Introduction
There are various kidney-related malignancies, yet,
renal cell carcinoma is the most frequenty occurring tumor among
these. These tumor types are not responsive to chemotherapy or
radiation therapy (1,2), thus, it is extremely hard to treat
them and this is the motivation why different groups of researchers
are attempting to acquaint some novel chemotherapeutic agent that
can treat this disease. Although, there have been many efforts to
treat this carcinoma with molecular or gene therapy, for example,
tyrosine kinase inhibitors, mammalian target of rapamycin (mTOR)
and vascular endothelial growth factor (VEGF) inhibitors (3), the pharmacological agents that can
play an important role in inhibition of these genes are the most
important. However, the mechanism underlying the initiation to the
progression of most cancers is similar to each other for example,
there are various master regulators of anti-apoptotic mechanism
such as Bcl-2 and c-FLIP that limit the potential of a drug to
initiate apoptosis mechanism within the cell (4,5).
In light of the fact that renal carcinoma is one of
the most deadly types of cancers, some candidates have been
introduced to stop and/or limit the progression of the metastasis
of this carcinoma, for example, sunitinib and pazopanib, drugs that
are reported to be the inhibitors of tyrosine kinase (6,7).
Indeed, these agents were found to be able to stop the growth of
cancer cells but extensive research on these agents revealed that
they are associated with drug resistance and they have various
side-effects as well (8,9). Therefore, there is a need to introduce
potent candidates that are originated from a natural source and are
not destructive to the normal physiological condition of cells or
tissue.
Thymoquinone is a natural polyphenolic compound
found abundantly in black cumin (Nigella sativa L.) seeds
and is grouped as monoterpenes. Thymoquinone has recently been
reported for its various medicinal properties, for example, it is
effective to treat gastroenteritis and various other organ and
cell-associated inflammation (10,11).
Moreover, few recent studies explored its efficacy to kill cancer
cells via induction of apoptosis mechanism, and some researchers
demonstrated that thymoquinone has potential to reduce the volume
of breast, colon and gastric cancer using in vivo model
(12,13), however, very limited studies have
been carried out to uncover its mechanism of action in renal
carcinoma cell lines.
In the present study, we have evaluated the efficacy
of thymoquinone against Caki cells, a renal carcinoma cell line and
uncovered its molecular mechanism of action against this cell
line.
Materials and methods
Cells and materials
Caki, A498 and ACHN cells were obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA). The
culture medium used throughout these experiments was Dulbecco's
modified Eagle's medium (DMEM) containing 10% fetal bovine serum
(FBS), 20 mM HEPES buffer and 100 µg/ml gentamycin. The
mouse kidney cells, TMCK-1, were a gift from Dr T.J. Lee (Yeungnam
University, Korea). z-VAD-fmk was purchased from R&D Systems.
Thymoquinone was purchased from Sigma Chemical Co. (St. Louis, MO,
USA). Anti-Bcl-2 (sc-783), anti-Bcl-xL (sc-634), anti-Mcl-1
(sc-819) and anti-cIAP2 (sc-7944) were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Anti-XIAP (610762) was
purchased from BD Biosciences (Bedford, MA, USA). Anti-PARP (#9542)
antibody was obtained from Cell Signaling Technology (Beverly, MA,
USA). Anti-actin (A5441) antibody was obtained from Sigma (St.
Louis, MO, USA). Other reagents were purchased from Sigma Chemical
Co.
Flow cytometric analysis
For flow cytometry, the cells were resuspended in
100 µl of phosphate-buffered saline (PBS), and 200 µl
of 95% ethanol was added while the cells were being vortexed. The
cells were then incubated at 4°C for 1 h, washed with PBS,
resuspended in 250 µl of 1.12% sodium citrate buffer (pH
8.4) with 12.5 µg of RNase and incubated for an additional
30 min at 37°C. The cellular DNA was then stained by adding 250
µl of a propidium iodide solution (50 µg/ml) to the
cells for 30 min at room temperature (14). The stained cells were analyzed by
fluorescent-activated cell sorting on a FACScan flow cytometer to
determine the relative DNA content, which was based on the red
fluorescence intensity.
Western blot analysis
For the western blot experiments, the cells were
washed with cold PBS and lysed on ice in modified RIPA buffer (50
mM Tris-HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1
mM Na3VO4 and 1 mM NaF) containing protease
inhibitors (100 µM phenylmethylsulfonyl fluoride, 10
µg/ml leupeptin, 10 µg/ml pepstatin and 2 mM EDTA).
The lysates were centrifuged at 10,000 × g for 10 min at 4°C and
the supernatant fractions were collected. The proteins were
separated by SDS-PAGE electrophoresis and transferred to
Immobilon-P membranes. The specific proteins were detected using an
enhanced chemiluminescence (ECL) western blotting kit according to
the manufacturer's instructions.
DNA fragmentation assay
DNA fragmentation was performed using the Cell Death
Detection ELISAPLUS kit (Boehringer Mannheim, Indianapolis, IN,
USA). Briefly, cells were centrifuged for 10 min at 200 × g, the
supernatant was removed, and pellet was lysed for 30 min. After
centrifuging the plate again at 200 × g for 10 min, and the
supernatant that contained the cytoplasmic histone-associated DNA
fragments was collected and incubated with an immobilized
anti-histone antibody. The reaction products were incubated with a
peroxidase substrate for 5 min and measured by spectrophotometry at
405 and 490 nm (reference wavelength) with a microplate reader. The
signals in the wells containing the substrate alone were subtracted
as background.
Asp-Glu-Val-Asp-ase (DEVDase) activity
assay
To evaluate DEVDase activity, cell lysates were
prepared after their respective treatments with thymoquinone.
Assays were performed in 96-well microtiter plates by incubating 20
mg of cell lysates in 100 µl of reaction buffer (1% NP-40,
20 mM Tris-HCl, pH 7.5, 137 mM NaCl, 10% glycerol) containing a
caspase substrate [Asp-Glu-Val-Asp-chromophore-p-nitroanilide
(DVAD-pNA)] at 5 mM. Lysates were incubated at 37°C for 2 h.
Thereafter, the absorbance at 405 nm was measured with a
spectrophotometer.
RNA isolation, reverse transcription
polymerase chain reaction (RT-PCR)
Total cellular RNA was extracted from cells using
TRIzol reagent (Life Technologies, Gaithersburg, MD, USA).
Complementary DNA was synthesized from 2 µg of total RNA
using M-MLV reverse transcriptase (Promega, Madison, WI, USA). The
cDNA for c-FLIP, Bcl-2 and actin were amplified by a PCR using
specific primers: c-FLIP (forward) 5′-CGGACTATAGAGTGCTGATGG-3′ and
(reverse) 5′-GATTATCAGGCAGATTCCTAG-3′; Bcl-2 (forward)
5′-GTCCTCAGCCCTCGCTCT-3′ and (reverse) 5′-CACCTAATTGGGCTCCATCT-3′;
actin (forward) 5′-GGCATCGTCACCAACTGGGAC-3′ and (reverse)
5′-CGATTTCCCGCTCGGCCGTGG-3′. PCR products were analyzed by agarose
gel electrophoresis and visualized using ethidium bromide
staining.
Transfection and promoter activity
assay
Transient transfection was performed in 6-well
plates. One day before the transfection, Caki cells were plated at
~60–80% confluence. The Bcl-2/-3254 promoter- or NF-κB-luciferase
plasmid was transfected into the cells using Lipofectamine™ 2000
(Invitrogen, Carlsbad, CA, USA). To assess the promoter-driven
expression of the luciferase gene, the cells were collected and
disrupted by sonication in lysis buffer (25 mM Tris-phosphate pH
7.8, 2 mM EDTA, 1% Triton X-100 and 10% glycerol), and aliquots of
the supernatants were used to analyze the luciferase activity
according to the manufacturer's instructions (Promega).
Measurement of reactive oxygen species
(ROS)
Intracellular accumulation of ROS was determined
using the fluorescent probes 2′,7′-dichlorodihydrofluorescein
diacetate (H2DCFDA). H2DCFDA is commonly used to measure ROS
generation. Caki cells were pretreated with NAC for 30 min, and
then added with thymoquinone. Cells were stained with the
fluorescent dye H2DCFDA for an additional 10 min. Then, cells were
observed using a fluorescence microscope (Axiovert 200M; Carl
Zeiss, Oberkochen, Germany).
Determination for the mitochondrial
membrane potential (MMP) by Rhodamine 123 and DiOC6
Rhodamine 123 and DiOC6 (Molecular
Probes, Carlsbad, CA, USA) uptake by mitochondria is directly
proportional to its membrane potential. Caki cells 4 h after
treatment were incubated with Rhodamine 123 (5 µM) and
DiOC6 for 30 min in the dark at 37°C (15). The cells were harvested and
suspended in PBS. The MMP was subsequently analyzed using a flow
cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
Analysis of cytochrome c release
Caki cells (1.2×106 cells/ml) were
harvested, washed once with ice-cold PBS and gently lysed for 2 min
in 80 µl ice-cold lysis buffer [250 mM sucrose, 1 mM EDTA,
20 mM Tris-HCl (pH 7.2), 1 mM DTT, 10 mM KCL, 1.5 mM
MgCl2, 5 µg/ml pepstatin A, 10 µg/ml
leupeptin and 2 µg/ml aprotinin]. Lysates were centrifuged
at 12,000 × g at 4°C for 10 min to obtain the supernatants
(cytosolic extracts free of mitochondria) and the pellets (fraction
that contains mitochondria). The resulting cytosolic fractions were
used for western blot analysis with an anti-cytochrome c
antibody.
Statistical analysis
The data were analyzed using a one-way ANOVA
followed by post hoc comparisons (Student-Newman-Keuls) using the
Statistical Package for Social Sciences version 22.0 (SPSS, Inc.,
Chicago, IL, USA).
Results
Thymoquinone efficiently induces
apoptosis in Caki cells
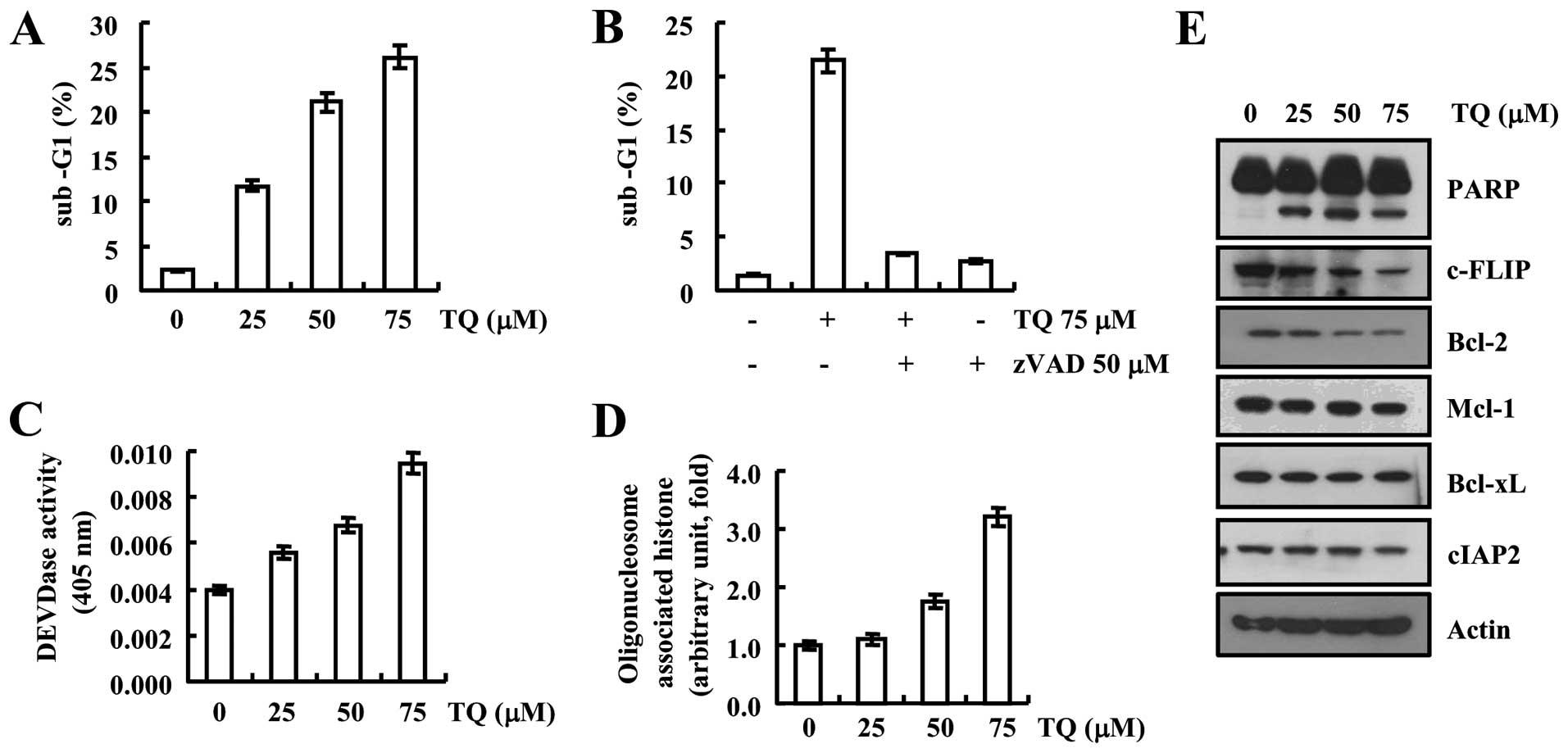
We first determined the cytotoxic effect of
thymoquinone against Caki cells and found that after treatment of
thymoquinone (25, 50 and 75 µM) for 24 h apoptotic
population (sub-G1) was increased in a dose-dependent manner
(Fig. 1A). Furthermore, we used
caspase inhibitor zVAD to examine the association of caspases in
apoptosis induced by thymoquinone, and we found that after
inhibition of caspase apoptotic population was decreased even at a
higher concentration of thymoquinone which was further validated by
DEVDase activity suggesting that thymoquinone induced
caspase-dependent apoptosis (Fig. 1B
and C). Moreover, the level of cytoplasmic histone was observed
to be increased that is an outcome of DNA fragmentation thus
thymoquinone-induced DNA fragmentation which resulted in increased
cytoplasmic histone (Fig. 1D). We
further determined the expression pattern of apoptotic and
anti-apoptotic proteins to understand the molecular mechanism
underlying the thymoquinone toxicity, as depicted in Fig. 1E, thymoquinone enhanced the cleavage
of PARP protein in a dose-dependent manner. Expression of
anti-apoptotic proteins such as c-FLIP and Bcl-2 was downregulated,
but expression of Mcl-1, Bcl-xL and cIAP2 did not change in western
blot examination (Fig. 1E).
Thymoquinone downregulates c-FLIP
expression to induce apoptosis in Caki cells
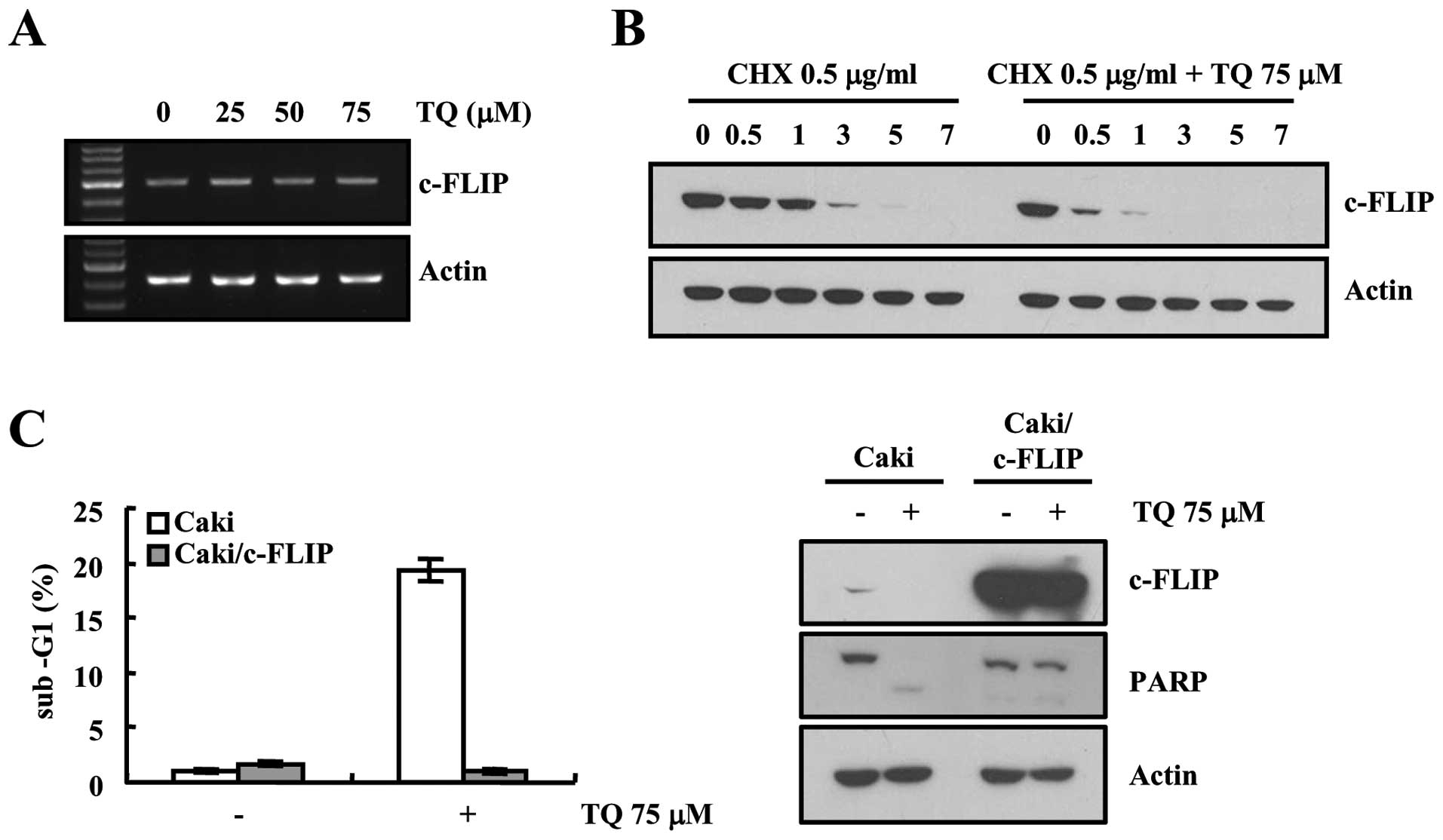
We found that thymoquinone downregulated the
expression of c-FLIP in our previous experiment, next we decided to
validate our data. We then examined the expression pattern of
c-FLIP at the transcriptional level, results of RT-PCR analysis
showed that there was no any change in the expression of c-FLIP at
the transcriptional level (Fig.
2A). Therefore, we investigated whether thymoquinone modulates
the protein stability of c-FLIP in Caki cells. Cells were treated
with cycloheximide (CHX), an inhibitor of de novo protein
synthesis, in the presence or absence of thymoquinone. CHX
gradually decreased c-FLIP, but co-treatment with CHX and
thymoquinone reduced more c-FLIP protein expression (Fig. 2B). To investigate the importance of
down-regulation of c-FLIP expression on thymoquinone-induced
apoptosis, c-FLIP protein was overexpressed in Caki cells.
Overexpression of c-FLIP markedly inhibited thymoquinone-induced
increase of sub-G1 cell population (Fig. 2C) and PARP cleavage (Fig. 2D). These results indicate that
downregulation of c-FLIP may be involved in thymoquinone-mediated
apoptosis.
Expression of Bcl-2 is downregulated
after treatment with thymoquinone through hindering the NF-κB
cascade
We evaluated the effect of thymoquinone on the
expression pattern of Bcl-2, another major regulator of the
anti-apoptotic mechanism. Results are depicted in Fig. 3, where we first determined the
expression of Bcl-2 at the transcriptional level and found that
Bcl-2 mRNA expression was reduced in a dose-dependent manner
(Fig. 3A). Furthermore, we
validated our data with Bcl-2 promoter assay and found that
thymoquinone reduced the Bcl-2 promoter activity in a
dose-dependent manner (Fig. 3B).
Moreover, we determined the effect of thymoquinone on NF-κB
expression as it is associated with expression of Bcl-2 protein.
NF-κB luciferase activity was reduced after treatment of
thymoquinone. In addition, overexpression of p65, a subunit of
NF-κB heightened the Bcl-2 promoter activity in case of
thymoquinone treated cells, which was further confirmed by
immunoblot analysis (Fig. 3D). We
examined the efficacy of thymoquinone to induce apoptosis in Bcl-2
overexpressed Caki cells and observed that overexpression of Bcl-2
reversed the apoptotic potential of thymoquinone as evidenced by
flow cytometric analysis (Fig. 3E).
Moreover, thymoquinone-induced cleavage of PARP protein was blocked
in Bcl-2 overexpressed cells (Fig.
3E), suggesting the downregulation of Bcl-2 plays a critical
role in thymoquinone-induced apoptosis.
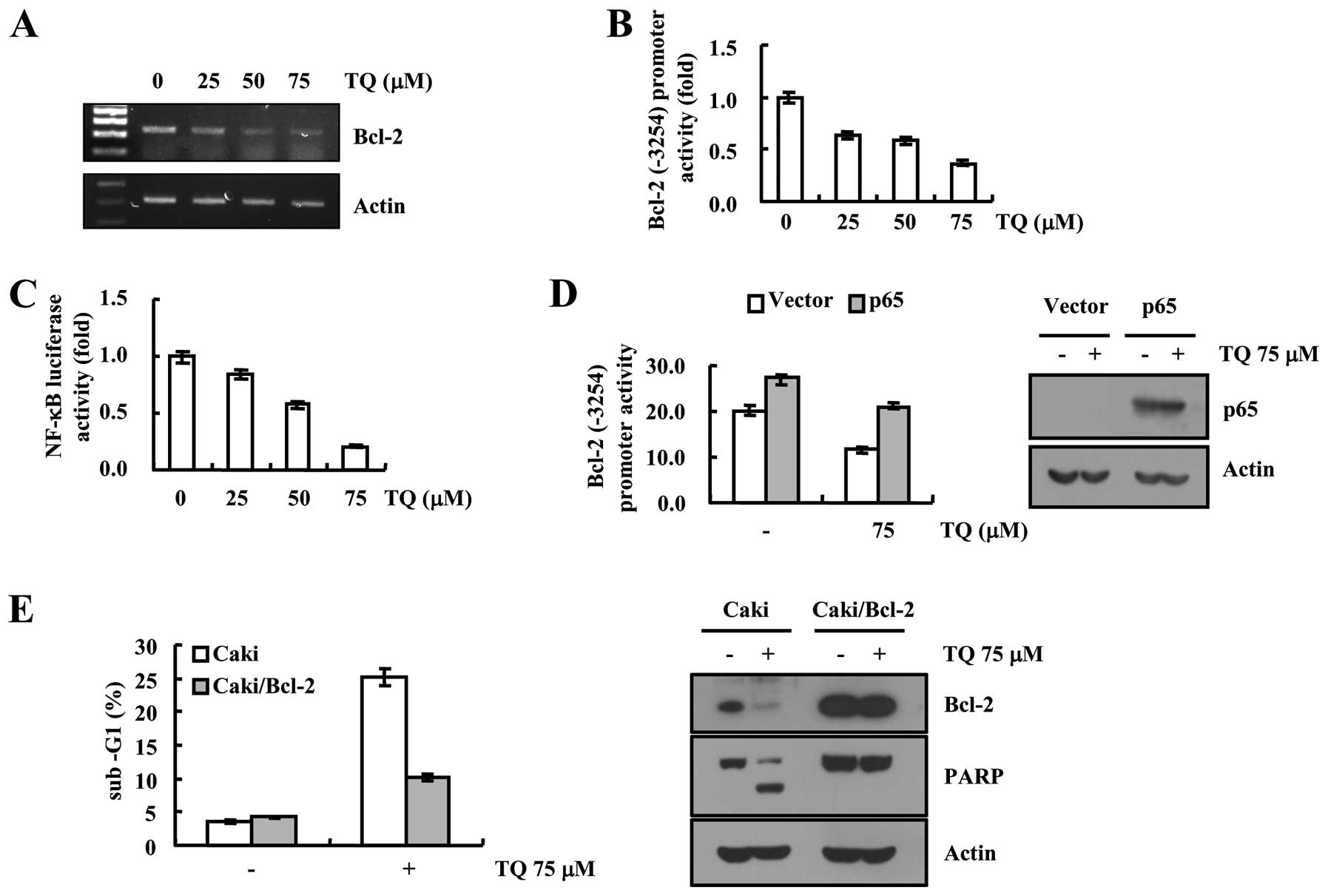 | Figure 3Thymoquinone downregulates Bcl-2
expression at the transcriptional level in Caki cells. (A) Caki
cells were treated with the indicated concentrations of
thymoquinone for 24 h. Bcl-2 and actin mRNA expression were
determined using RT-PCR. (B) Caki cells were transiently
transfected with a plasmid harboring the luciferase gene under the
control of the Bcl-2/-3254 promoter. After transfection, the Caki
cells were treated with the indicated concentrations of
thymoquinone for 24 h. After treatment, the cells were lysed, and
the luciferase activity was analyzed. (C) Caki cells were
transiently transfected with NF-κB-luciferase construct. After
transfection, the Caki cells were treated with the indicated
concentrations of thymoquinone for 24 h. After treatment, the cells
were lysed and the luciferase activity was analyzed. (D) Caki cells
were transiently co-transfected with NF-κB subunit (p65) and
Bcl-2-luciferase construct. After transfection, the Caki cells were
treated with 75 µM thymoquinone for 24 h. After treatment,
the cells were lysed, and the luciferase activity was analyzed.
Equal amounts of cell lysates (60 µg) were subjected to
electrophoresis and analyzed by western blotting for p65 and actin
as a control for protein loading. (E) Vector (Caki/Vec) and Bcl-2
overexpressed cells (Caki/Bcl-2) were treated with 75 µM
thymoquinone for 24 h. Apoptosis was analyzed as a sub-G1 fraction
by FACS. Equal amounts of cell lysates (60 µg) were
subjected to electrophoresis and analyzed by western blotting for
PARP, Bcl-2 and actin as a control for protein loading. The values
in (B, C, D and E) represent the mean ± SD from three independent
samples. |
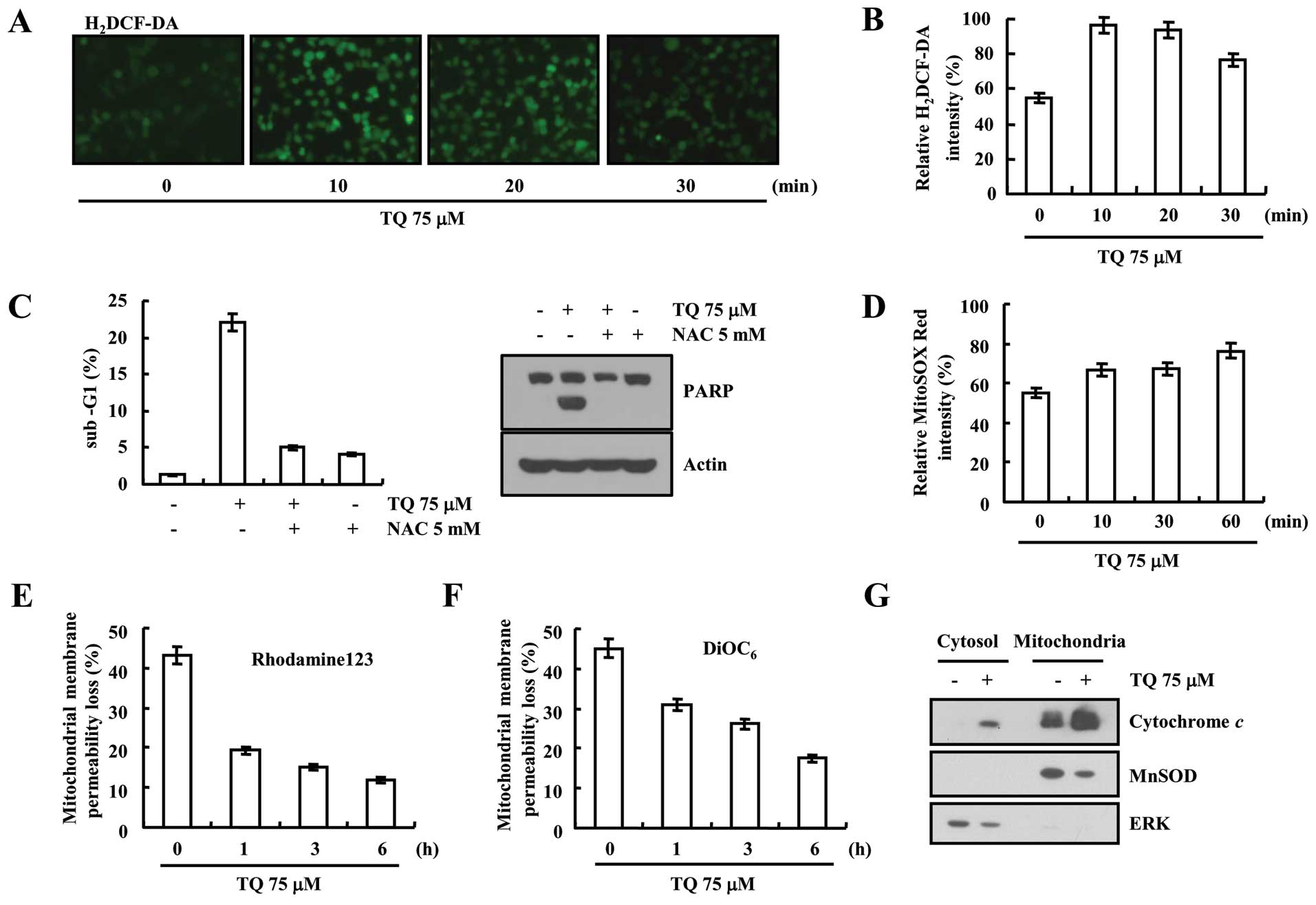
Thymoquinone-induced ROS generation
resulting in loss of MMP in Caki cells
The induction of ROS production plays an important
role in apoptosis. Therefore, we carried out H2DCFDA staining to
determine the level of intracellular ROS. Thymoquinone (75
µM) potentially induced the generation of intracellular ROS
within 10 min, and then slightly reduced after 30 min of treatment
(Fig. 4A and B). Furthermore, we
determined the effect of thymoquinone-induced ROS on cell death
with or without using the ROS scavenger N-acetylcysteine
(NAC). Thymoquinone-induced ROS generation caused apoptosis in Caki
cells which was reduced after using the ROS scavenger (Fig. 4C). We observed increased intensity
of MitoSOX Red dye which detects mitochondrial ROS production
(Fig. 4D). Furthermore, we
determined the MMP, as the exaggerated production of ROS leads to
the mitochondrial damage. Rhodamine 123 and DiOC6
fluorometry data revealed the loss of MMP in time-dependent manner
at 75 µM thymoquinone treatment (Fig. 4E and F). Treatment with thymoquinone
caused cytochrome c release into cytoplasm (Fig. 4G). These results suggest that
thymoquinone reduces the MMP levels and induces cytochrome c
release.
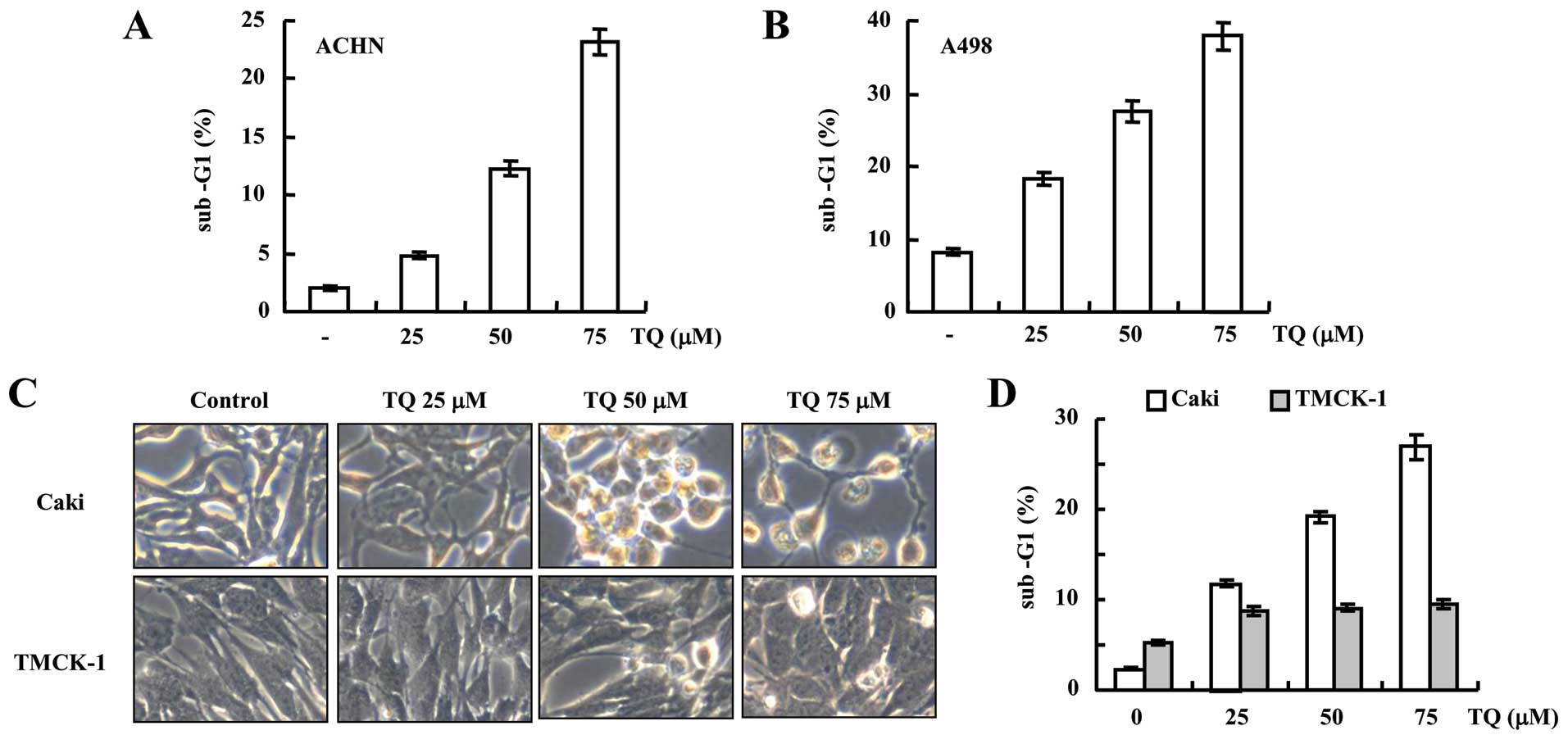
Thymoquinone exhibits cytotoxic effect
against various renal carcinoma cells, but not in normal cells
Although, our results demonstrated that thymoquinone
potentially induced apoptosis in Caki cells, we further examined
the efficacy of thymoquinone against other renal carcinoma cell
line for example ACHN and A498 and found that thymoquinone has
efficacy to induce apoptosis in all the renal carcinoma cells used
in the present study (Fig. 5A and
B). However, there was the insignificant cytotoxic effect of
thymoquinone on normal TMCK-1 cells neither on both morphology and
cell population analysis, even at a higher concentration of 75
µM which induced the apoptosis in Caki cells (Fig. 5C and D) suggesting the safe use of
thymoquinone.
Discussion
There is an intricate balance between pro- and
anti-apoptotic proteins in normal cells; however, in cancer cells
there is a dysregulation of this balance and that may be due to
overexpression of anti-apoptotic proteins which demise the process
of apoptosis (16), for instance,
overexpression of Bcl-2, an anti-apoptotic protein, protects the
apoptosis of prostate cancer cells (17). Consequently, in the present study we
evaluated for example the apoptotic PARP as well as the
anti-apoptotic (c-FLIP, Bcl-2, Mcl-1, Bcl-xL and cIAP2) markers
through immunoblot analysis and observed that thymoquinone
treatment induced the cleavage of PARP and downregulation of c-FLIP
and Bcl-2 proteins.
c-FLIP is a master regulator of anti-apoptotic
mechanism and is found to be abundantly expressed in a variety of
cancer cells (18). Overexpression
of c-FLIP interferes with caspase-8 protein and is able to block
the cleavage of caspase-8 which ultimately terminates the apoptotic
machinery (4). Thymoquinone
downregulated the expression of c-FLIP at translation level but
surprisingly there was no effect of thymoquinone on transcriptional
regulation of c-FLIP, however, it is well documented in literature
that ROS contribute to post-translation modification of c-FLIP,
through upregulation of proteasomal activity (19,20).
Notably, we found that thymoquinone potentially induced production
of intracellular ROS in Caki cells, thus, we hypothesize that this
property of thymoquinone may be responsible for the downregulation
of c-FLIP.
Another key regulator protein of anti-apoptotic
machinery is Bcl-2 (21–23), and in the present study, we found
that thymoquinone potentially suppressed the expression of this
protein at both transcriptional and translation level resulting in
the initiation of the apoptotic cascade. However, recently numerous
studies have explored the association of NF-κB with Bcl-2, for
example, expression of the NF-κB targets BCL-2 expression on the
transcriptional level (24,25). We, therefore, examined the possible
role of thymoquinone in the regulation of NF-κB activity and
notably, a highly decreased NF-κB activity was observed in
luciferase reporter assay in a concentration-dependent manner
suggesting NF-κB inhibitory efficacy of thymoquinone. In contrast,
when we overexpressed p65, a subunit of NF-κB it caused increased
promoter activity of Bcl-2 even at a higher concentration of
thymoquinone which clearly indicates that downregulation of Bcl-2
by thymoquinone was associated with NF-κB activity. Heckman et
al (25) reported similar
results in lymphoma cells where overexpression of NF-κB was
responsible for the Bcl-2 overexpression, taken together, we can
conclude that hindrance in NF-κB activity may be an effective tool
to target the Bcl-2 expression which was accomplished by
thymoquinone in our case.
We further evaluated the effect of thymoquinone on
MMP, as we found that thymoquinone potentially downregulated Bcl-2
expression which in turn is associated with the loss of MMP leading
to mitochondrial death, a potent and common anticancer target for
many pharmacological agents (26–28).
In addition, a well-known inducer of mitochondrial stress is
generation of exaggerated ROS inside the cell which eventually kill
the mitochondria (29-31). Interestingly, both the phenomenon
associated with mitochondrial stress (Bcl-2 downregulation and ROS
generation) were found to be followed by thymoquinone in the
present study, thus, we examined the mitochondrial membrane
potential (MMP) in the presence of thymoquinone and as expected,
the loss of MMP was the outcome of the experiment with an intense
release of cytochrome c into cytosol, a defining feature of
mitochondrial damage (31).
However, undoubtedly, a potent candidate for anticancer therapy
should be one which possess toxic effect against cancer cells but
not to normal cells and we in the case of thymoquinone found
similar results showing insignificant toxic effect on normal cells
(TMCK-1 cells).
On account of the results obtained in the present
study, we conclude that thymoquinone has potential to induce
apoptosis in renal cell carcinoma with a major mechanism of c-FLIP
and Bcl-2 downregulation. In addition, it is able to induce
mitochondrial dysfunctioning through the generation of
intracellular ROS in Caki cells which eventually leads to cell
death. Moreover, it originates from natural plants and its
insignificant toxicity to the normal cells warrant its safe use as
a drug against renal cell carcinoma. However, further experiments
on an animal model is needed to evaluate its efficacy and precise
mechanism in vivo.
Acknowledgments
The present study was supported by an NRF grant
funded by the Korea Government (MSIP) (2014R1A5A2010008) and a 2015
Scholar Research Grant from Keimyung University.
References
|
1
|
Hodorová I, Rybárová S, Solár P, Vecanová
J, Mihalik J, Bohus P, Mellová Y and Kluchová D: Multidrug
resistance proteins in renal cell carcinoma. Folia Biol.
54:187–192. 2008.
|
|
2
|
Motzer RJ, Russo P, Nanus DM and Berg WJ:
Renal cell carcinoma. Curr Probl Cancer. 21:185–232. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rini BI and Atkins MB: Resistance to
targeted therapy in renal-cell carcinoma. Lancet Oncol.
10:992–1000. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park EJ, Min KJ, Choi KS and Kwon TK:
Dicoumarol sensitizes renal cell carcinoma Caki cells to
TRAIL-induced apoptosis through down-regulation of Bcl-2, Mcl-1 and
c-FLIP in a NQO1-independent manner. Exp Cell Res. 323:144–154.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han MA, Woo SM, Min KJ, Kim S, Park JW,
Kim DE, Kim SH, Choi YH and Kwon TK: 6-Shogaol enhances renal
carcinoma Caki cells to TRAIL-induced apoptosis through reactive
oxygen species-mediated cytochrome c release and down-regulation of
c-FLIP(L) expression. Chem Biol Interact. 228:69–78. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Motzer RJ, Hutson TE, Cella D, Reeves J,
Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, et
al: Pazopanib versus sunitinib in metastatic renal-cell carcinoma.
N Engl J Med. 369:722–731. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keisner SV and Shah SR: Pazopanib: The
newest tyrosine kinase inhibitor for the treatment of advanced or
metastatic renal cell carcinoma. Drugs. 71:443–454. 2011.PubMed/NCBI
|
|
8
|
Gotink KJ, Rovithi M, de Haas RR,
Honeywell RJ, Dekker H, Poel D, Azijli K, Peters GJ, Broxterman HJ
and Verheul HM: Cross-resistance to clinically used tyrosine kinase
inhibitors sunitinib, sorafenib and pazopanib. Cell Oncol.
38:119–129. 2015. View Article : Google Scholar
|
|
9
|
Juengel E, Kim D, Makarević J, Reiter M,
Tsaur I, Bartsch G, Haferkamp A and Blaheta RA: Molecular analysis
of sunitinib resistant renal cell carcinoma cells after sequential
treatment with RAD001 (everolimus) or sorafenib. J Cell Mol Med.
19:430–441. 2015. View Article : Google Scholar
|
|
10
|
Salomi MJ, Nair SC and Panikkar KR:
Inhibitory effects of Nigella sativa and saffron (Crocus sativus)
on chemical carcinogenesis in mice. Nutr Cancer. 16:67–72. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tiruppur Venkatachallam SK, Pattekhan H,
Divakar S and Kadimi US: Chemical composition of Nigella sativa L.
seed extracts obtained by supercritical carbon dioxide. J Food Sci
Technol. 47:598–605. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lei X, Lv X, Liu M, Yang Z, Ji M, Guo X
and Dong W: Thymoquinone inhibits growth and augments
5-fluorouracil-induced apoptosis in gastric cancer cells both in
vitro and in vivo. Biochem Biophys Res Commun. 417:864–868. 2012.
View Article : Google Scholar
|
|
13
|
Gali-Muhtasib H, Roessner A and
Schneider-Stock R: Thymoquinone: A promising anti-cancer drug from
natural sources. Int J Biochem Cell Biol. 38:1249–1253. 2006.
View Article : Google Scholar
|
|
14
|
Mi Y, Zhang C, Bu Y, Zhang Y, He L, Li H,
Zhu H, Li Y, Lei Y and Zhu J: DEPDC1 is a novel cell cycle related
gene that regulates mitotic progression. BMB Rep. 48:413–418. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Seo K, Ki SH and Shin SM: Methylglyoxal
induces mitochondrial dysfunction and cell death in liver. Toxicol
Res. 30:193–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raffo AJ, Perlman H, Chen MW, Day ML,
Streitman JS and Buttyan R: Overexpression of bcl-2 protects
prostate cancer cells from apoptosis in vitro and confers
resistance to androgen depletion in vivo. Cancer Res. 55:4438–4445.
1995.PubMed/NCBI
|
|
18
|
Shirley S and Micheau O: Targeting c-FLIP
in cancer. Cancer Lett. 332:141–150. 2013. View Article : Google Scholar
|
|
19
|
Wilkie-Grantham RP, Matsuzawa S and Reed
JC: Novel phosphorylation and ubiquitination sites regulate
reactive oxygen species-dependent degradation of anti-apoptotic
c-FLIP protein. J Biol Chem. 288:12777–12790. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Willis S, Day CL, Hinds MG and Huang DC:
The Bcl-2-regulated apoptotic pathway. J Cell Sci. 116:4053–4056.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar
|
|
22
|
Labi V, Grespi F, Baumgartner F and
Villunger A: Targeting the Bcl-2-regulated apoptosis pathway by BH3
mimetics: A breakthrough in anticancer therapy? Cell Death Differ.
15:977–987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tracey L, Pérez-Rosado A, Artiga MJ,
Camacho FI, Rodríguez A, Martínez N, Ruiz-Ballesteros E, Mollejo M,
Martinez B, Cuadros M, et al: Expression of the NF-kappaB targets
BCL2 and BIRC5/Survivin characterizes small B-cell and aggressive
B-cell lymphomas, respectively. J Pathol. 206:123–134. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Catz SD and Johnson JL: Transcriptional
regulation of bcl-2 by nuclear factor kappa B and its significance
in prostate cancer. Oncogene. 20:7342–7351. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Heckman CA, Mehew JW and Boxer LM:
NF-kappaB activates Bcl-2 expression in t(14;18) lymphoma cells.
Oncogene. 21:3898–3908. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang S, Hu J, Meng Q, Dong X, Wang K, Qi
Y, Chu C, Zhang X and Hou L: Daidzein induced apoptosis via
down-regulation of Bcl-2/Bax and triggering of the mitochondrial
pathway in BGC-823 cells. Cell Biochem Biophys. 65:197–202. 2013.
View Article : Google Scholar
|
|
27
|
Chen QY, Lu GH, Wu YQ, Zheng Y, Xu K, Wu
LJ, Jiang ZY, Feng R and Zhou JY: Curcumin induces mitochondria
pathway mediated cell apoptosis in A549 lung adenocarcinoma cells.
Oncol Rep. 23:1285–1292. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Slimen IB, Najar T, Ghram A, Dabbebi H,
Ben Mrad M and Abdrabbah M: Reactive oxygen species, heat stress
and oxidative-induced mitochondrial damage. A review. Int J
Hyperthermia. 30:513–523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cai J, Yang J and Jones DP: Mitochondrial
control of apoptosis: The role of cytochrome c. Biochim Biophys
Acta. 1366:139–149. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Banjerdpongchai R, Kongtawelert P,
Khantamat O, Srisomsap C, Chokchaichamnankit D, Subhasitanont P and
Svasti J: Mitochondrial and endoplasmic reticulum stress pathways
cooperate in zearalenone-induced apoptosis of human leukemic cells.
J Hematol Oncol. 3:502010. View Article : Google Scholar : PubMed/NCBI
|