened for putative mutations by
sequencing of products obtained by reverse transcription followed
by polymerase chain reaction (RT-PCR) from the L02 cell line. cDNA
from L02 cells was amplified with 6 sets of primers covering the
entire coding region (exons 1–9) of the LKB1 gene. These primers
are described in Table I. As shown
in Fig. 1C, compared with the
HEK-293T cells and HUVECs, LKB1 transcription was severely affected
in the L02 cells particularly for the 5′ end region. LKB1 mRNA
expression was reduced to ~5% of the HEK-293T cells with upstream
primer P1, 10% with midstream primer P2, and 25% with downstream
primer P3. The majority of the LKB1 coding sequence (93%) was
amplified from the L02 cells at very low levels by the primers
chosen, except for the region containing the initial 89 bases,
which was unable to be obtained by RT-PCR. Direct sequencing of the
PCR products did not detect any abnormalities in nucleotides
90-1302 of the LKB1 open reading frame, suggesting the putative
mutation region may be narrowed down to 89 bases downstream of the
start codon. Another possibility for silencing of LKB1 expression
is epigenetic inactivation caused by promoter hypermethylation
(22). However, treatment with the
demethylating agent 5-aza-2′-deoxycytidine could not restore mRNA
or protein expression of LKB1 in our research (data not shown).
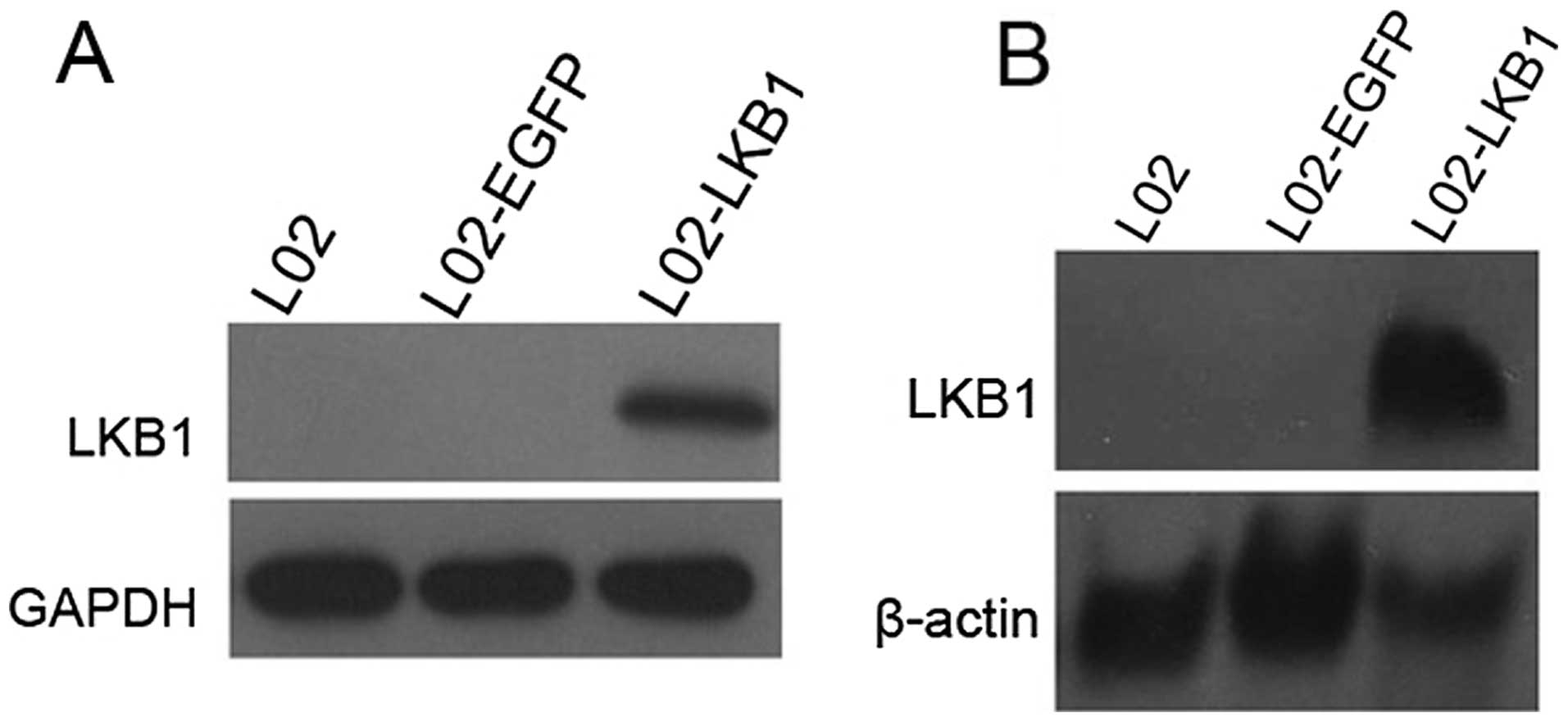
Construction of a stable L02 cell line
constitutively expressing LKB1
To obtain further insights into the biological role
of LKB1, we attempted to reconstruct the in vivo situation
of L02 cells by stably expressing wild-type LKB1. L02 cells were
transfected with an expression vector encoding both LKB1 and a
neomycin resistance gene or a vector encoding the selection marker
only. Transfectants were subjected to G418 selection for 16 days.
After the selection, a strongly reduced number of colonies were
detected in the LKB1 transfectants compared with the mock vector
transfectants of the L02 cells. Individual colonies were selected
and analyzed for inducible expression of LKB1 by northern blot and
western blot analyses. As shown in Fig.
2, recombinant LKB1 was readily detected at high levels in the
L02 cells stably transfected with the LKB1 construct, but not in
the cells transfected with the control vector. Thus, the
reconstruction of LKB1 gene expression was successfully achieved in
the L02 cells.
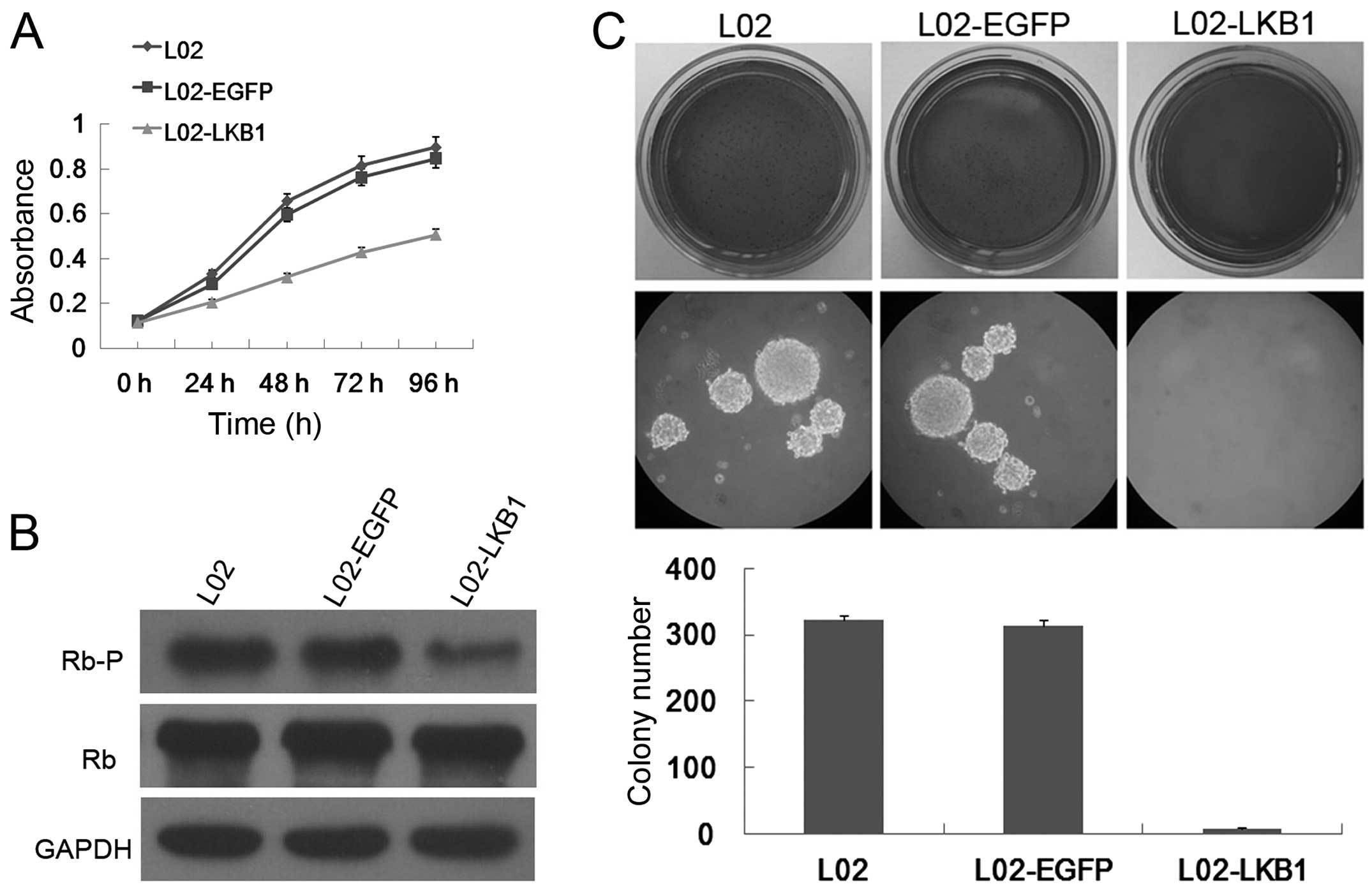
Ectopic LKB1 expression inhibits cell
proliferation of L02 cells
Downregulation of LKB1 in HeLa and G361 cells
provided a growth advantage to these cells (21,23).
To investigate this possibility in L02 cells, we performed MTS
assay, which detects the activity of mitochondrial dehydrogenase
enzymes of viable cells and is used as a measure for cell
proliferation in cell cultures (24). Indeed, growth curve experiments
clearly demonstrated that LKB1 expression conferred a proliferative
disadvantage to L02 cells when compared with the parental L02 and
mock vector-transfected L02 cells (Fig.
3A), which suggests that ectopic LKB1 expression strongly
inhibited the growth of L02 cells. No difference was noted in the
cellular growth between the parental L02 and the vector-transfected
L02 cells. Thus, introducing wild-type LKB1 into the L02 cells led
to growth suppression.
Ectopic LKB1 expression blocks Rb
phosphorylation in the L02 cells
Cell cycle machinery controls cell proliferation,
and the retinoblastoma (Rb) protein is a well-known gate keeper of
cell cycle progression (25).
Compatible with the decrease in cell proliferation rate in
vitro, stable transfection with the LKB1 plasmid, but not with
the mock vector, considerably reduced phosphorylation of Rb at
Ser807 and Ser811 in the L02 cells, with no differences observed in
the total Rb protein level (Fig.
3B). These results are consistent with our previous study
(19), and indicate that activation
of LKB1 signaling leads to hypophosphorylation of Rb, which in turn
contributes to reduced cell cycle progression and cell
proliferation in L02 cells.
Ectopic LKB1 expression rescues
anchorage-independent growth of L02 cells
Anchorage-independent growth in soft agar is one of
the hallmark characteristics of cellular transformation and
uncontrolled cell growth, with normal cells typically not capable
of growth in semi-solid matrices (26). Recent studies have provided
compelling evidence that loss of LKB1 activity is a critical step
in oncogenesis (27–30). To explore the oncogenic potential of
LKB1-deficient L02 cells, a soft agar colony formation assay was
carried out. Notably, L02 cells and the control vector
transfectants grew efficiently in soft agar and formed numerous
colonies, whereas the L02 cells with constitutive LKB1 expression
exhibited a significant reduction in anchorage-independent growth
on soft agar (Fig. 3C). These
results unambiguously suggest that L02 cells gained induced
tumorigenic phenotypes in vitro, and LKB1 expression is an
obstacle for anchorage-independent growth of L02 cells.
Ectopic LKB1 expression abrogates
subcutaneous tumor growth
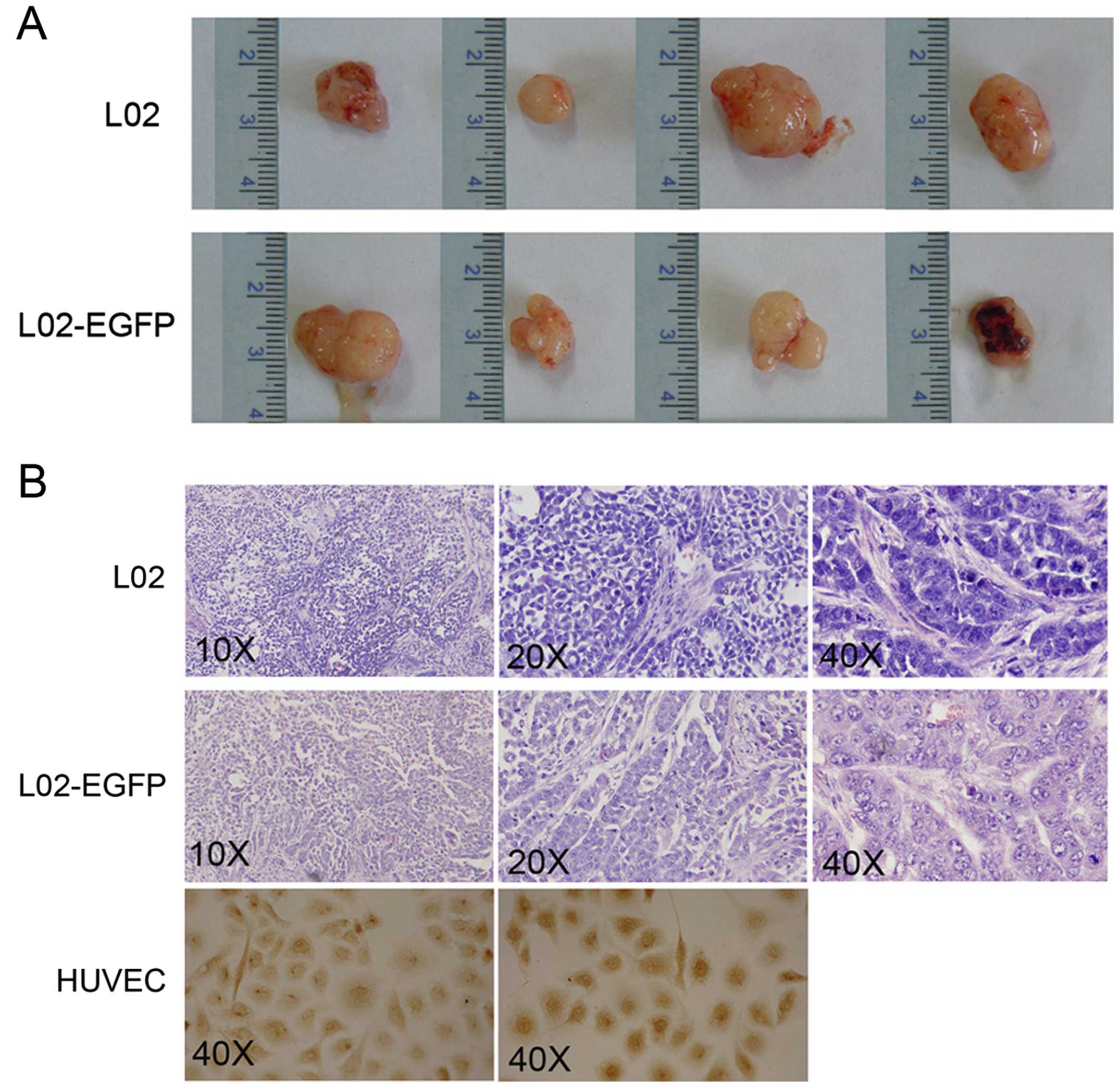
These encouraging results prompted us to further
study the potential carcinogenesis of L02 cells in vivo. A
limiting dilution tumorigenicity assay was conducted utilizing
subcutaneous injections into athymic nude mice without any protein
support. As shown in Fig. 4A, both
L02 parental cells and mock vector transfected L02 cells were
capable of developing subcutaneous tumors in all of the inoculated
nude mice. In contrast, L02 cells with stable LKB1 expression were
unable to form any tumor in the mouse model. These data
demonstrated that the L02 cell line is tumorigenic in vivo,
which could be completely abolished by sustained LKB1 expression.
No distant metastases to spleen, liver, kidneys, lungs or intestine
in the sets of mice were observed upon necropsy.
H&E staining of tumor tissues revealed a
trabecular pattern of cell growth, where tumor cells were arranged
in plates of various thickness, separated by sinusoid vascular
spaces. There was no discernable normal lobular architecture,
although vascular structures were present. Cytologic features of
malignancy were noted, with irregular nuclear contours, giant
multinucleated cells, and increased nuclear/cytoplasmic ratio
(Fig. 4B). These morphological
appearances indicated the hepatic histogenesis of the malignant
tumors in agreement with the cell lines inoculated, excluding the
formation of the primary tumors. All histologic sections were
analyzed by one of our expert pathologists.
A major question concerning the mechanism of tumor
formation of the L02 cells was whether the tumors still expressed
LKB1. Immunohistology revealed that the LKB1 protein was not
detected in the subcutaneous tumors from the nude mice (Fig. 4B), with HUVECs grown on coverslips
as a positive control. These results suggest that LKB1 inactivation
had occurred prior to tumor emergence and loss of LKB1 expression
is required as a precondition of tumor formation.
Discussion
An immortalized cell line gains the ability to
overcome cellular senescence and proliferate indefinitely, which is
acquired either through random mutation or deliberate modification
such as ectopic expression of different genes (31). The L02 cell line was generated by
Yeh et al in the early 1980's from normal adult human liver
cells and is able to be spontaneously immortalized when cultured
under certain conditions (14), but
the underlying mechanisms have not been fully described. Tumor
suppressor LKB1 is active and expressed ubiquitously in normal
adult and embryonic tissues with high expression in the liver
(32,33). Regarded as a normal hepatic cell
line, L02 cells are not believed to be an exception to the wide
spectrum of LKB1 expression. In the present study, severely reduced
LKB1 mRNA and protein expression was identified in the L02 cell
line, which was somewhat striking but may provide a plausible
explanation for the immortalization of L02 cells, since our
previous study indicated that endogenous LKB1 knockdown in normal
cells accelerates cell cycle progression by a decline in the p53
and p16 pathways (20). Our results
were substantiated by a previous study which showed that an
immortalized hepatocyte line TPH1 beared a homozygous deletion of
the LKB1 gene (34).
In the present study, we first demonstrated that
LKB1 expression was downregulated at both the mRNA and protein
levels in the L02 cell line, suggesting that LKB1 function could be
impaired at the level of transcription. There are several potential
mechanisms that may underlie the decreased LKB1 expression in L02
cells. Knusdson's two-hit model explains that tumorigenesis is
mainly caused by tumor-suppressor gene inactivation (35). Even though RT-PCR analysis did not
show sequence variants in a sizable fraction of the LKB1-coding
region, the putative mutation side may be narrowed down to the
initial 89 bases in the 5′ end region. The absence of mutations in
the present study may be due to incomplete screening of the gene,
insensitivity of our mutation-detection methods, transcript
instability as a result of the presence of a mutation, or large
genomic deletions that are undetectable by the PCR-based approach.
Therefore, beyond the scope of the present study, further research
consisting of detailed mutational analysis of LKB1 is needed to
clarify the mechanisms underlying LKB1 inactivation in L02
cells.
There is ample evidence that deregulation of the
LKB1 signaling pathway appears to play a major role in tumor
pathogenesis, since it facilitates malignant development through
suppression of cell apoptosis and growth arrest (27–30).
To investigate the oncogenic state of the L02 cell line, functional
assays were carried out including evaluation of the clonogenicity
and tumor-initiating potential. Our results indicated that L02
cells were capable of forming colonies in soft agar with high
efficiency, and developing subcutaneous tumors in immunocompromised
mice. The present study provided functional evidence for the first
time that the L02 cell line is tumorgenic in vivo and in
vitro, and illustrated a need for cautious interpretation of
previous studies in which it was assumed that L02 cells resemble
typical normal human hepatic cells.
Several studies have demonstrated that
re-introduction of LKB1 into various cancer cell lines lacking its
expression results in G1 cell cycle arrest and cell growth
suppression (21,23,36).
Our previous study also showed that ectopic LKB1 expression
activated the LKB1/AMPK signaling pathway and led to G1 arrest even
in cells with endogenous LKB1 protein in a LKB1 kinase-dependent
manner (19). In the present study,
constitutive LKB1 expression was restored in the L02 cells through
stable transfection, and led to significantly inhibited cellular
growth, hypophosphorlation of Rb, and decreased colony formation.
Subcutaneous tumorigenicity was ablated completely in nude mice by
ectopic LKB1 stable expression. These results indicate that LKB1
has growth-suppressing activity when re-introduced into L02 cells,
and has the capacity of reversing the tumorigenesis of L02 cells
in vivo and in vitro, which is in good agreement with
previous observations and further confirms the tumor-suppressor
role of LKB1 in cell proliferation.
Hepatocellular carcinoma (HCC) is ranked as the
fifth most common human cancer, and the third leading cause of
cancer-related death worldwide, with over 500,000 new cases
annually diagnosed resulting in >90% mortality (37). HCC is a very heterogeneous tumor
type whose precise molecular mechanisms still await further
studies. Notably, Nakau et al reported a high HCC prevalence
in LKB1 (+/−) mice (38),
suggestive of a strong LKB1 involvement in liver tumorigenesis. We
demonstrated that the L02 cells possessed the capability to
initiate tumor formation in immunocompromised mice after
subcutaneous inoculation, and histology of the tumor tissues was
markedly similar to the representative histopathology of human
HCCs, including malignant cellular growth in trabecular patterns
recapitulating liver cords (39).
No expression of LKB1 protein was observed in these subcutaneous
tumors, indicating that complete loss of LKB1 expression was
required for the development of the carcinomas. The present study
provides the first evidence that the L02 cell line is capable of
initiating tumors which share histological features with human HCC.
Thus, the L02 cell line may be an important and valuable model to
better define the cellular and molecular mechanisms of liver
transformation, and to study the mechanisms by which LKB1 exerts
its growth-suppressive effect. However, the present study only
crudely described the sequential cues that transform L02 cells into
malignant tumors. Future research will be required to characterize
in greater detail the underlying molecular mechanisms.
In summary, the results of the present study
indicate that endogenous LKB1 expression is severely impaired in
the L02 cell line, which, in turn, confers accelerated cell growth
and malignant transformation of L02 cells, and ectopic LKB1
expression antagonized the tumorigenic properties of the L02
cells.
Abbreviations:
|
LKB1
|
liver kinase B1
|
|
STK11
|
serine/threonine kinase 11
|
|
PJS
|
Peutz-Jeghers syndrome
|
|
HEK-293T cells
|
human embryonic kidney 293T cells
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
DIG
|
digoxigenin
|
|
RT-PCR
|
reverse transcription PCR
|
|
H&E
|
hematoxylin and eosin
|
|
HCC
|
hepatocellular carcinoma
|
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81201544).
References
|
1
|
McGarrity TJ and Amos C: Peutz-Jeghers
syndrome: Clinicopathology and molecular alterations. Cell Mol Life
Sci. 63:2135–2144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rustgi AK: The genetics of hereditary
colon cancer. Genes Dev. 21:2525–2538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hemminki A, Markie D, Tomlinson I,
Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M,
Höglund P, et al: A serine/threonine kinase gene defective in
Peutz-Jeghers syndrome. Nature. 391:184–187. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hearle N, Schumacher V, Menko FH,
Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott
RJ, Lim W, et al: Frequency and spectrum of cancers in the
Peutz-Jeghers syndrome. Clin Cancer Res. 12:3209–3215. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ji H, Ramsey MR, Hayes DN, Fan C, Mcnamara
K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al:
LKB1 modulates lung cancer differentiation and metastasis. Nature.
448:807–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou J, Huang W, Tao R, Ibaragi S, Lan F,
Ido Y, Wu X, Alekseyev YO, Lenburg ME, Hu GF, et al: Inactivation
of AMPK alters gene expression and promotes growth of prostate
cancer cells. Oncogene 2. 8:1993–2002. 2009. View Article : Google Scholar
|
|
7
|
Fenton H, Carlile B, Montgomery EA,
Carraway H, Herman J, Sahin F, Su GH and Argani P: LKB1 protein
expression in human breast cancer. Appl Immunohistochem Mol
Morphol. 14:146–153. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sanchez-Cespedes M: A role for LKB1 gene
in human cancer beyond the Peutz-Jeghers syndrome. Oncogene.
26:7825–7832. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ylikorkala A, Rossi DJ, Korsisaari N,
Luukko K, Alitalo K, Henkemeyer M and Mäkelä TP: Vascular
abnormalities and deregulation of VEGF in Lkb1-deficient mice.
Science. 293:1323–1326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jishage K, Nezu J, Kawase Y, Iwata T,
Watanabe M, Miyoshi A, Ose A, Habu K, Kake T, Kamada N, et al: Role
of Lkb1, the causative gene of Peutz-Jegher's syndrome, in
embryogenesis and polyposis. Proc Natl Acad Sci USA. 99:8903–8908.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Williams T and Brenman JE: LKB1 and AMPK
in cell polarity and division. Trends Cell Biol. 18:193–198. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Partanen JI, Tervonen TA, Myllynen M, Lind
E, Imai M, Katajisto P, Dijkgraaf GJ, Kovanen PE, Mäkelä TP, Werb
Z, et al: Tumor suppressor function of Liver kinase B1 (Lkb1) is
linked to regulation of epithelial integrity. Proc Natl Acad Sci
USA. 109:E388–E397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yeh HJ, Chu TH and Shen TW: Ultrastructure
of continuously cultured adult human liver cells. Acta Biol Exp
Sinica. 14:361–365. 1980.
|
|
15
|
Li CY, Cao CZ, Xu WX, Cao MM, Yang F, Dong
L, Yu M, Zhan YQ, Gao YB, Li W, et al: Recombinant human hepassocin
stimulates proliferation of hepatocytes in vivo and improves
survival in rats with fulminant hepatic failure. Gut. 59:817–826.
2010. View Article : Google Scholar
|
|
16
|
Tao YM, Huang JL, Zeng S, Zhang S, Fan XG,
Wang ZM, Yang HX, Yuan XH, Wang P, Wu F, et al: BTB/POZ
domain-containing protein 7: Epithelial-mesenchymal transition
promoter and prognostic biomarker of hepatocellular carcinoma.
Hepatology. 57:2326–2337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wan X, Xu C, Lin Y, Lu C, Li D, Sang J, He
H, Liu X, Li Y and Yu C: Uric acid regulates hepatic steatosis and
insulin resistance through the NLRP3 inflammasome-dependent
mechanism. J Hepatol. 64:925–932. 2016. View Article : Google Scholar
|
|
18
|
Qiu X, Dong S, Qiao F, Lu S, Song Y, Lao
Y, Li Y, Zeng T, Hu J, Zhang L, et al: HBx-mediated miR-21
upregulation represses tumor-suppressor function of PDCD4 in
hepatocellular carcinoma. Oncogene. 32:3296–3305. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liang X, Wang P, Gao Q and Tao X:
Exogenous activation of LKB1/AMPK signaling induces G1
arrest in cells with endogenous LKB1 expression. Mol Med Rep.
9:1019–1024. 2014.PubMed/NCBI
|
|
20
|
Liang X, Wang P, Gao Q, Xiang T and Tao X:
Endogenous LKB1 knockdown accelerates G1/S transition
through p53 and p16 pathways. Cancer Biol Ther. 9:156–160. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tiainen M, Ylikorkala A and Mäkelä TP:
Growth suppression by Lkb1 is mediated by a G1 cell
cycle arrest. Proc Natl Acad Sci USA. 96:9248–9251. 1999.
View Article : Google Scholar
|
|
22
|
Lee SM, Choi JE, Na YK, Lee EJ, Lee WK,
Choi YY, Yoon GS, Jeon HS, Kim DS and Park JY: Genetic and
epigenetic alterations of the LKB1 gene and their associations with
mutations in TP53 and EGFR pathway genes in Korean non-small cell
lung cancers. Lung Cancer. 81:194–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tiainen M, Vaahtomeri K, Ylikorkala A and
Mäkelä TP: Growth arrest by the LKB1 tumor suppressor: Induction of
p21WAF1/CIP1. Hum Mol Genet. 11:1497–1504. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gopinath P and Ghosh SS: Apoptotic
induction with bifunctional E. coli cytosine deaminase-uracil
phosphoribosyltransferase mediated suicide gene therapy is
synergized by curcumin treatment in vitro. Mol Biotechnol.
39:39–48. 2008. View Article : Google Scholar
|
|
25
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee CJ, Jang JH, Lee JY, Lee MH, Li Y, Ryu
HW, Choi KI, Dong Z, Lee HS, Oh SR, et al: Aschantin targeting on
the kinase domain of mammalian target of rapamycin suppresses
epidermal growth factor-induced neoplastic cell transformation.
Carcinogenesis. 36:1223–1234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
George SH, Milea A, Sowamber R, Chehade R,
Tone A and Shaw PA: Loss of LKB1 and p53 synergizes to alter
fallopian tube epithelial phenotype and high-grade serous
tumorigenesis. Oncogene. 35:59–68. 2016. View Article : Google Scholar
|
|
28
|
Okon IS, Coughlan KA, Zhang C, Moriasi C,
Ding Y, Song P, Zhang W, Li G and Zou MH: Protein kinase LKB1
promotes RAB7-mediated neuropilin-1 degradation to inhibit
angiogenesis. J Clin Invest. 124:4590–4602. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao Y, Zhang W, Han X, Li F, Wang X, Wang
R, Fang Z, Tong X, Yao S, Li F, et al: YAP inhibits squamous
transdifferentiation of Lkb1-deficient lung adenocarcinoma through
ZEB2-dependent DNp63 repression. Nat Commun. 5:46292014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanwar PS, Mohapatra G, Chiang S, Engler
DA, Zhang L, Kaneko-Tarui T, Ohguchi Y, Birrer MJ and Teixeira JM:
Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface
epithelium induces papillary serous ovarian cancer. Carcinogenesis.
35:546–553. 2014. View Article : Google Scholar :
|
|
31
|
Kavsan VM, Iershov AV and Balynska OV:
Immortalized cells and one oncogene in malignant transformation:
Old insights on new explanation. BMC Cell Biol. 12:232011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sebbagh M, Olschwang S, Santoni M-J and
Borg J-P: The LKB1 complex-AMPK pathway: The tree that hides the
forest. Fam Cancer. 10:415–424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alessi DR, Sakamoto K and Bayascas JR:
LKB1-dependent signaling pathways. Annu Rev Biochem. 75:137–163.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pineau P, Marchio A, Nagamori S, Seki S,
Tiollais P and Dejean A: Homozygous deletion scanning in
hepatobiliary tumor cell lines reveals alternative pathways for
liver carcinogenesis. Hepatology. 37:852–861. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Berger AH, Knudson AG and Pandolfi PP: A
continuum model for tumour suppression. Nature. 476:163–169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Qiu W, Schönleben F, Thaker HM, Goggins M
and Su GH: A novel mutation of STK11/LKB1 gene leads to the loss of
cell growth inhibition in head and neck squamous cell carcinoma.
Oncogene. 25:2937–2942. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gomaa AI, Khan SA, Toledano MB, Waked I
and Taylor-Robinson SD: Hepatocellular carcinoma: Epidemiology,
risk factors and pathogenesis. World J Gastroenterol. 14:4300–4308.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakau M, Miyoshi H, Seldin MF, Imamura M,
Oshima M and Taketo MM: Hepatocellular carcinoma caused by loss of
heterozygosity in Lkb1 gene knockout mice. Cancer Res.
62:4549–4553. 2002.PubMed/NCBI
|
|
39
|
Jain D: Tissue diagnosis of hepatocellular
carcinoma. J Clin Exp Hepatol. 4(Suppl 3): S67–S73. 2014.
View Article : Google Scholar
|