Introduction
Lung cancer, mostly caused by smoking and exposure
to pollutants, is one of the leading causes of death worldwide
(1). According to its histological
types, lung cancer is classified into small cell lung cancer and
non-small cell lung cancer (NSCLC). For many years, the existing
therapeutic strategies for lung cancer have been several
traditional therapies, including surgery, chemotherapy and
radiotherapy. Despite the advances in these traditional therapies,
the mortality rates remain high, with an overall 5-year survival of
only 15% (2). Therefore,
comprehensive understanding of the molecular mechanisms underlying
lung cancer progression may contribute to the effectiveness of
anticancer therapy and thereby, the overall survival of lung
cancer.
Centrosomal protein 55 (CEP55), also named C10orf3
(3) and URCC6, is the latest member
found in the centrosomal relative protein family, which upon
observation was revealed to localize to the centrosome in
interphase cells, midzone during anaphase, and the midbody during
cytokinesis by tagging with GFP-C (4–6).
Furthermore, CEP55 has been identified as a microtubule-bundling
protein and plays an important role in cell mitosis through
cooperation with CDK1, ERK2 and PLK1 (4). Accumulated evidence has shown that
CEP55 overexpression occurs in a wide range of solid tumors,
including human colon (3), bladder
cancer (7) and hepatocellular
carcinoma tumorigenesis (8). In
addition, CEP55 has been demonstrated to regulate critical cell
functions including cell growth, transformation and cytokinesis.
Overexpression of CEP55 was found to enhance cell cycle transition
by activation of p21, whereas knockdown of CEP55 was found to
inhibit cell growth in gastric (9)
and breast cancer (10). Moreover,
CEP55 was found to be upregulated by VEGFA in lung cancer tissues
and associated with metastasis (11). In spite of these functional
observations, the role of CEP55 in lung cancer has remained largely
unclear.
The present study aimed to further investigate the
biological function of CEP55 in lung cancer. We found that CEP55
was overexpressed in lung cancer tissues and cell lines. Our in
vitro studies demonstrated that CEP55 promoted cell viability
and proliferation through cell cycle progression and inhibition of
apoptosis. These findings suggest that CEP55 overexpression is
associated with tumor growth and may serve as a potential new
therapeutic target for lung cancer.
Materials and methods
Clinical tissue specimens
A total of 20 fresh tumor tissue samples with paired
non-cancerous lung tissue samples were derived from lung cancer
patients who had undergone surgery at our institution. None of
these patients had received radiotherapy or chemotherapy prior to
surgical treatment. Dissected samples were frozen immediately after
surgery and stored at −80°C until use. All patients had signed
written informed consents prior to the use of the clinical
materials for this study.
Cell lines and culture
The human NSCLC cell lines H1299, A549, H128 and 95D
were purchased from the Cell Bank of the Type Culture Collection of
the Chinese Academy of Sciences (Shanghai, China). These cells were
cultured in RPMI-1640 (Hyclone and Biowest) supplemented with 10%
fetal bovine serum (FBS) and incubated in a humidified atmosphere
containing 5% CO2. The medium was replaced every 2–3
days as indicated.
CEP55 short hairpin RNA (shRNA) stable
transfection
Two CEP55 shRNA constructs, as well as a scrambled
negative control (NC) shRNA were purchased from OriGene
Technologies, Inc. (Rockville, MD, USA) and cloned into the
pLKO.1-EGFP vector between AgeI and EcoRI restriction
sites. Lentivirus particles were generated by co-transfecting
recombined and packing vectors into 293T cells via Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA). A549 and 95D cells were then
cultured in 6-well plates and transfected with CEP55
shRNA-expression lentiviruses (sh-1 or -2) according to the
manufacturer's instructions. All transfected cell lines were
further evaluated for knockdown efficiency of the target gene
(CEP55) using quantitative real-time PCR (qRT-PCR) and western blot
analysis.
Quantitative RT-PCR
Total RNA was isolated using the TRIzol reagent
(Invitrogen). Complementary DNA (cDNA) was synthesized from 2
µg of total RNA using 200 U/ml SuperScript II Reverse
Transcriptase (Invitrogen). Real-time PCR amplification reactions
for CEP55 were carried out using the Bio-Rad Connect Real-Time PCR
platform as previously reported (12). All reagents were purchased from
Thermo Fisher Scientific (Waltham, MA, USA). The relative mRNA
expression of CEP55 was normalized by using human GAPDH mRNA levels
as an internal control. Data were analyzed using the
2−ΔΔCt method as previously described (13). Each experiment was performed in
triplicate.
Protein isolation and western blot
analysis
Total protein was isolated from cells using RIPA
lysis buffer containing protease inhibitors (Sigma-Aldrich, St.
Louis, MO, USA). The suspension was collected after centrifugation
at 12,000 × g for 15 min at 4°C, followed by protein concentration
determination using BCA assay. Equal amounts of proteins (20–40
µg) were separated on 10% sodium dodecyl
sulfate-polyacrylamide gel (SDS-PAGE) and transferred to a PVDF
membrane using a Bio-Rad semi-dry transfer system. Then the
membrane was blocked with TBST (Tris-buffered saline, 0.1%
Tween-20) containing 5% non-fat dry milk for 1 h at room
temperature, and probed with the corresponding primary antibodies
overnight. Subsequently, the membrane was incubated with
appropriated horseradish peroxidase-conjugated secondary antibodies
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 2 h at
room temperature. Immunoreactive bands were visualized using Super
ECL detection reagent (Applygen Technologies, Inc., Beijing,
China).
Cell viability assay
MTT assay was used to determine cell viability in
the A549 and 95D cells. Briefly, the cells were seeded into 96-well
plates in triplicates at a density of 2,000 cells/well and cultured
for 24 h. Then 50 µl MTT was added into each well at 1, 2,
3, 4, and 5 days after transfection. After 4 h of incubation, 200
µl dimethyl sulfoxide (DMSO) was added into each well. The
absorbance value was measured using a microplate reader at 595 nm.
Each experiment was performed in triplicate.
Colony formation assay
To evaluate the effect of CEP55 on monolayer colony
formation, stably transfected cells were seeded into 6-well plates
at a density of 1,500 cells/well and allowed to grow for 7 days to
form colonies. After being cultured for 7 days, the cells were
fixed with methanol and stained with crystal violet. The number of
colonies consisting of >50 cells/colony was counted under a
microscope. Each experiment was performed in triplicate and
repeated three times.
Cell cycle and apoptosis analysis
Cells were washed with cold PBS twice after a 48-h
transfection and re-inoculated into 6-cm dishes at a density of
80,000 cells/dish. Then the cells were collected and stained with
propidium iodide (PI). The number of cells in each cell cycle phase
was analyzed using a flow cytometer (FACSCalibur; BD Biosciences).
In addition, apoptosis was determined by dual staining with an
Annexin V-APC and PI apoptosis detection kit (Nanjing KeyGen
Biotech, Co., Ltd., Nanjing, China) according to the manufacturer's
instructions.
Statistical analysis
All statistical analyses were performed by SPSS
software version 10.0 (SPSS, Inc., Chicago, IL, USA) and expressed
as the mean ± standard deviation (SD) of three independent
experiments. Paired Student's t-test was used to compare
differences between the groups. Statistically significant
differences were accepted at p<0.05.
Results
CEP55 is upregulated in lung cancer
tissues and cell lines
To investigate the expression of CEP55 in lung
cancer, we performed quantitative RT-PCR to detect CEP55 mRNA
expression in 20 fresh tumor tissue samples with paired
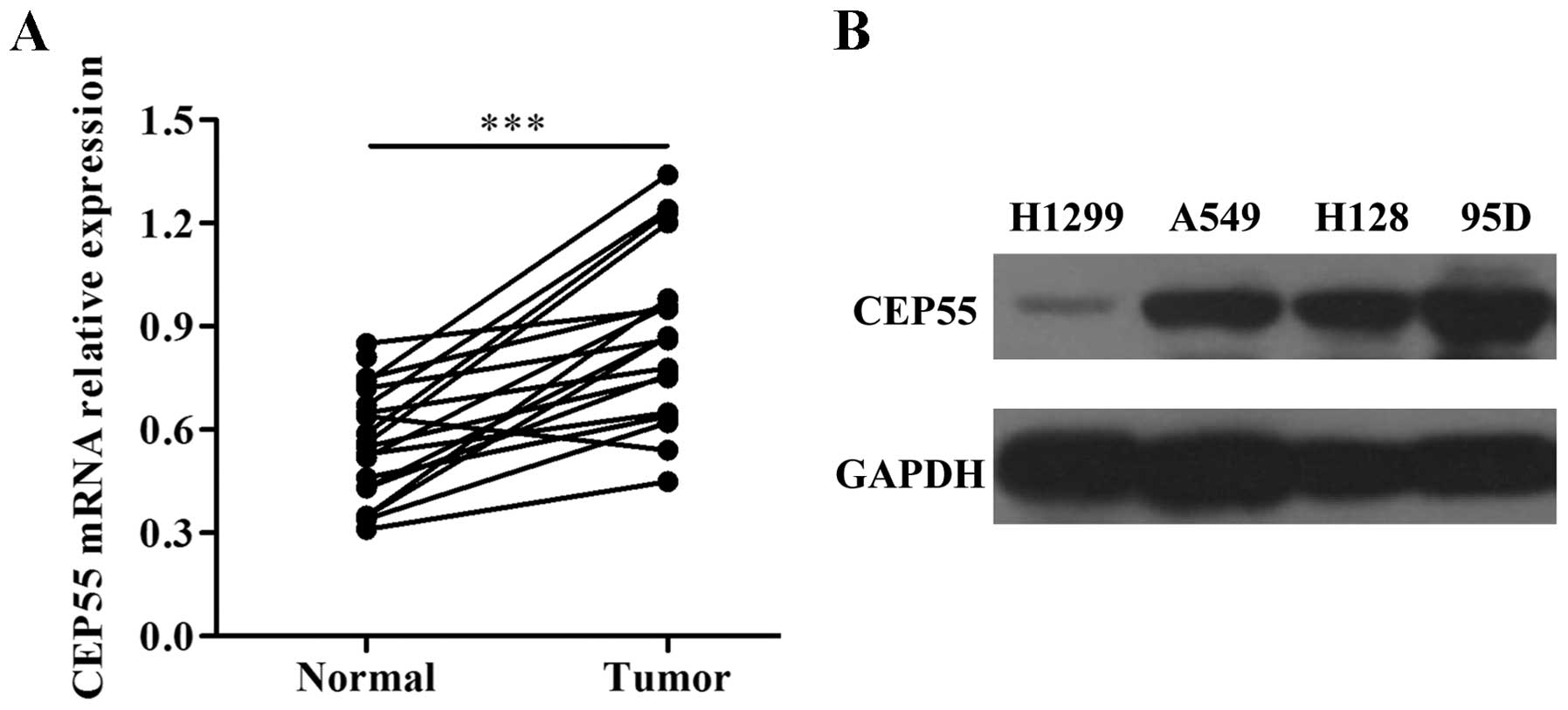
non-cancerous lung tissues. As shown in Fig. 1A, the mRNA level of CEP55 was
significantly elevated in the lung cancer tissues compared with
that noted in the adjacent normal tissues (p<0.001). Next, we
further determined CEP55 expression in four lung cancer cell lines
by western blot analysis. The results indicated that the protein
level of CEP55 was obviously increased in all four lung cancer cell
lines, among which it was expressed at a higher level in the A459
and 95D cells (Fig. 1B). Thus, A459
and 95D cells were chosen for the following studies.
The expression of CEP55 is significantly
suppressed in lung cancer cells
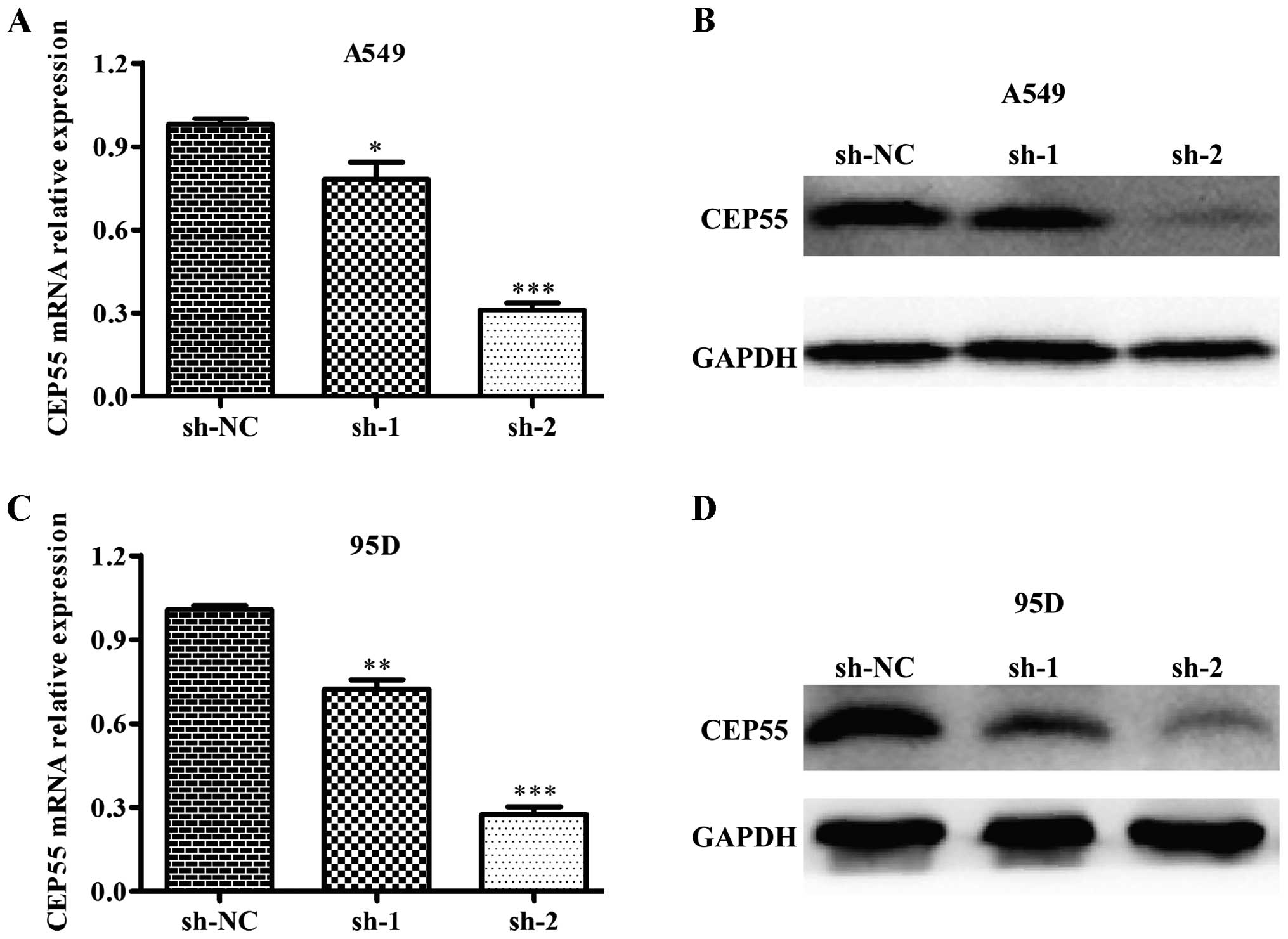
To study the potential function of CEP55, we
designed two shRNA constructs to specifically silence CEP55
expression in the A549 and 95D cells using a lentiviral system.
Subsequently, we determined the knockdown efficiency on CEP55 mRNA
and protein levels in the A549 and 95D cells. As shown in Fig. 2A and C, the CEP55 mRNA level was
significantly decreased in the sh-CEP55-infected cells. Notably, we
found that the second construct (sh-2) (p<0.001) showed better
inhibiting efficiency than sh-1 (p<0.05, p<0.01) in both cell
lines compared with the first constructs (sh-1). Similarly, sh-2
suppressed the CEP55 protein level more markedly than sh-1 in the
A549 and 95D cells (Fig. 2B and D).
Therefore, sh-2 was selected for further experiments.
CEP55 knockdown inhibits lung cancer cell
viability and colony formation ability
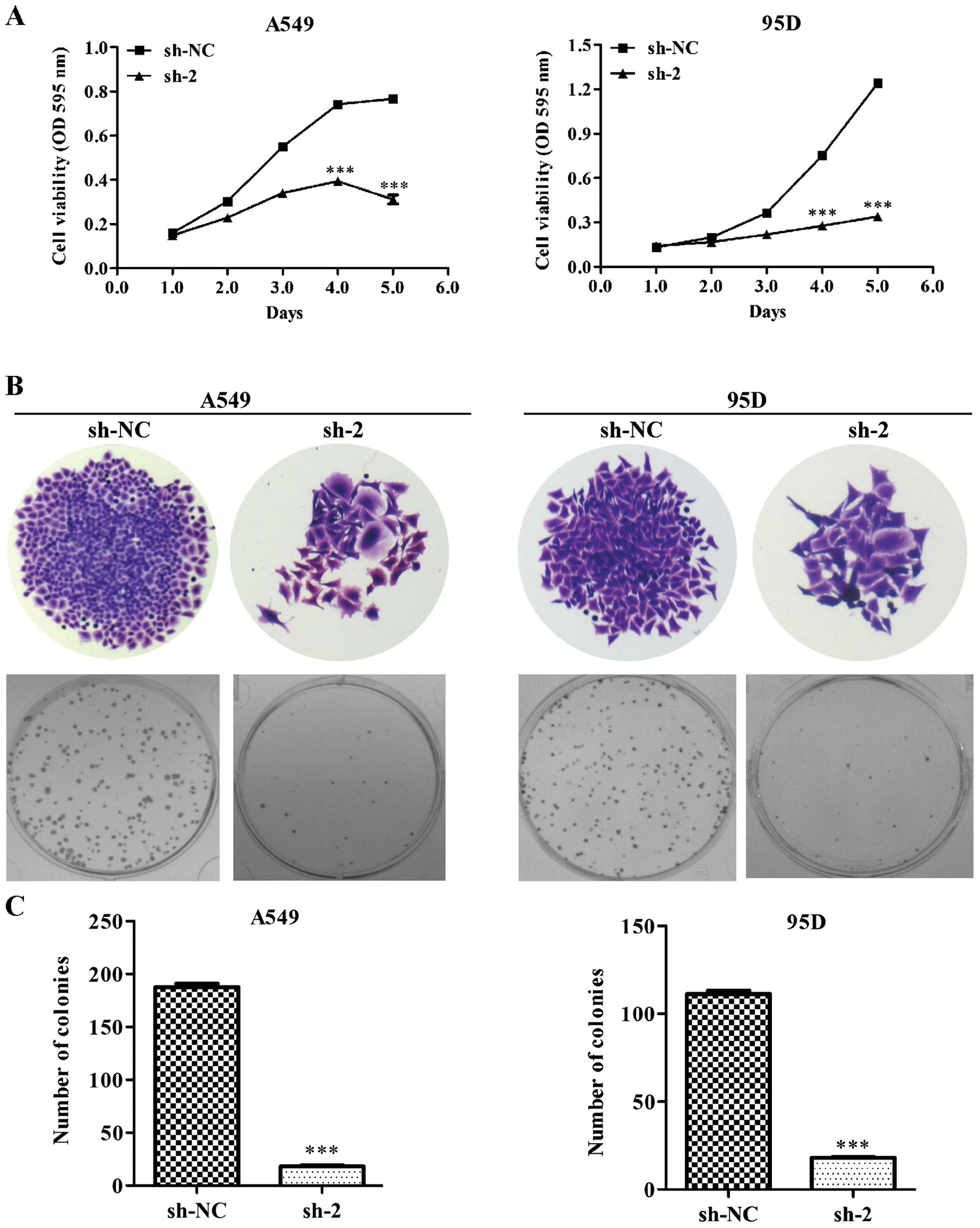
To investigate the inhibitory effect of CEP55 on
cell growth, we measured cell viability using an MTT assay and
found that the cell viability was significantly lower after five
consecutive days in the CEP55-knockdown (sh-2) cells compared with
that noted in the non-target scramble control shRNA-transfected
cells (sh-NC) in both the A549 and 95D cell lines (Fig. 3A, p<0.001). Furthermore, we
determined the colony formation ability of the lung cancer cells
after knockdown of CEP55. As shown in Fig. 3B, knockdown of CEP55 obviously
reduced the size of the single colonies and the number of colonies
formed in the A549 and 95D cells. The number of colonies in the
sh-2 group was apparently smaller than that in the sh-NC group in
both cell lines (Fig. 3C,
p<0.001). These results further indicate that CEP55 acts as a
potential tumor gene in lung cancer.
Cell cycle arrest and apoptosis is
induced by knockdown of CEP55 in lung cancer cells
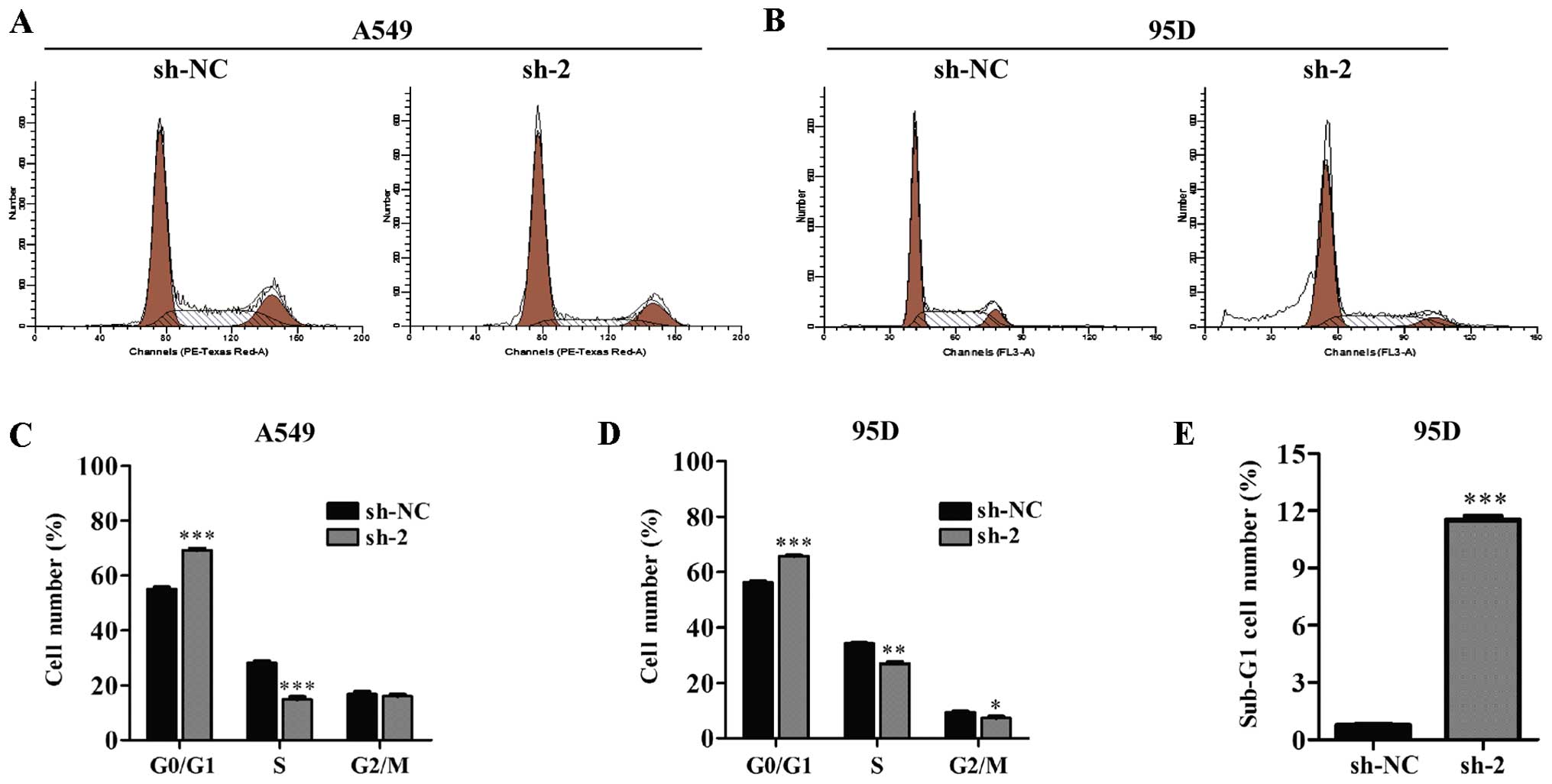
To explore the underlying mechanism of reduced cell
viability by CEP55 inhibition, flow cytometry was used to analyze
the cell cycle distribution in lung cancer cells. The results
showed that the percentage of cells in the G1 phase (p<0.001)
was greatly increased in the CEP55-knockdown cells compared to the
control cells in both the A549 (Fig. 4A
and C) and 95D cell lines (Fig. 4B
and D). In addition, more cells were accumulated in the sub-G1
phase in the sh-2 infected 95D cells; much more than those in the
sh-NC infected cells (Fig. 4E,
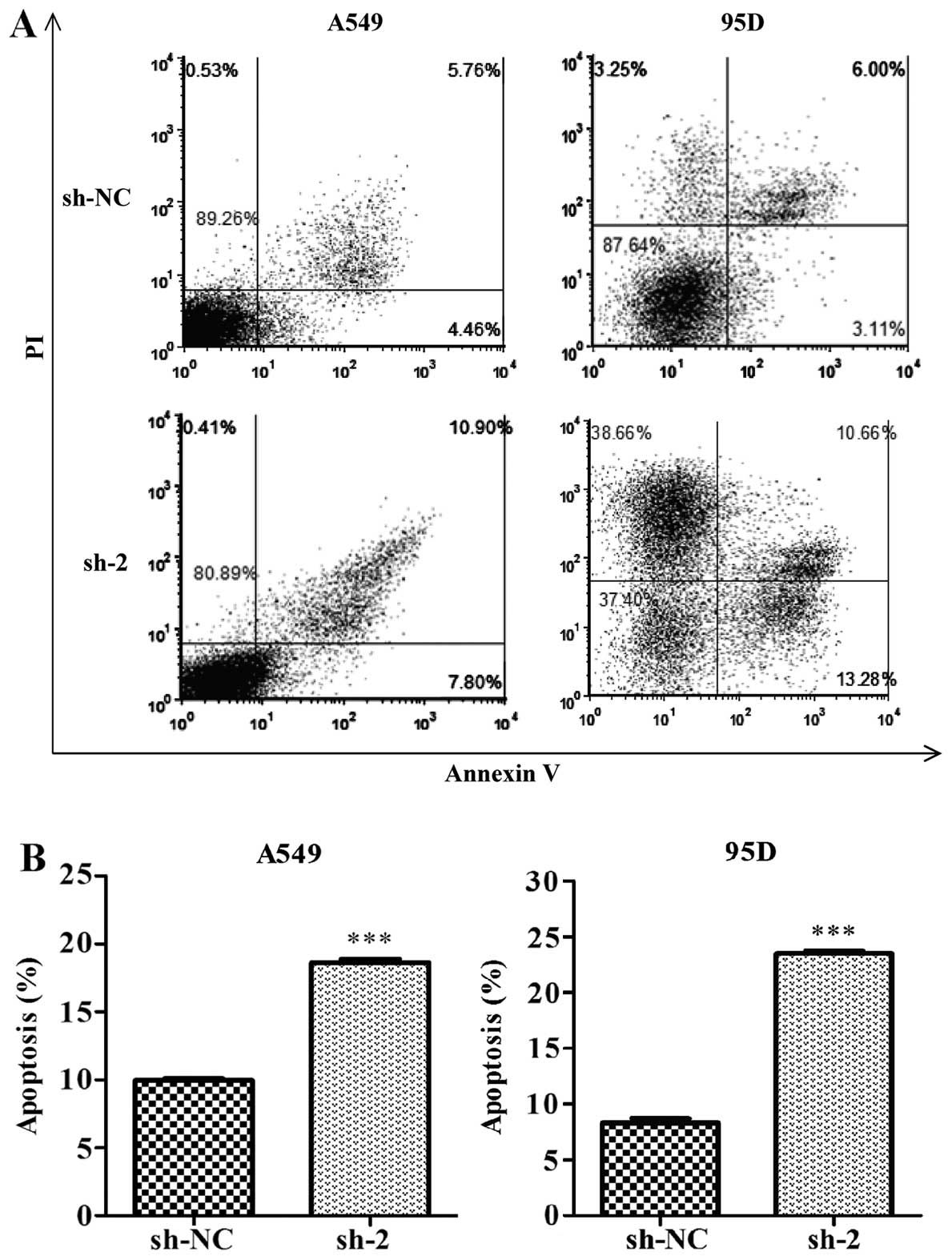
p<0.001). Next, we evaluated cell apoptosis using Annexin V
staining and flow cytometry. Our results showed that, the
percentages of both early apoptotic (Annexin
V+/PI−) and late apoptotic (Annexin
V+/PI+) cells in the CEP55 sh-2-transfected
cells were increased significantly compared with those in the
control shRNA-transfected cells (Fig.
5A). Further analysis indicated that knockdown of CEP55
markedly increased the overall apoptotic cells in the A549 and 95D
cell lines (Fig. 5B,
p<0.001).
Molecular targets of CEP55 in lung cancer
cells
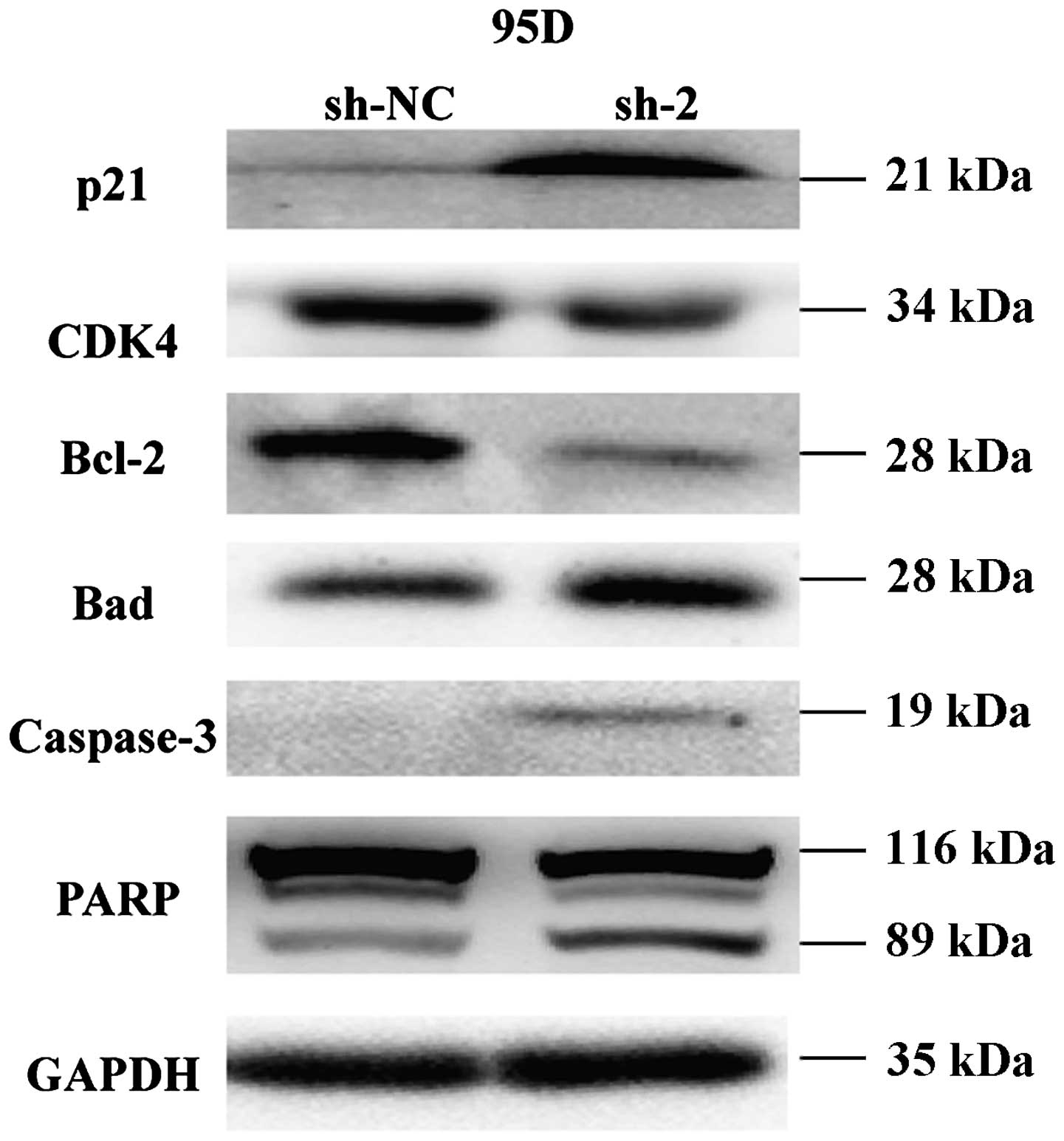
To gain insight into the molecular mechanisms of
CEP55 silencing on cell cycle and apoptotic regulation, several
molecular targets associated with cell cycle and apoptotic
regulation were assessed by western blot analysis in the 95D cells
stably transfected with sh-2. As shown in Fig. 6, knockdown of CEP55 inhibited
expression of CDK4 and Bcl-2 and enhanced expression of p21, cell
cycle regulators. Moreover, Bad, caspase-3 and PARP with
pro-apoptotic effects were significantly upregulated in the
sh-2-transfected cells compared with that in the sh-NC-transfected
cells. These results suggest that CEP55 acts as an oncogene by
regulating the expression of important genes involved in the
proliferation and apoptotic pathways.
Discussion
NSCLC, as a major type, accounts for >80% of all
lung cancers. Molecular targeted therapy has been widely applied
for the treatment of lung cancer with higher efficacy and lower
toxicity compared with traditional chemotherapy. Therefore, it is
of great importance to identify specific tumor genes and
therapeutic targets to improve personalized cancer therapy. CEP55
has been reported to be overexpressed in various human cancer
tissues like epithelial ovarian (14) and gastric (9) carcinomas. For these studies, we
hypothesized that CEP55 was overexpressed in lung cancer tissues
and cell lines. To confirm this hypothesis, we firstly determined
the expression of CEP55 in tumor tissues from lung cancer patients
and found that it was highly expressed in tumor tissues compared
with normal lung tissues. Herein, previous study indicated that
VEGFA upregulated the expression of the CEP55 protein, which
subsequently resulted in the complex formation with PI3K (11). Therefore, these results suggest a
potential oncogenic role of CEP55 in lung cancer, and motivated us
to explore its functional significance in lung cancer.
In the present study, the in vitro studies
demonstrated that CEP55 knockdown inhibited cell viability and
proliferation through the induction of cell cycle arrest and
apoptosis in the NSCLC cell lines A549 and 95D, indicating
antitumor activity of CEP55 inhibition. Evidence points to the
contribution of CEP55 in cell proliferation involved in gastric
(9) and breast (10) cancers, as well as nasopharyngeal
carcinomas (15). To the best of
our knowledge, dysregulation of cell proliferation is the key
characteristic of cancer cells, which is closely associated with
cell cycle regulation (16,17). As one of the main checkpoints of the
cell cycle, the G1/S transition plays a crucial role in the
initiation and completion of DNA replication, which is strongly
regulated by the CDK/cyclin complex activity (18,19).
In addition CDKs, cyclin-dependent kinase inhibitors (CDKIs) also
suppress the G1/S transition in cell cycle regulation (20). Our results showed that cells were
arrested in the G1 phase in the CEP55 knocked-down cells.
Consistently, further analysis revealed that knockdown of CEP55
downregulated the expression of CDK4 and upregulated the expression
of CDKI p21. It has been widely accepted that centrosome-associated
proteins play an essential role in cell cycle progression (21,22).
Thus, we conclude that CEP55, as a member of the
centrosome-associated protein family promotes cell cycle
progression in cancer occurrence.
Additionally, we further found that knockdown of
CEP55 induced cell apoptosis in the NSCLC cell lines A549 and 95D.
Caspase cascade plays a central role in apoptosis, whose activation
is regulated by various factors, among which the Bcl-2 family of
proteins, as anti-apoptotic mitochondria proteins, are the
principle regulators in the intrinsic mitochondrial apoptotic
pathway (23). In addition,
pro-apoptotic proteins, such as Bad and caspase-3 may be activated
in cell apoptosis (24,25). As the specificity substrate of
caspases, PARP plays an important role in DNA repair and can be
cleaved by activated caspase-3 resulting in cell apoptosis
(26). Recently, it has been
reported that CEP55 silencing promoted breast cancer cell apoptosis
(10). Consistent with this report,
we found that silencing of CEP55 resulted in a significant increase
in apoptotic cells in the NSCLC cells. Moreover, the mechanisms
involved activation of anti-apoptotic factors, such as Bcl-2, Bad
and caspase-3, as well as subsequent amplification of PARP
cleavage.
In conclusion, our in vitro data provided
enough evidence that the CEP55 level was elevated in lung cancer
and its inhibition significantly suppressed lung cancer cell
survival and proliferation. These findings suggested that CEP55 may
be a potential therapeutic target in lung cancer treatment.
References
|
1
|
Al Zeyadi M, Dimova I, Ranchich V, Rukova
B, Nesheva D, Hamude Z, Georgiev S, Petrov D and Toncheva D: Whole
genome microarray analysis in non-small cell lung cancer.
Biotechnol Biotechnol Equip. 29:111–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ni M, Shi XL, Qu ZG, Jiang H, Chen ZQ and
Hu J: Epithelial mesenchymal transition of non-small-cell lung
cancer cells A549 induced by SPHK1. Asian Pac J Trop Med.
8:142–146. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sakai M, Shimokawa T, Kobayashi T,
Matsushima S, Yamada Y, Nakamura Y and Furukawa Y: Elevated
expression of C10orf3 (chromosome 10 open reading frame 3) is
involved in the growth of human colon tumor. Oncogene. 25:480–486.
2006.
|
|
4
|
Fabbro M, Zhou BB, Takahashi M, Sarcevic
B, Lal P, Graham ME, Gabrielli BG, Robinson PJ, Nigg EA, Ono Y, et
al: Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome
protein, Cep55, is required for its recruitment to midbody and
cytokinesis. Dev Cell. 9:477–488. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao WM, Seki A and Fang G: Cep55, a
microtubule-bundling protein, associates with centralspindlin to
control the midbody integrity and cell abscission during
cytokinesis. Mol Biol Cell. 17:3881–3896. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martinez-Garay I, Rustom A, Gerdes HH and
Kutsche K: The novel centrosomal associated protein CEP55 is
present in the spindle midzone and the midbody. Genomics.
87:243–253. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singh PK, Srivastava AK, Rath SK, Dalela
D, Goel MM and Bhatt ML: Expression and clinical significance of
Centrosomal protein 55 (CEP55) in human urinary bladder
transitional cell carcinoma. Immunobiology. 220:103–108. 2015.
View Article : Google Scholar
|
|
8
|
Chen CH, Lu PJ, Chen YC, Fu SL, Wu KJ,
Tsou AP, Lee YC, Lin TC, Hsu SL, Lin WJ, et al: FLJ10540-elicited
cell transformation is through the activation of PI3-kinase/AKT
pathway. Oncogene. 26:4272–4283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tao J, Zhi X, Tian Y, Li Z, Zhu Y, Wang W,
Xie K, Tang J, Zhang X, Wang L, et al: CEP55 contributes to human
gastric carcinoma by regulating cell proliferation. Tumour Biol.
35:4389–4399. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Y, Jin T, Dai X and Xu J:
Lentivirus-mediated knockdown of CEP55 suppresses cell
proliferation of breast cancer cells. Biosci Trends. 10:67–73.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen CH, Lai JM, Chou TY, Chen CY, Su LJ,
Lee YC, Cheng TS, Hong YR, Chou CK, Whang-Peng J, et al: VEGFA
upregulates FLJ10540 and modulates migration and invasion of lung
cancer via PI3K/AKT pathway. PLoS One. 4:e50522009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang W, Liang Z and Li J: Inhibition of
rhotekin exhibits antitumor effects in lung cancer cells. Oncol
Rep. 35:2529–2534. 2016.PubMed/NCBI
|
|
13
|
Eskander RN, Randall LM, Sakai T, Guo Y,
Hoang B and Zi X: Flavokawain B, a novel, naturally occurring
chalcone, exhibits robust apoptotic effects and induces G2/M arrest
of a uterine leiomyosarcoma cell line. J Obstet Gynaecol Res.
38:1086–1094. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang W, Niu C, He W, Hou T, Sun X, Xu L
and Zhang Y: Upregulation of centrosomal protein 55 is associated
with unfavorable prognosis and tumor invasion in epithelial ovarian
carcinoma. Tumour Biol. 37:6239–6254. 2016. View Article : Google Scholar :
|
|
15
|
Chen CH, Shiu LY, Su LJ, Huang CY, Huang
SC, Huang CC, Yin YF, Wang WS, Tsai HT, Fang FM, et al: FLJ10540 is
associated with tumor progression in nasopharyngeal carcinomas and
contributes to nasopharyngeal cell proliferation, and metastasis
via osteopontin/CD44 pathway. J Transl Med. 10:932012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nguyen-Ba G and Vasseur P: Epigenetic
events during the process of cell transformation induced by
carcinogens (Review). Oncol Rep. 6:925–932. 1999.PubMed/NCBI
|
|
17
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cicenas J, Kalyan K, Sorokinas A, Jatulyte
A, Valiunas D, Kaupinis A and Valius M: Highlights of the latest
advances in research on CDK Inhibitors. Cancers (Basel).
6:2224–2242. 2014. View Article : Google Scholar
|
|
20
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: Roles beyond cell cycle regulation. Development.
140:3079–3093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Delattre M and Gönczy P: The arithmetic of
centrosome biogenesis. J Cell Sci. 117:1619–1630. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Srsen V and Merdes A: The centrosome and
cell proliferation. Cell Div. 1:262006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dejean LM, Martinez-Caballero S, Manon S
and Kinnally KW: Regulation of the mitochondrial apoptosis-induced
channel, MAC, by BCL-2 family proteins. Biochim Biophys Acta.
1762:191–201. 2006. View Article : Google Scholar
|
|
24
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu SW, Andrabi SA, Wang H, Kim NS, Poirier
GG, Dawson TM and Dawson VL: Apoptosis-inducing factor mediates
poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad
Sci USA. 103:18314–18319. 2006. View Article : Google Scholar : PubMed/NCBI
|