Introduction
Non-small cell lung cancer (NSCLC) is one of the
most common cancers in many countries, and treatment outcomes for
NSCLC patients are still disappointing. Most patients receiving
chemotherapy do not respond, resulting in disease progression.
Thus, inherent or acquired drug-resistance leads to treatment
failure. In recent years, progress in chemotherapy and molecular
target-based therapy have altered the standard therapy for NSCLC
(1–3). Gene therapy has become an attractive
regimen, in addition to conventional therapy (4). Adenovirus is a useful agent for cancer
gene therapy due to its high potential for gene transfer, ease of
high titer production and demonstrated safety in clinical trials
(5). Thus, recombinant adenovirus
vectors are widely used in preclinical and clinical gene therapy,
particularly in NSCLC gene therapy (6). Moreover, results have suggested
synergism between chemotherapy and adenovirus p53 gene therapy with
no increased side effects (7).
However, in a multicenter phase II study, intratumoral adenoviral
p53 gene therapy appears to provide no additional benefit in
patients receiving an effective first line chemotherapy for
advanced NSCLC (8).
The combination of adenovirus with its receptor is a
key step for virus infection and sequent biological effect.
Adenovirus can infect cells since it uses the knob domain of the
fiber binding to its cellular receptor, the coxsackie and
adenovirus receptor (9). Several
lines of evidence showed the relationships between CAR expression
and adenovirus infection (10,11),
and low levels of CAR in tumors are thought to be one of the
reasons for poor adenovirus infection (12–14).
It has become evident that CAR expression is often low in various
types of tumors, and some biological or chemical agents could
result in an increase in CAR expression in several tumor cell
lines, making them more susceptible to adenovirus infectivity
(15–17).
Adenoviral vector-mediated gene transfer is highly
effective, but large differences regarding transduction
efficiencies among different cell lines and between in vitro
and in vivo gene transfer have been reported. To evaluate
the potential of adenovirus vector-based gene therapy on
cisplatin-resistant lung cancer, the present study examined the
adenovirus infection rates and CAR expressions in A549 and its
cisplatin-resistant subline A549/DDP.
Materials and methods
Cells culture, antibodies and
chemicals
The human lung adenocarcinoma cell lines A549 and
its cisplatin-resistant subline A549/DDP, were propagated in
monolayer cultures in RPMI-1640 medium supplemented with 10% fetal
bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin.
The transformed embryonic kidney cell line 293 was grown in
Dulbecco's modified Eagle's medium (DMEM) containing high glucose
(4.5 g/l), 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin.
Cells were grown at 37°C in a 5% CO2 humidified
incubator. Antibodies were commercially available, including mouse
monoclonal antibody against CAR (Upstate Biotechnology,
Charlottesville, VA, USA), rabbit polyclonal antibody against CAR,
mouse monoclonal antibody against p53, mouse monoclonal antibody
against p21, mouse monoclonal antibody against α-tubulin and mouse
monoclonal antibody conjugated with HRP against actin (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). Histone deacetylase inhibitor
trichostatin A (TSA) and proteasome inhibitor MG-132 was purchased
from Sigma Chemicals (St. Louis, MO, USA). Each agent was prepared
as a stock solution in dimethyl sulphoxide (DMSO) medium such that
the final concentration of DMSO exposed to cells was <0.1%. In
all experiments, control cells were treated with 0.1% DMSO. Each
experiment was repeated at least three times.
MTT assay
Cells were plated into 96-well tissue culture plates
overnight. Then, cells were treated with cisplatin, MG-132 or TSA
at the indicated concentrations, respectively, for 72 h. Four hours
before the end point, a medium containing 0.5 mg/ml MTT was added.
Finally DMSO was added to each well and mixed thoroughly to
dissolve the crystals of MTT formazan. Results were quantified
using a LabSystems Multiskan MS at 540 nm wavelength. Control
absorbance was designated as 100%, and cell survival was expressed
as a percentage of control absorbance.
FACS assay
The effect of MG-132 and TSA on CAR or GFP
expression in cell lines was examined with FACS (Becton-Dickinson,
Franklin Lakes, NJ, USA). Cells were incubated with MG-132 or TSA
for the indicated hours. Before analysis, cells were washed,
trypsinized and resuspended in phosphate-buffered saline (PBS).
Cells (2×105) were incubated with antibody against CAR
at a 1:200 dilution for 1 h at 4°C, followed by FITC-labeled goat
anti-mouse IgG for 30 min at 4°C. After fixation, 3×104
cells were analyzed by FACS. Analysis was performed using LYSYS II
software (Becton-Dickinson). To analyze the GFP expression, cells
were trypsinized, washed and fixed for analysis.
Transfer assay
Recombinant adenovirus encoding enhanced green
fluorescent protein (rAd.EGFP) was purchased from Sino-Gene
(Shanghai, China). The cytomegalovirus promoter was used to drive
the transcription of EGFP. Titers of recombinant viruses were
determined by plaque forming assay in the 293 cells. All vectors
were prepared, purified and stored at −80°C.
The rAd.EGFP transfer assay for the potential of
adenovirus transfer detected with a fluorescence microscope and
FACS. Cells (5×104) were plated into 24-well plates
overnight at 37°C. Next day, fresh medium or fresh medium
containing MG-132 or TSA cells were replaced. Twenty-four hours
later, medium was removed and cells were washed once with PBS.
Then, cells were infected with rAd.EGFP at indicated MOI for 2 h.
Subsequently, cells were washed and overlaid with growth medium for
the indicated time periods.
For fluorescence microscope analysis, a Leica
fluorescence stereo microscope equipped with a 50 W mercury lamp
was used. Selective excitation of GFP was produced through a
D425/60 bandpass filter. Emitted fluorescence was collected through
a long pass filter on a Hamamatsu cooled charged coupled device
camera. FACS analysis and MTT assay were as mentioned above.
Western blot analysis
Cell lysates were prepared by SDS lysis buffer.
Protein concentration was measured using the BCA protein assay kit
(Pierce, Rockford, IL, USA). Equal amount of protein was separated
by electrophoresis on a 10.5% SDS polyacrylamide gel. The proteins
were electrotransferred from gel to nitrocellulose membrane. The
membrane was blocked with 5% dry milk solution for 30 min, and then
incubated with primary antibody for 2 h at room temperature. After
washed, the membrane was incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies. Finally, membrane was
detected with the enhanced chemiluminescence (ECL) detection system
(Amersham Biosciences Europe, Freiburg, Germany) according to the
manufacturer's instructions.
RT-PCR analysis
Total RNAs were isolated from the cells using TRIzol
procedure (Invitrogen Life Technologies, Carlsbad, CA, USA). cDNA
was synthesized from 4 µg of total RNA with SuperScript RT
kit (Invitrogen Life Technologies). The reverse transcription PCR
exponential phase was determined from 18 to 36 cycles to allow
semi-quantitative comparisons among cDNAs developed from identical
reactions. Each PCR regime involved an initial denaturation at 94°C
for 5 min followed by cycles predetermined for each type of cDNA:
28 cycles for GAPDH and 30 cycles for CAR. Primer sequences were
designed as follows: sense primer, 5′-ATAAAGCCACCACCGCCACC-3′ and
antisense primer, 5′-TGGTCACAGCTTTCGCAGCC-3′ for human GAPDH
(internal control); sense primer, 5′-AGCCTTCAGGTGCGAGATGTTACG-3′
and antisense primer, 5′-TACGACAGCAAAAGATGATAAGAC-3′ for human CAR.
The thermal cycle was defined by denaturation at 94°C for 5 min,
followed by indicated cycles with denaturation at 94°C for 30 sec,
annealing at 60°C for 60 sec and extension at 72°C for 45 sec, and
then a final elongation at 72°C for 5 min. The PCR products were
analyzed on 1% agarose gels and visualized by ethidium bromide
staining.
Results
Cytotoxicity studies
Cell lines were plated at 7,000 cells/well into
96-well plates. Cisplatin was added the next day, and the plates
were assayed after 72 h. The analysis was performed by the MTT
assay for the cisplatin-resistant subline cells and its parental
cells. The results of MTT assays showed that the IC50
(inhibitory concentration 50%) of cisplatin to A549 is 1.47
µg/ml, and the IC50 of cisplatin to A549/DDP is
15.11 µg/ml. Based on these results, we recorded that the
resistant factor of A549/DDP was 10.28, and the model of
cisplatin-resistant subline was established.
CAR expressions and rAd.EGFP infection
rates of A549 and A549/DDP
A549 and A549/DDP cells were infected with rAd. EGFP
at different MOIs, and GFP expression was analyzed in the two cell
lines. Compared with A549, A549/DDP showed resistance to adenovirus
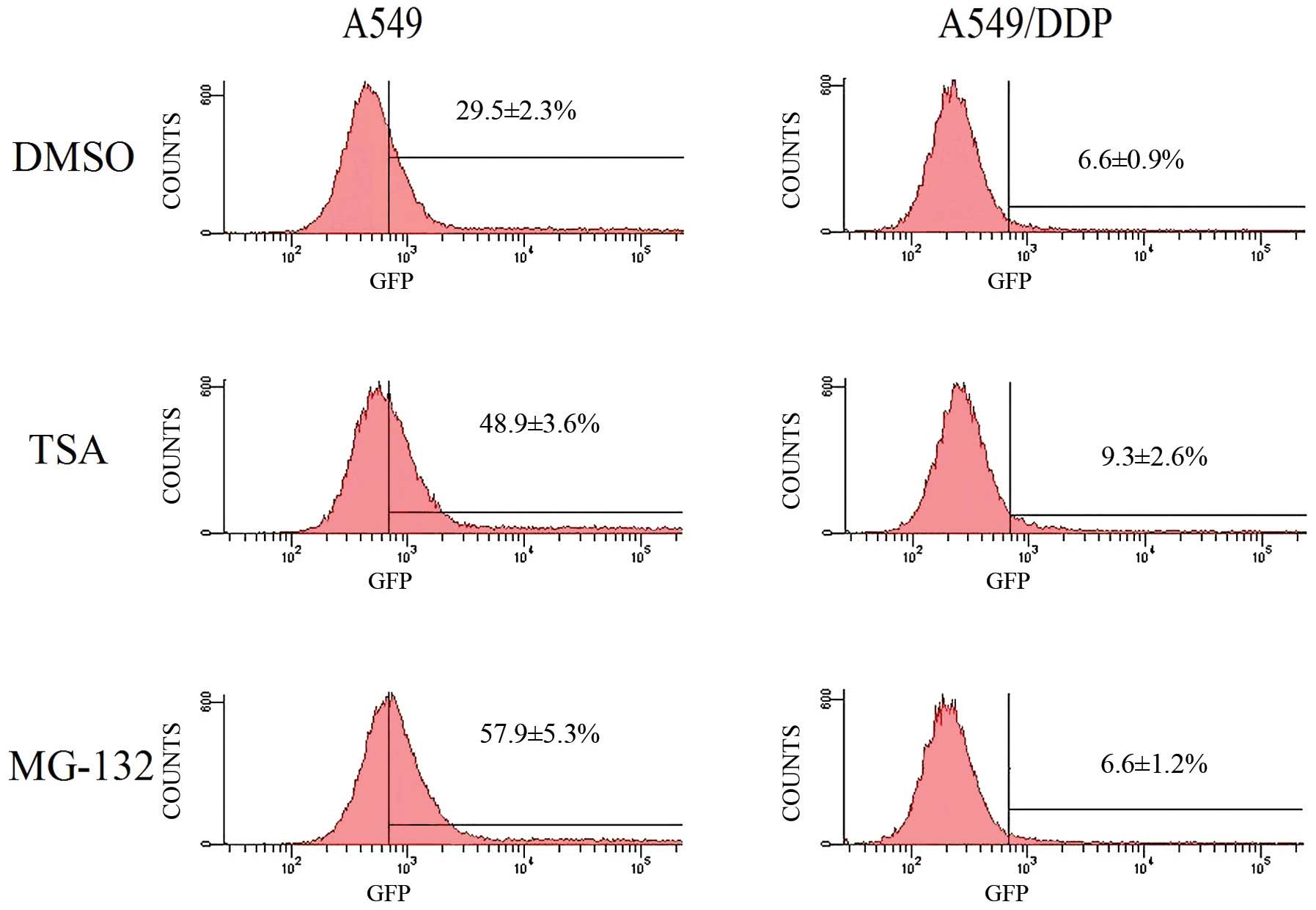
infection. For example, with 2 MOI of rAd.EGFP, transfer rate of
adenovirus was (29.5±2.3%) in A549 and (6.6±0.9%) in A549/DDP cells
(Figs. 1 and 2). The difference was significant in
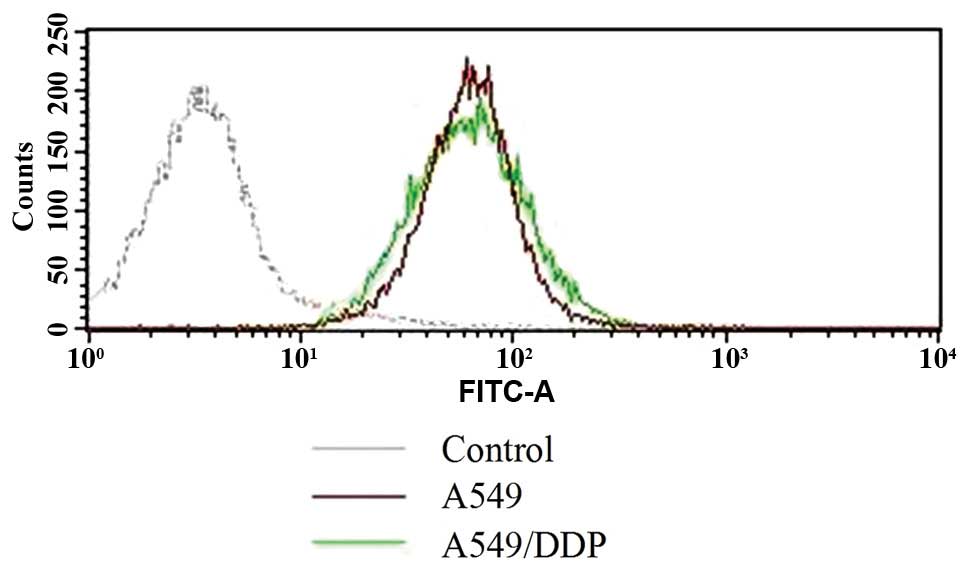
statistical analysis (p=0.004). Nevertheless, FACS analysis of the
two cancer cell lines indicated that CAR expression levels of A549
and A549/DDP cells were not different (Fig. 3).
Induction of CAR expression with MG-132
and TSA
FACS analysis showed that both A549 and A549/DDP
cells were CAR-positive cell lines. Thus, we further studied
whether the CAR expression could be affected by drugs, such as
MG-132 or TSA. Cytotoxicity studies were performed to determine the
minimally cytotoxic concentration of MG-132 or TSA for the two cell
lines. For A549 cells, the MG-132 concentration, showing no or
minimal cytotoxicity that was selected for these studies were 0.7
µmol/l, and the TSA concentration was 30 ng/ml. Whereas, the
concentrations of MG-132 for A549/DDP cells was 0.5 µmol/l,
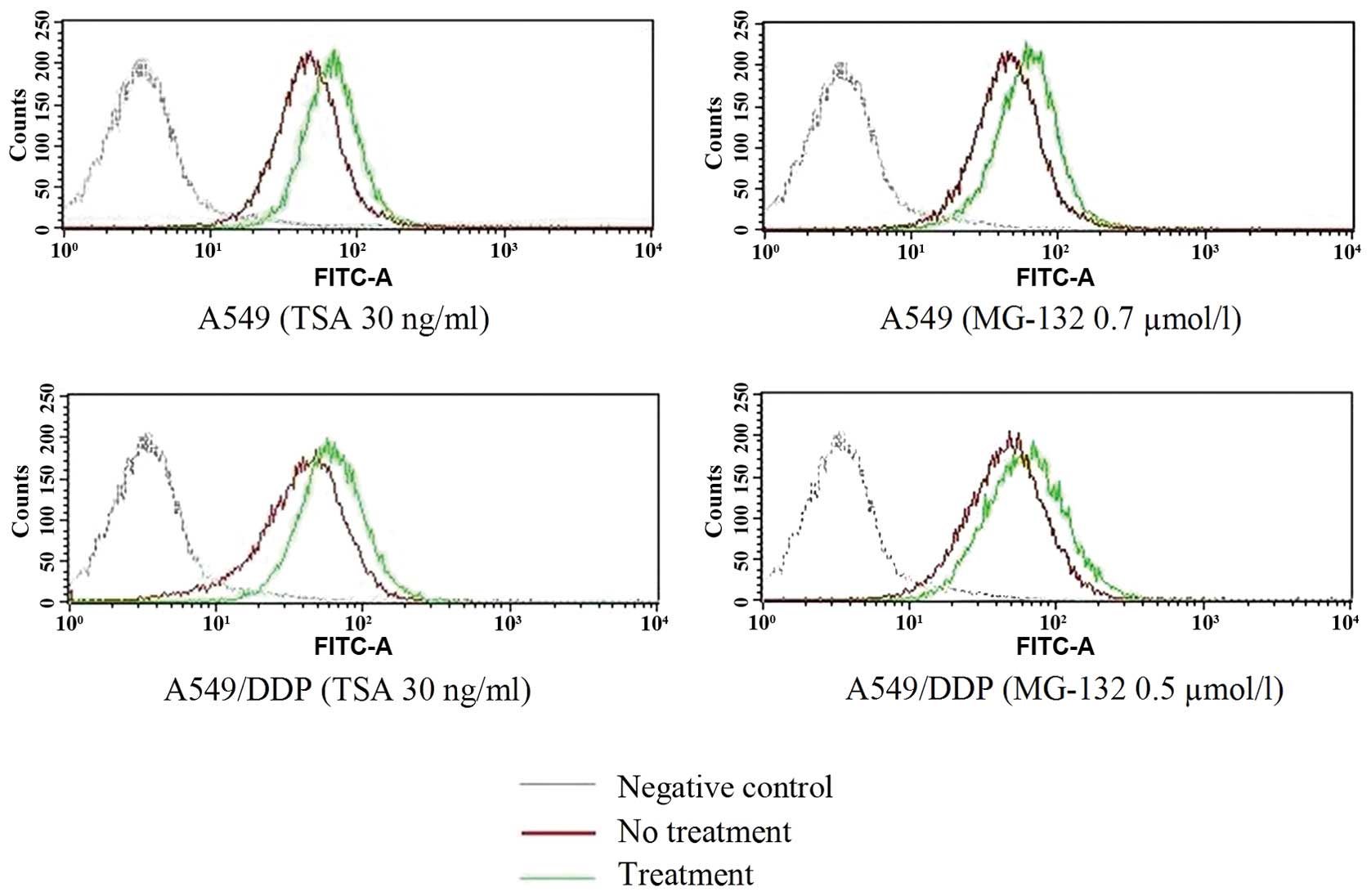
and the TSA concentration was also 30 ng/ml. When A549 cells were
incubated with 0.7 µmol/l MG-132 or 30 ng/ml TSA for 48 h,
average CAR density was apparently increased, and the same as
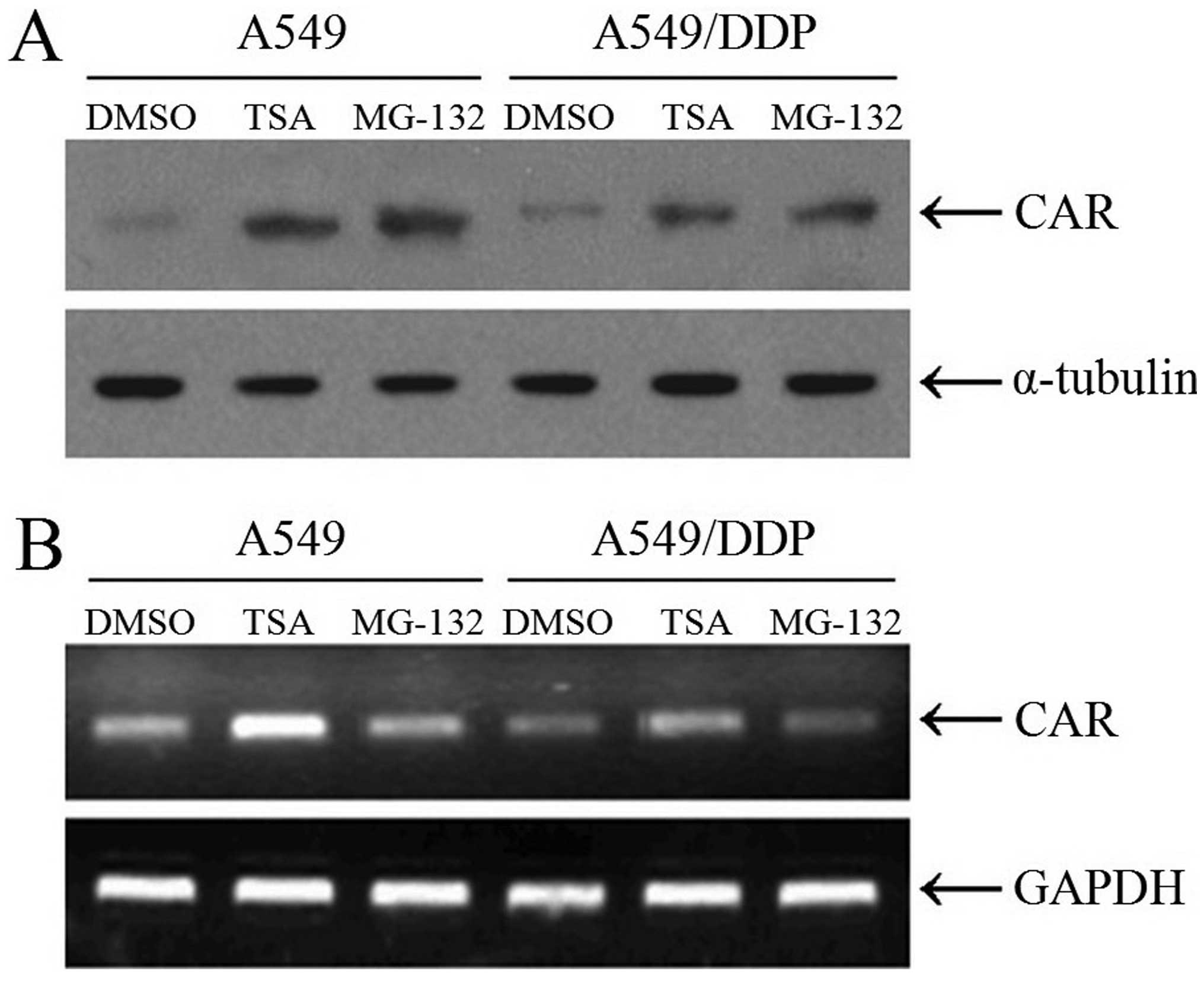
A549/DDP cells incubated with MG-132 or TSA (Fig. 4).
We further confirmed the CAR expression levels in
A549 and A549/DDP cells with western blotting and RT-PCR analysis.
The results showed that TSA rather than MG-132 enhanced the CAR
mRNA transcription, which indicated that MG-132 modified the CAR
expression with post-transcriptional mechanism and TSA with
transcriptional mechanism, consist with the fact that MG-132 is a
proteasome inhibitor and TSA a histone deacetylase inhibitor
(Fig. 5).
Adenovirus infection affected by MG-132
and TSA
To evaluate the effect of MG-132 or TSA on the
adenovirus infectious efficiency, GFP density was compared. Treated
with MG-132 or TSA at the indicated concentration for 48 h, A549 or
A549/DDP cells were infected with rAd.EGFP at multiplicities of
infection (MOI) of 0, 0.5, 1, 2, 4 or 8, and incubated in normal
medium for another 24 h when analyses was performed. At the
indicated time, adenovirus infected cells were photographed under a
fluorescent microscope. Then, the infected cells were trypsinized,
and further analyzed for GFP expression by flow cytometry.
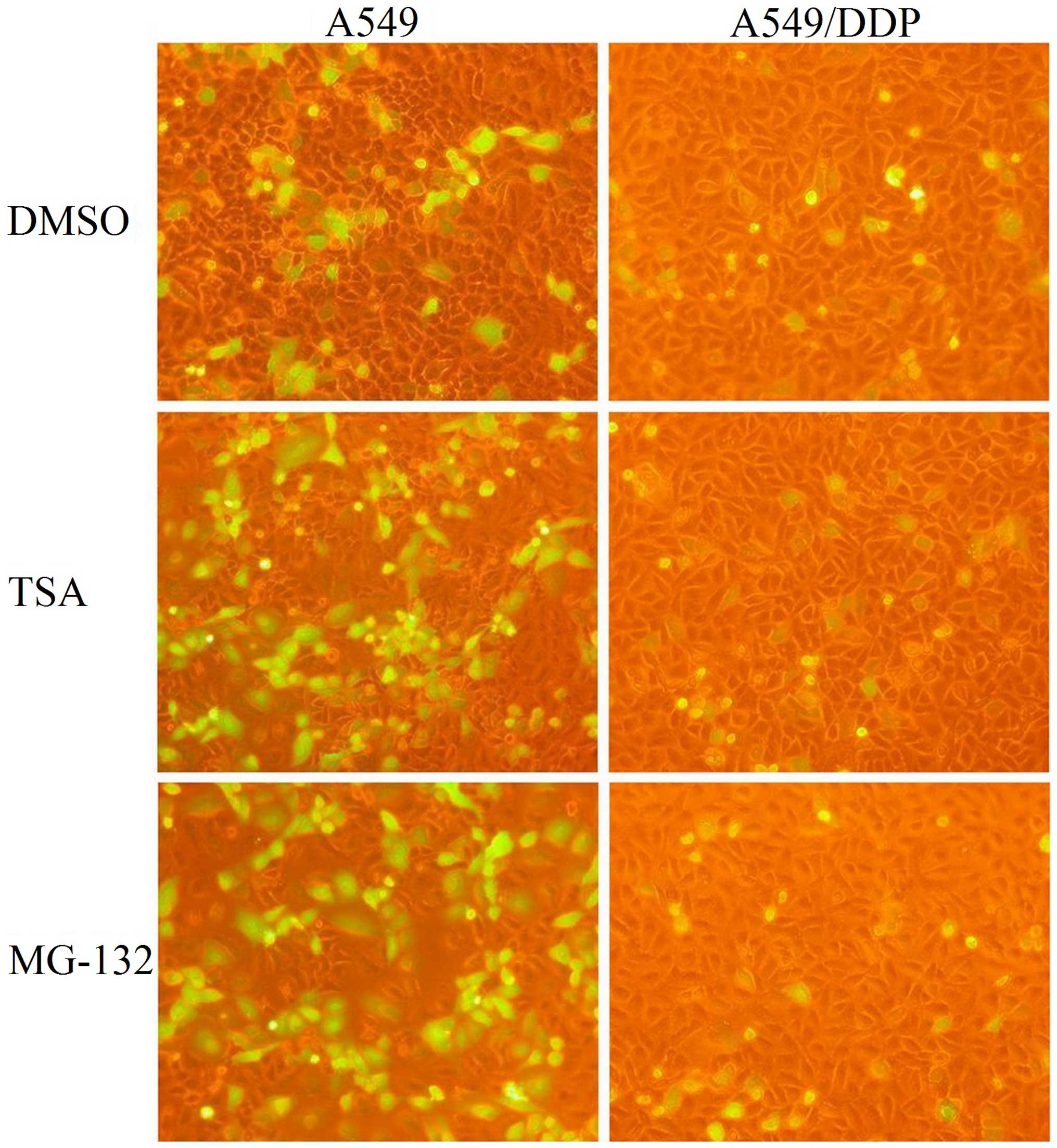
After MG-132 or TSA treatment, a marked increase in
GFP expression of A549 cells occurred with every multiplicity of
rAd.EGFP, but the GFP expression was not different in A549/DDP
cells after drugs treatments (Fig.
1). For example, with 2 MOI of rAd.EGFP, transfer rate of
adenovirus in A549 cells was (29.5±2.3%) in control group
(57.9±5.3%) in MG-132 treated group and (48.9±3.6%) in TSA-treated
group. In A549/DDP cells, transfer rate of adenovirus was
(6.6±0.9%) in control group, (6.6±1.2%) in MG-132-treated group and
(9.3±2.6%) in TSA-treated group (Fig.
2).
Discussion
Previously, various studies have suggested that
adenoviral vector-mediated gene therapy is more effective in the
drug resistant breast or bladder cancer cell lines compared to the
parental cell line, which may partially be explained by the
efficiency of adenoviral gene transfer (18,19).
In the present study, we employed a human lung cancer cell line and
its drug resistant sublines to study adenovirus infection
sensibility of the two cell lines. The results shown that rAd.EGFP
induced higher GFP gene expression in the parental cell line
compared to the drug-resistant cell line, indicating greater
efficiency of adenovirus-mediated gene transfer in the parental
cell lines compared to the drug-resistant line.
Although patients with advanced non-small cell lung
cancer (NSCLC) can be effectively treated initially with
combination chemotherapy, many patients receiving chemotherapy do
not respond, resulting in disease progression. Furthermore, second
line chemotherapy provides little benefit to patients who have
relapsed after an initial response. Thus, inherent or acquired
drug-resistance leads to treatment failure. Much is known
concerning the molecular mechanisms by which tumor cells acquire
drug-resistance, but the treatment of drug-resistant tumors remains
a significant problem. Gene therapy has become an attractive
regimen, in addition to conventional therapy, and recombinant
adenovirus vectors are widely used in preclinical and clinical gene
therapy (4). The p53 gene is
abnormal in 40–74% of NSCLC samples tested (20). Thus, therapeutic approaches
involving gene therapy targeting the p53 gene have been explored in
preclinical models and clinical trials.
The recombinant adenovirus encoding human p53 tumor
suppressor gene (rAd.p53) phase I single agent trials in NSCLC
demonstrated effective p53 gene transfer, minimal toxicity and
transient injected lesion tumor regression (21). Preclinical studies had previously
demonstrated synergistic antitumor effect between adenovirus gene
therapy and radiotherapy or chemotherapy, which may lead to
enhanced antitumoral activity without increased toxicity (22,23).
Combination of p53 gene therapy with radiotherapy has been
evaluated in NSCLC patients who were not eligible for
chemoradiotherapy or surgery (24).
Radiation toxicity was not enhanced by adenoviral vector.
Intratumoral injection of rAd. p53 in combination with radiation
therapy is well tolerated and demonstrates evidence of tumor
regression at the primary injected tumor. Since chemotherapy could
not be administered to these high risk patients, systemic control
of disease was poor, with over 50% of patients experiencing
metastatic progression within 1 year. The activity and toxicity of
rAd.p53 combined with platinum-based chemotherapy were also
evaluated in advanced NSCLC. There was no evidence of increased
chemotherapy-related toxicity by adenoviral vector, and some
evidence of clinical activity could be observed (7). However, in a multicenter phase II
study, intratumoral adenoviral p53 gene therapy appears to provide
no additional benefit in patients receiving an effective first line
chemotherapy for advanced NSCLC (8). One possible explanation for this
observation may be the relatively high efficacy of the first line
chemotherapy, and gene therapy should be used in a coordinated
fashion in the proper clinical context.
Adenovirus can infect cells since it uses the knob
domain of the fiber to bind to its cellular receptor, the coxsackie
and adenovirus receptor (CAR), and the efficiency of adenoviral
gene transfer is critical for adenoviral vector-based gene therapy
(10). The efficiency of adenoviral
gene transfer to several tumors, including lung cancer, correlates
with the expression of CAR, and low levels of CAR in tumors are
thought to be one of the reasons for poor adenovirus infection
(12-14). We analyzed CAR expressions in the
two cell lines, but no significant differences of CAR expression
were observed between the A549/DDP and its parental cell line,
suggesting that the differences in gene transfer efficiency between
the drug resistant cell lines and the parental cell line may be
independent of CAR expression. Despite the known importance of CAR
for successful transduction of cells, other unknown mechanism of
cell virus interaction may also be important. Variable CAR
expressions in tumors may interfere with the interpretation of
results of clinical trials, but it is too early to determine
whether expressions of CAR in tumor cells are appropriate
additional criteria for the enrollment of patients in adenovirus
gene therapy trials (25).
It is demonstrated that CAR expression could be
induced with biological or chemical agents, which would lead to
increased adenovirus-mediated transgene expression (15). Histone acetylation inhibitors could
restore the gene expression of the tumor-associated genes that have
been transcriptionally silenced by promoter associated histone
deacetylation, and use of certain histone deacetylase inhibitors,
such as FR901228 (26–28), trichostatin A (29,30),
CHAP31, FK228 (31,32), SAHA, MS275 and LBH589 (33–35),
resulted in an increase in CAR expression in several tumor cell
lines, making them more susceptible to adenovirus infectivity.
Other agents, such as chemotherapeutics (15), cytokines (36) and inhibitors of the Raf/MEK/ERK
pathway (37) were also reported to
have the ability to induce CAR expression in some tumor cell lines.
Based on the above experiments, we further modified the CAR
expression in the two cell lines with proteasome inhibitors MG-132
or histone deacetylase inhibitors TSA, and the results indicated
that the CAR expression in both A549 and A549/DDP could be
upregulated. In the parental cell lines, upregulated CAR expression
with MG-132 or TSA brought about higher GFP gene expression after
rAd.EGFP infection, but the upregulated CAR expression in the
drug-resistant cell lines had no help in GFP gene expression, which
also certifies that other unknown mechanism of cell virus
interaction beside CAR expression may also be important for the
gene transfer efficiency.
Though the CAR expression in A549/DDP cells is no
less than its parental cells, the cisplatin-resistant subline shows
obvious rejection of the adenovirus infection, which implies that
the rejection of adenovirus infection is independent of CAR
expression in A549/DDP cells. The mechanism of high efficiency of
adenoviral gene transfer in the parental cells is unclear, thus
needing further investigation. Inherent or acquired drug-resistance
is one of the reasons for antitumor chemotherapy treatment failure.
Considering that patients enrolled in adenoviral cancer gene
therapy trials usually have received chemotherapy or radiotherapy,
the tumor cells in those people usually have multiple
drug-resistance (MDR) to a certain degree. After MDR has taken
place, the impact of MDR on gene therapy is unknown, and requires
investigation.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81272341).
Abbreviations:
|
CAR
|
coxsackie and adenovirus receptor
|
|
rAd.EGFP
|
recombinant adenovirus encoding
enhanced green fluorescent protein
|
|
MOI
|
multiplicities of infection
|
References
|
1
|
Lu S, Yu Y, Chen Z, Ye X, Li Z and Niu X:
Maintenance therapy improves survival outcomes in patients with
advanced non-small cell lung cancer: A meta-analysis of 14 studies.
Lung. 193:805–814. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Casadio C, Guarize J, Donghi S, Di Tonno
C, Fumagalli C, Vacirca D, Dell'Orto P, De Marinis F, Spaggiari L,
Viale G, et al: Molecular testing for targeted therapy in advanced
non-small cell lung cancer: Suitability of endobronchial ultrasound
trans-bronchial needle aspiration. Am J Clin Pathol. 144:629–634.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zarogoulidis P, Domvri K, Huang H and
Zarogoulidis K: Gene therapy for lung cancer malignant pleural
effusion: Current and future nano-biotechnology. Transl Lung Cancer
Res. 1:234–237. 2012.PubMed/NCBI
|
|
4
|
Cavazzana-Calvo M, Thrasher A and Mavilio
F: The future of gene therapy. Nature. 427:779–781. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma A, Tandon M, Bangari DS and Mittal
SK: Adenoviral vector-based strategies for cancer therapy. Curr
Drug Ther. 4:117–138. 2009. View Article : Google Scholar :
|
|
6
|
Toloza EM, Morse MA and Lyerly HK: Gene
therapy for lung cancer. J Cell Biochem. 99:1–22. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nemunaitis J, Swisher SG, Timmons T,
Connors D, Mack M, Doerksen L, Weill D, Wait J, Lawrence DD, Kemp
BL, et al: Adenovirus-mediated p53 gene transfer in sequence with
cisplatin to tumors of patients with non-small-cell lung cancer. J
Clin Oncol. 18:609–622. 2000.PubMed/NCBI
|
|
8
|
Schuler M, Herrmann R, De Greve JL,
Stewart AK, Gatzemeier U, Stewart DJ, Laufman L, Gralla R, Kuball
J, Buhl R, et al: Adenovirus-mediated wild-type p53 gene transfer
in patients receiving chemotherapy for advanced non-small-cell lung
cancer: Results of a multicenter phase II study. J Clin Oncol.
19:1750–1758. 2001.PubMed/NCBI
|
|
9
|
Schreiber J, Langhorst H, Jüttner R and
Rathjen FG: The IgCAMs CAR, BT-IgSF, and CLMP: Structure, function,
and diseases. Adv Neurobiol. 8:21–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim M, Zinn KR, Barnett BG, Sumerel LA,
Krasnykh V, Curiel DT and Douglas JT: The therapeutic efficacy of
adeno-viral vectors for cancer gene therapy is limited by a low
level of primary adenovirus receptors on tumour cells. Eur J
Cancer. 38:1917–1926. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma J, Zhao J, Lu J, Jiang Y, Yang H, Li P,
Zhao M, Liu K and Dong Z: Coxsackievirus and adenovirus receptor
promotes antitumor activity of oncolytic adenovirus H101 in
esophageal cancer. Int J Mol Med. 30:1403–1409. 2012.PubMed/NCBI
|
|
12
|
Li Y, Pong RC, Bergelson JM, Hall MC,
Sagalowsky AI, Tseng CP, Wang Z and Hsieh JT: Loss of adenoviral
receptor expression in human bladder cancer cells: A potential
impact on the efficacy of gene therapy. Cancer Res. 59:325–330.
1999.PubMed/NCBI
|
|
13
|
Pearson AS, Koch PE, Atkinson N, Xiong M,
Finberg RW, Roth JA and Fang B: Factors limiting
adenovirus-mediated gene transfer into human lung and pancreatic
cancer cell lines. Clin Cancer Res. 5:4208–4213. 1999.
|
|
14
|
Qin M, Chen S, Yu T, Escuadro B, Sharma S
and Batra RK: Coxsackievirus adenovirus receptor expression
predicts the efficiency of adenoviral gene transfer into non-small
cell lung cancer xenografts. Clin Cancer Res. 9:4992–4999.
2003.PubMed/NCBI
|
|
15
|
Hemminki A, Kanerva A, Liu B, Wang M,
Alvarez RD, Siegal GP and Curiel DT: Modulation of
coxsackie-adenovirus receptor expression for increased adenoviral
transgene expression. Cancer Res. 63:847–853. 2003.PubMed/NCBI
|
|
16
|
Zhang NH, Song LB, Wu XJ, Li RP, Zeng MS,
Zhu XF, Wan DS, Liu Q, Zeng YX and Zhang XS: Proteasome inhibitor
MG-132 modifies coxsackie and adenovirus receptor expression in
colon cancer cell line lovo. Cell Cycle. 7:925–933. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee CH, Kasala D, Na Y, Lee MS, Kim SW,
Jeong JH and Yun CO: Enhanced therapeutic efficacy of an
adenovirus-PEI-bile-acid complex in tumors with low coxsackie and
adenovirus receptor expression. Biomaterials. 35:5505–5516. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shirakawa T, Sasaki R, Gardner TA, Kao C,
Zhang ZJ, Sugimura K, Matsuo M, Kamidono S and Gotoh A:
Drug-resistant human bladder-cancer cells are more sensitive to
adenovirus-mediated wild-type p53 gene therapy compared to
drug-sensitive cells. Int J Cancer. 94:282–289. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ingemarsdotter CK, Tookman LA, Browne A,
Pirlo K, Cutts R, Chelela C, Khurrum KF, Leung EY, Dowson S, Webber
L, et al: Paclitaxel resistance increases oncolytic adenovirus
efficacy via upregulated CAR expression and dysfunctional cell
cycle control. Mol Oncol. 9:791–805. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marchetti A, Buttitta F, Merlo G, Diella
F, Pellegrini S, Pepe S, Macchiarini P, Chella A, Angeletti CA,
Callahan R, et al: p53 alterations in non-small cell lung cancers
correlate with metastatic involvement of hilar and mediastinal
lymph nodes. Cancer Res. 53:2846–2851. 1993.PubMed/NCBI
|
|
21
|
Swisher SG, Roth JA, Nemunaitis J,
Lawrence DD, Kemp BL, Carrasco CH, Connors DG, El-Naggar AK,
Fossella F, Glisson BS, et al: Adenovirus-mediated p53 gene
transfer in advanced non-small-cell lung cancer. J Natl Cancer
Inst. 91:763–771. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gurnani M, Lipari P, Dell J, Shi B and
Nielsen LL: Adenovirus-mediated p53 gene therapy has greater
efficacy when combined with chemotherapy against human head and
neck, ovarian, prostate, and breast cancer. Cancer Chemother
Pharmacol. 44:143–151. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishizaki M, Meyn RE, Levy LB, Atkinson
EN, White RA, Roth JA and Ji L: Synergistic inhibition of human
lung cancer cell growth by adenovirus-mediated wild-type p53 gene
transfer in combination with docetaxel and radiation therapeutics
in vitro and in vivo. Clin Cancer Res. 7:2887–2897. 2001.PubMed/NCBI
|
|
24
|
Swisher SG, Roth JA, Komaki R, Gu J, Lee
JJ, Hicks M, Ro JY, Hong WK, Merritt JA, Ahrar K, et al: Induction
of p53-regulated genes and tumor regression in lung cancer patients
after intratumoral delivery of adenoviral p53 (INGN 201) and
radiation therapy. Clin Cancer Res. 9:93–101. 2003.PubMed/NCBI
|
|
25
|
Hasenburg A, Fischer DC, Tong XW,
Rojas-Martinez A, Kaufman RH, Ramzy I, Kohlberger P, Orlowska-Volk
M, Aguilar-Cordova E and Kieback DG: Adenovirus-mediated thymidine
kinase gene therapy for recurrent ovarian cancer: Expression of
coxsackie-adenovirus receptor and integrins alphavbeta3 and
alphavbeta5. J Soc Gynecol Investig. 9:174–180. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pong RC, Lai YJ, Chen H, Okegawa T,
Frenkel E, Sagalowsky A and Hsieh JT: Epigenetic regulation of
coxsackie and adenovirus receptor (CAR) gene promoter in urogenital
cancer cells. Cancer Res. 63:8680–8686. 2003.PubMed/NCBI
|
|
27
|
Kitazono M, Goldsmith ME, Aikou T, Bates S
and Fojo T: Enhanced adenovirus transgene expression in malignant
cells treated with the histone deacetylase inhibitor FR901228.
Cancer Res. 61:6328–6330. 2001.PubMed/NCBI
|
|
28
|
Watanabe T, Hioki M, Fujiwara T, Nishizaki
M, Kagawa S, Taki M, Kishimoto H, Endo Y, Urata Y, Tanaka N, et al:
Histone deacetylase inhibitor FR901228 enhances the antitumor
effect of telomerase-specific replication-selective adenoviral
agent OBP-301 in human lung cancer cells. Exp Cell Res.
312:256–265. 2006.
|
|
29
|
Bieler A, Mantwill K, Dravits T,
Bernshausen A, Glockzin G, Köhler-Vargas N, Lage H, Gansbacher B
and Holm PS: Novel three-pronged strategy to enhance cancer cell
killing in glioblastoma cell lines: Histone deacetylase inhibitor,
chemotherapy, and oncolytic adenovirus dl520. Hum Gene Ther.
17:55–70. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
El-Zawahry A, Lu P, White SJ and
Voelkel-Johnson C: In vitro efficacy of AdTRAIL gene therapy of
bladder cancer is enhanced by trichostatin A-mediated restoration
of CAR expression and downregulation of cFLIP and
Bcl-XL. Cancer Gene Ther. 13:281–289. 2006. View Article : Google Scholar
|
|
31
|
Sasaki Y, Negishi H, Idogawa M, Suzuki H,
Mita H, Toyota M, Shinomura Y, Imai K and Tokino T: Histone
deacetylase inhibitor FK228 enhances adenovirus-mediated p53 family
gene therapy in cancer models. Mol Cancer Ther. 7:779–787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Earel JK Jr, VanOosten RL and Griffith TS:
Histone deacetylase inhibitors modulate the sensitivity of tumor
necrosis factor-related apoptosis-inducing ligand-resistant bladder
tumor cells. Cancer Res. 66:499–507. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Berghauser Pont LM, Kleijn A, Kloezeman
JJ, van den Bossche W, Kaufmann JK, de Vrij J, Leenstra S, Dirven
CM and Lamfers ML: The HDAC inhibitors scriptaid and LBH589
combined with the oncolytic virus delta24-RGD exert enhanced
anti-tumor efficacy in patient-derived glioblastoma cells. PLoS
One. 10:e01270582015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim DR, Park MY, Lim HJ, Park JS, Cho YJ,
Lee SW, Yoon HI, Lee JH, Kim YS and Lee CT: Combination therapy of
conditionally replicating adenovirus and histone deacetylase
inhibitors. Int J Mol Med. 29:218–224. 2012.
|
|
35
|
MacTavish H, Diallo JS, Huang B, Stanford
M, Le Boeuf F, De Silva N, Cox J, Simmons JG, Guimond T, Falls T,
et al: Enhancement of vaccinia virus based oncolysis with histone
deacetylase inhibitors. PLoS One. 5:e144622010. View Article : Google Scholar
|
|
36
|
Vincent T, Pettersson RF, Crystal RG and
Leopold PL: Cytokine-mediated downregulation of
coxsackievirus-adenovirus receptor in endothelial cells. J Virol.
78:8047–8058. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Anders M, Christian C, McMahon M,
McCormick F and Korn WM: Inhibition of the Raf/MEK/ERK pathway
up-regulates expression of the coxsackievirus and adenovirus
receptor in cancer cells. Cancer Res. 63:2088–2095. 2003.PubMed/NCBI
|