Introduction
Malignant gliomas account for close to 50% of all
CNS tumors. The median survival of patients with glioblastoma
multiforme (GBM) remains close to one year from the time of
diagnosis in spite of surgical resection followed by radiotherapy
and chemotherapy (1). Such poor
outcome has led to the exploration of a wide variety of novel
therapies, and some of them have been incorporated as a standard
treatment for patients with this GBM. Temozolomide (TMZ) is
approved to use as first-line treatment for patients with primary
and recurrent high-grade gliomas. It has been shown that TMZ
combined with radiotherapy can improved the 2-year survival rate
from 10.4% with radiotherapy alone to 26.5% in patients with GBM
(1). Whereas the efficacy of TMZ is
encouraging, additional prolongation of survival remains a
challenge. GBM shows chemoresistance shortly after the initiation
of treatment. Additionally, recent studies suggested that 60–75% of
patients with GBM derive no benefit from treatment with TMZ
(2). There is a critical need for
means to overcome this drug resistance and expand the limited
therapeutic benefit of TMZ.
TMZ is an alkylating agent which binds to DNA and
interferes with replication, resulting in the insertion of DNA
strand breaks and, ultimately, cell death (3). p53 status in addition to MGMT plays a
role in chemotherapy resistance to TMZ (4,5). GBM
patients with low mutant p53 expression have higher
progression-free survival time and may have longer life expectancy
in comparison with the high mutant p53 expression group (6). p53 facilitates favorable antitumor
drug response through a variety of key cellular functions,
including cell cycle arrest, senescence, and apoptosis (5). Wip1 (wild-type p53-induced phosphatase
1, or PPM1D), initially identified as a p53-regulated allele
located on 17q23-24, is a member of the protein phosphatase 2C
(PP2C) family and expressed in a p53-dependent manner (7). PPM1D is frequently amplified and
overexpressed in many cancers, including gliomas, breast cancers,
neuroblastomas, ovarian clear cell adenocarcinomas, and
medulloblastomas (8–13). In addition, it has been shown that
PPM1D may serve as an oncogene important to astrocytoma
progression, especially in astrocytomas with wild-type p53
(8). PPM1D overexpression inhibits
p53 functions and reduces selection for p53 mutations during cancer
progression. However, whether PPM1D has a role in chemotherapy
resistance to TMZ through regulating p53 functions remains
uncertain.
In our previous studies, the lentiviral shRNA
expression vector capable of stable PPM1D gene silencing at both
mRNA and protein levels in glioma cells was constructed (14). In the present study, we demonstrated
PPM1D silencing can improve the effect of TMZ on inhibiting the
growth and inducing cell apoptosis in glioma cells. The possible
mechanisms were also detected.
Materials and methods
Cell culture
The human glioma cell line U87-MG cells were
cultured in complete medium consisting of DMEM (Gibco, Grand
Island, NY, USA) containing high glucose and pyruvate, supplemented
with 10% FBS, 2 mmol/l L-glutamine, 100 U/ml penicillin G and 100
ng/ml streptomycin. Cells were maintained at 37°C in a humidified
5% CO2 atmosphere. The cells were dissociated using
0.25% trypsin and 0.02% EDTA solution and subcultured once in 3
days.
Lentiviral vector infection and PPM1D
silencing
The target shRNAs against human PPM1D gene (GenBank
accession NM_003620) for RNAi were designed as:
5′-GCATAGACGAAATGGCTTA-3′. The sequence 5′-TTCTCCGAACGTGTCACGT-3′,
which had no significant homology to any known human or mouse
genes, was used as a negative control. Lentiviral vector infection,
assessment of infection rate and silencing efficacy determination
were performed as previously described (14). Cells treated with PBS instead of
lentiviral vector infected were used as control. PPM1D mRNA
expression levels in U87-MG cells were detected by real-time
quantitative PCR. Primer sequences used to amplify PPM1D mRNA for
RT-qPCR were as follows: forward 5′-CTACACCACCAGTCAAGTCAC-3′, and
reverse 5′-AGAAGGCATTGCTACGAACC-3′; these amplified a 93-bp
product. Primer sequences of the control amplification of GAPDH
mRNA were as follows: forward 5′-TGACTTCAACAGCGACACCCA-3′ and
reverse 5′-CACCCTGTTGCTGTAGCCAAA-3′; these amplified a fragment of
121 bp. The relative mRNA level was calculated comparing normalized
gene expression values in treated versus untreated control U87-MG
cells with the 2−ΔΔCt method.
Drug treatment
Three days after cells were infected with lentiviral
vector and the silencing efficacy of PPM1D was verified, cells were
harvested and approximately 1.0×105 cells were plated in
60-mm petri dishes 24 h before treatment with 0.1 mmol/l TMZ
(Schering-Plough, Madison, NJ, USA). The IC50 value of
TMZ, defined as the concentration that reduces the global growth of
cells by 50%, was previously determined to be approximately 200
mmol/l (15). Therefore, the
concentration of TMZ used in this study was set at 0.1 mmol/l,
which was considered to be approximately similar to the plasma
concentration found in human subjects (16) and was the same as published data of
TMZ used for glioma cells (17,18).
TMZ stocks were prepared by dissolving the drug in DMSO, diluting
with sterile H2O, filtering, and storing at −80°C. After
treatment, cells were gently washed, incubated in fresh media at
37°C, and harvested at various time points. Cells were divided into
3 groups: CON (infection with PBS alone+DMSO), NC (infection with
negative control vector+TMZ) and KD (infection with PPM1D RNAi
lentiviral vector+TMZ).
Cell proliferation assay
Cell proliferation assay was performed with
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT). In brief, when cells were treated with TMZ and incubated for
1, 2, 3, 4 and 5 days, 10 μl of the 5 mg/ml MTT was added.
The liquid was disgarded and 100 μl DMSO was added. The
absorbance (A) at 490 nm was measured using a microplate reader
after the plate was incubated. Then a calibration curve was
prepared using the data obtained from the wells that contained
known numbers of viable cells. Experiments were performed three
times with representative data presented. The experiment was
performed in triplicate and repeated three times.
Cell cycle analysis
Cells were trypsinized and collected together with
the cells floating in the media 4 days after treated with TMZ.
These cells were washed in PBS, fixed in 70% (v/v) ethanol for more
than 1 h. Then the cells were washed and resuspended in PBS
containing 50 mg/ml propidium iodine (PI, Sigma Chemical Co.) and
100 μg/ml RNase A (Fermentas Co., Glen Burnie, MD, USA) for
1 h at room temperature in the dark. Then a total of 20,000
PI-stained nuclei were analyzed using a Becton Dickinson FACScan
(BD Biosciences, San Jose, CA, USA). Analysis was used DNA content
as a measure of progression in the cell cycle.
Apoptosis assay
After infected with lentiviral vector and treated
with TMZ, both floating and adherent cells were collected 4 days
after TMZ treatment. The cell aliquots were combined, washed with
ice-cold PBS, resuspended in 1X binding buffer, and stained using
the Annexin V-APC apoptosis detection kit (BD Biosciences),
following the protocols provided. Annexin V-APC staining cells were
quantified by using a Becton Dickinson FACScan, according to the
manufacturer's instructions.
Microarray-based gene expression
profiling
Total RNA was extracted from cells with TRIzol
(Invitrogen), according to the manufacturer's instructions. After
RNA yield and quality were assessed by Thremo Nanodrop 2000 and
Agilent 2100 Bioanalyzer, amplified RNA (aRNA) was obtained with
GeneChip 3′IVT Express kit (Affymetrix), following the instructions
provided. Then purified aRNA was fragmented and hybridized with
hybridization reaction mixture for 16 h at 45°C to human cDNA
microarrays. Preparation of microarrays for scanning was conducted
with Affymetrix wash protocols appropriately matched to the
specific chip type on a Model 450 Fluidics station. Microarrays
were scanned on a GeneChip Scanner 3000 (Affymetrix) after the last
wash. The Affymetrix GeneChip operating Software v1.3 operating
system controlled data acquisition functions and maintained the
mediated first-level data analysis and desktop data management for
the entire GeneChip System.
Data selection
Primary data collection was done and analyzed by
GeneChip Analysis Software (Affymetrix). The fold changes were
calculated relative to baseline controls. Data from three
independent replicate experiments were used to perform a paired
two-sample t-test for each gene. Regulated genes used to define
genes as significantly differentially expressed as fold change of
≥1.5 or ≤ −1.5, which signifies changes in the expression level
between KD and NC cells. A change must be in the P-value of
<0.05, which describes the likelihood of change of expression
for each transcript, where P-values indicated the level of
significance of the difference based on the paired two-sample
t-test. Pathway Architect Software (Stratagene) was used for gene
pathway enrichment analysis and ontology assessment.
Western blotting
After treated with TMZ for 4 days, the cells were
lysed using M-PER lysis buffer. Protein was extracted and
quantified using a BCA protein assay kit (Pierce Biotechnology,
Inc., Rockford, IL, USA). A total of 30 μg of each sample
was heated at 95°C for 10 min and loaded into 10% gel. Samples were
electrophoresed at 110 V for 60–90 min and then transferred to PVDF
membranes at 300 mA for 150 min using a semi-dry transfer
apparatus. Membranes were blocked in 5% non-fat dry milk for 2 h,
incubated with primary antibodies [anti-AKT3, anti-RB1, anti-MAPK8
and anti-PIK3R1 (Cell Signaling Technology, Inc., Danvers, MA,
USA)] overnight, washed with TBS containing 0.05% Triton-X 100
(TBST) followed by an incubation of 1 h in goat anti-rabbit
secondary antibody (1:5,000; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) conjugated with HRP. After final washing with TBST,
the membranes were developed using chemiluminescence and exposed to
X-ray film. The immunoblots were quantified with software quantity
one version 4.6.2. The expression levels of AKT3, RB1, MAPK8 and
PIK3R1 in each sample were internally normalized to GAPDH and
levels were given relative to expression in NC group.
Statistical analysis
Statistical analyses were performed using SPSS
software version 16.0. The relationships between various parameters
were analyzed statistically by the χ2-test, ANOVA,
Fisher's exact test, or Student's t-test as appropriate. P<0.05
was considered to indicate a statistically significant
difference.
Results
Lentiviral vector infection and PPM1D
silencing
Infection efficiency of lentiviral RNAi vector of
PPM1D and the negative control vector in U87-MG cells was >90%
(data not shown). The silencing efficacy of PPM1D in U87-MG cells
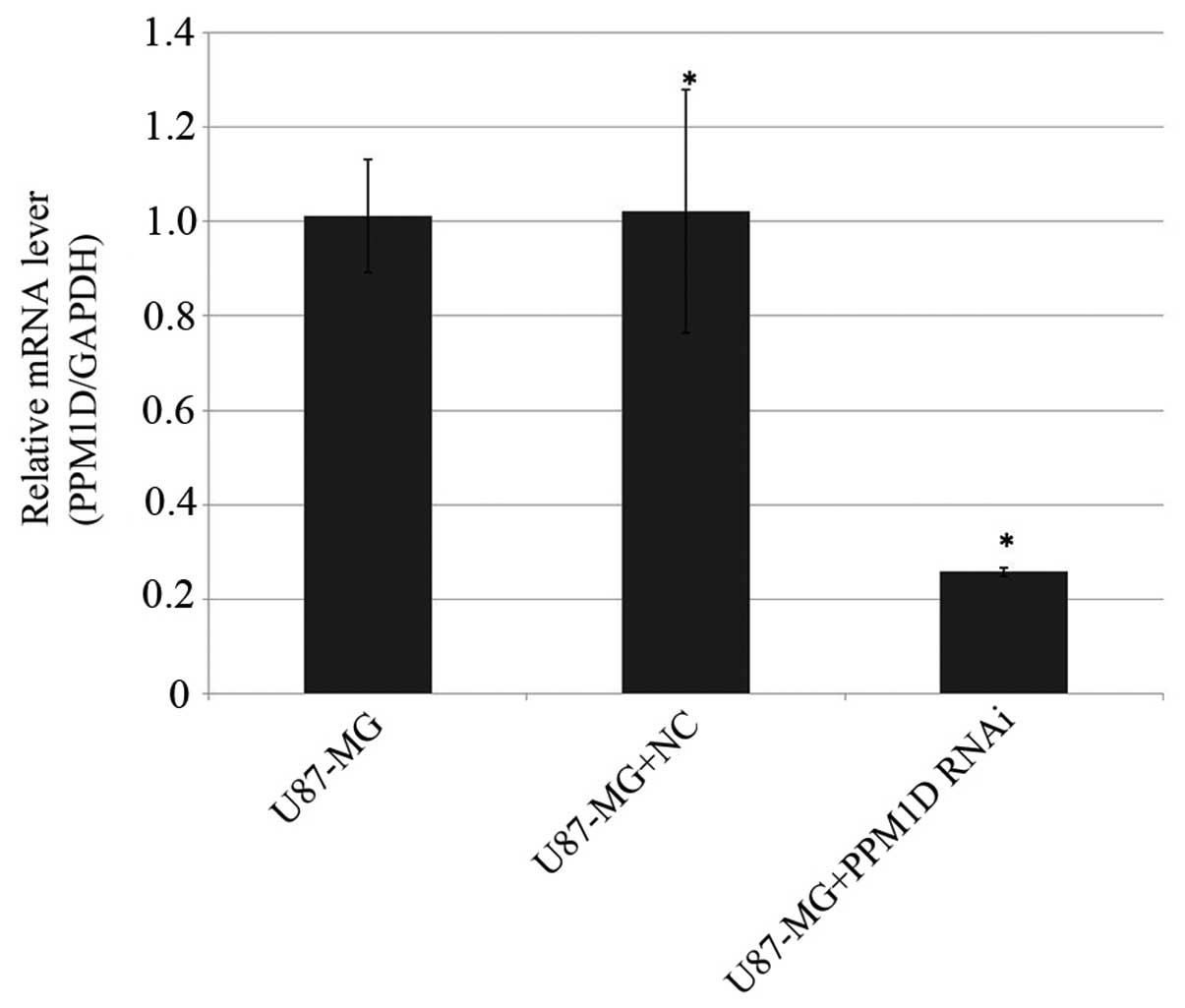
was determined by RT-qPCR. As shown in Fig. 1, the expression level of PPM1D in
U87-MG cells was remarkably reduced by 74.3% compared with that in
negative control cells (P<0.05).
PPM1D gene silencing improves TMZ induced
cell proliferation
The cell numbers of PPM1D silencing and control
siRNA transfection cells following TMZ administration were detected
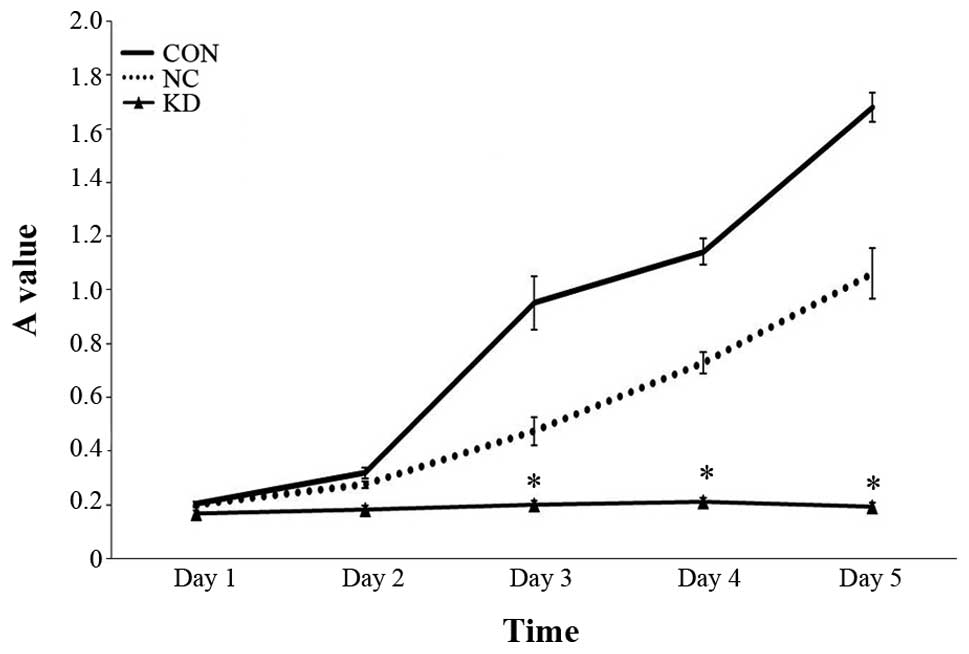
by MTT at 24, 48, 72, 96 and 120 h. A statistically significant
(P<0.05) reduction in cell growth was seen in KD group comparing
with NC group and CON group (Fig.
2). Cell proliferation ability in KD group has reduced by 57.7%
comparing with NC group 72 h after TMZ treatment. In contrast,
though cell growth in NC group showed a slightly reduced cell
proliferation, the differences did not reach statistical
significance.
Combination treatment of TMZ and PPM1D
gene silencing induce apoptosis and cell cycle arrest
To explore whether the reduction in cell number in
KD group was caused by reduced cell proliferation or by increased
cell death, we subjected the cells to cell cycle and apoptosis
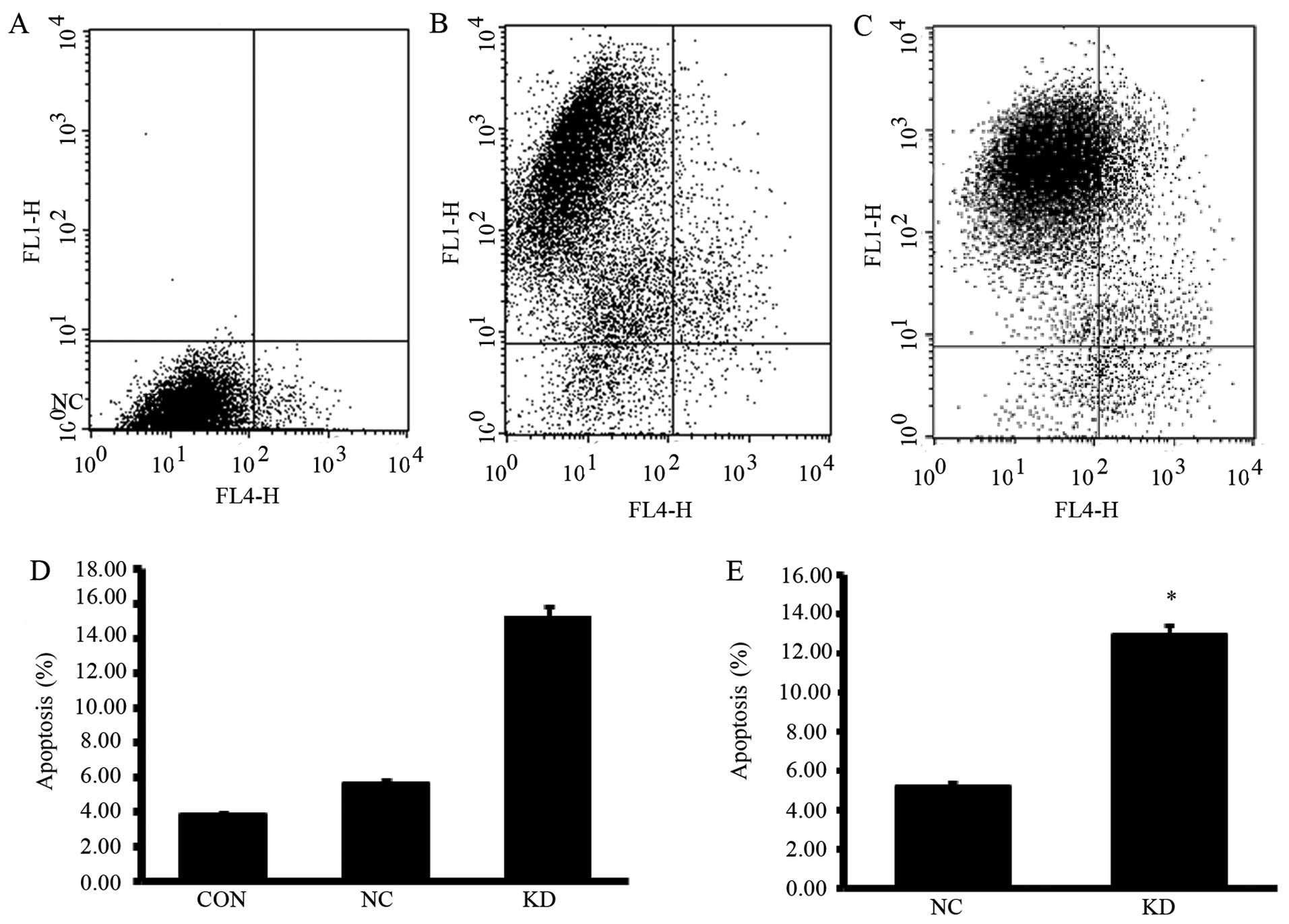
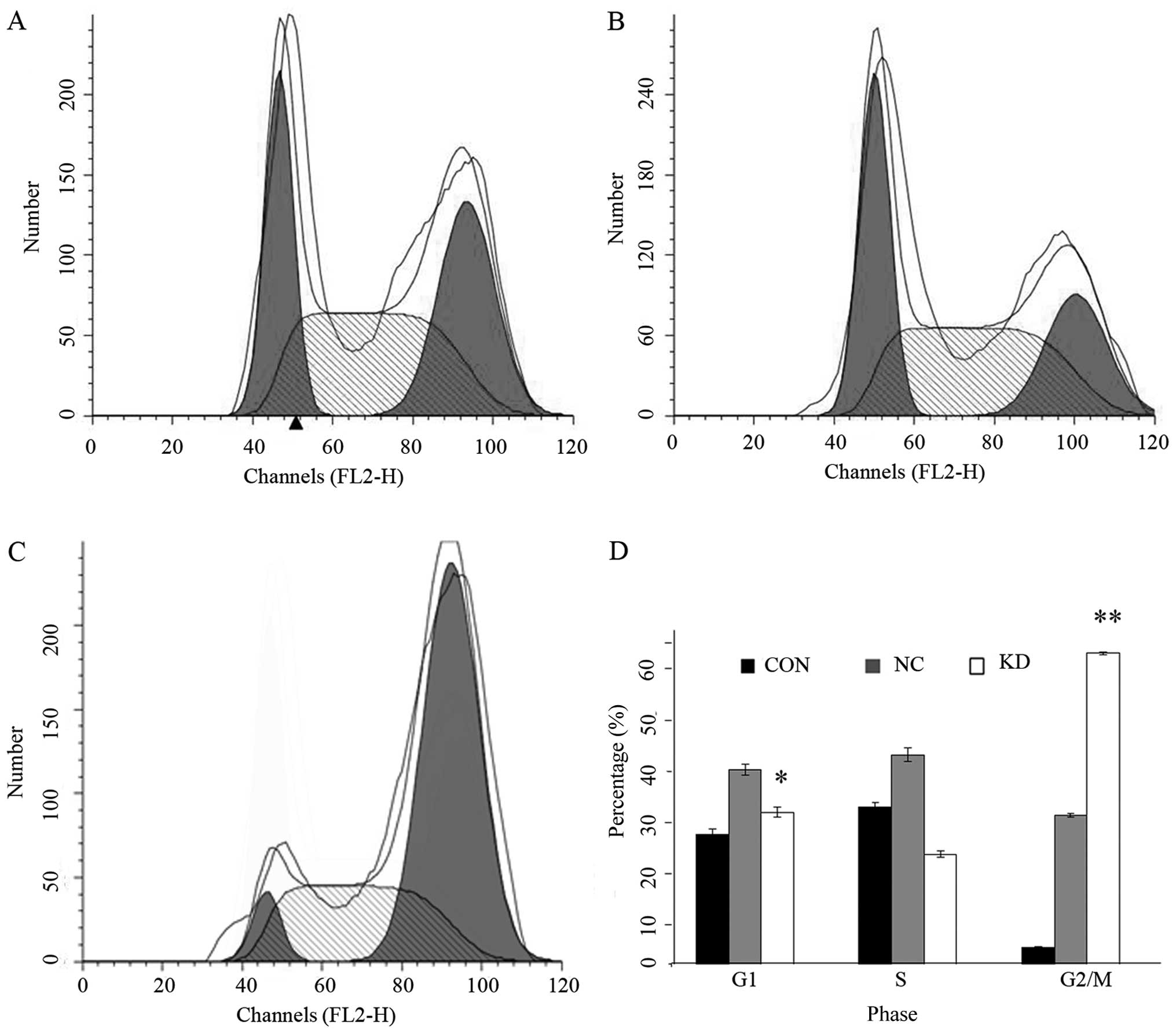
analyses. As shown in Fig. 3, cells
in KD group showed a significantly increased G2/M phase
and decreased G1 phase cells (P<0.05). Apoptosis
analyses showed that PPM1D silencing enhanced the sensitivity to
TMZ induced apoptosis inU87-MG cells (Fig. 4). Moreover, NC group cells which
were treated with TMZ alone showed slightly increased apoptotic
cells compared with CON group cells, but the differences did not
reach statistical significance.
Differential gene induction and pathway
enrichment analysis in PPM1D gene silencing U-87MG cells treated
with TMZ
To investigate the mechanisms of changes in cell
biology when PPM1D gene silenced U-87MG cells were treated with
TMZ, both KD and NC cells were conducted with a whole genome-based
transcriptional analysis. Compared with NC group, 367 genes were
upregulated and 444 genes were downregulated. Then all the
differentially expressed genes were confirmed with gene pathway
enrichment analysis, based on all known KEGG pathway genes. The
analysis algorithm ranks all genes by their significance of
differential expression and then looks for groups of biologically
relevant genes that are enriched at either the top or bottom of the
ranked list. The statistical significance of a pathway was examined
by a permutation procedure and showed as a nominal P-value
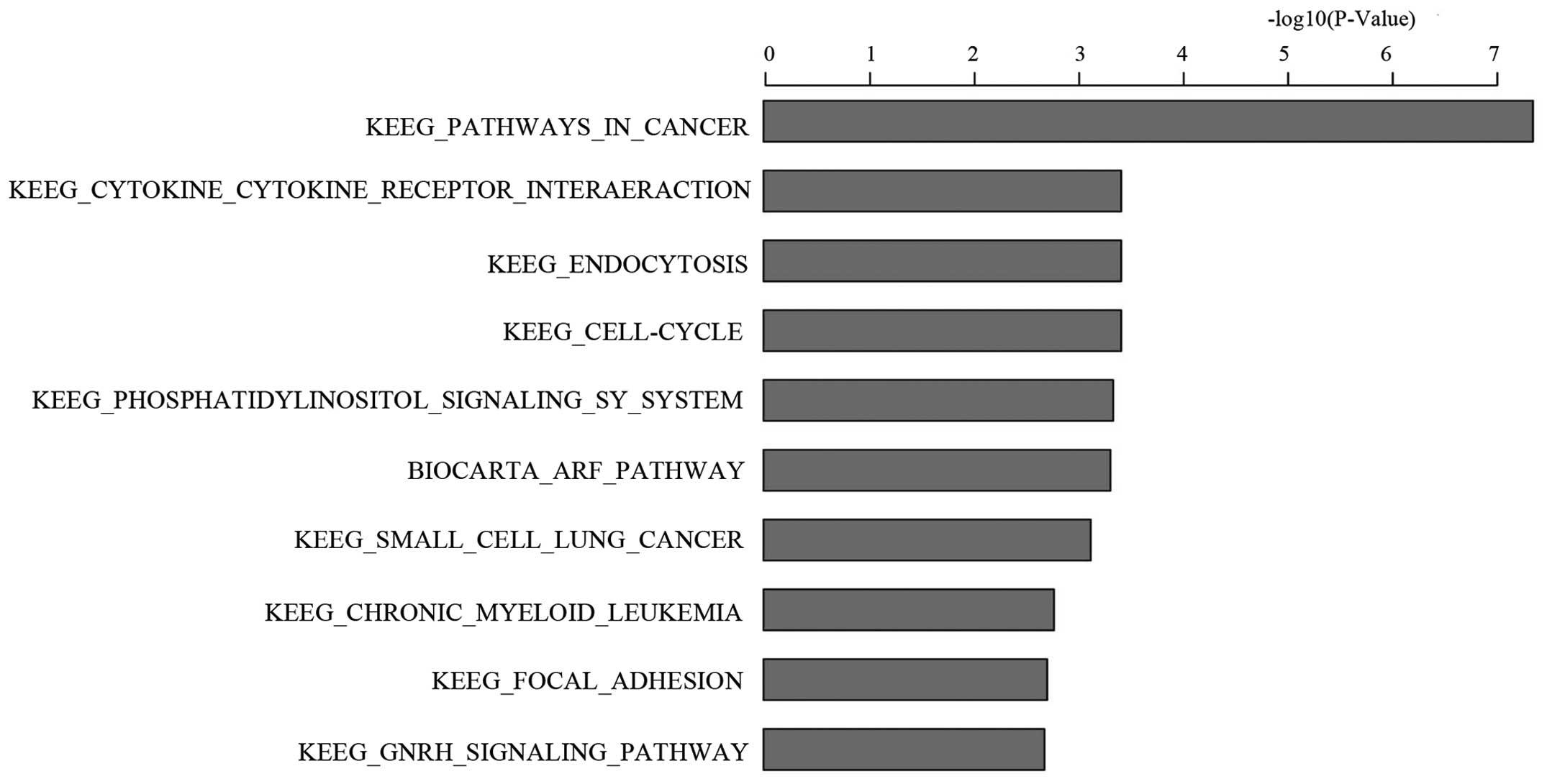
according to the enrichment score. The ten most significant
differential expression pathways and genes were listed in Fig. 5 and Table I, based on an ascending P-value.
Cancer in KEGG pathway was the most significant differential
expression pathway with a P-value of 4.36E-08.
 | Table IThe ten most significant differential
expression pathways and genes between KD and NC. |
Table I
The ten most significant differential
expression pathways and genes between KD and NC.
| Gene set name | Pathway analysis
|
|---|
| No. of genes | P-value | Gene names |
|---|
|
KEGG_PATHWAYS_IN_CANCER | 26 | 4.36E-08 | RB1, ABL1, CDK6,
SKP2, CBL, CBLB, PIK3R1, RAC1, AKT3, ITGA6, FN1, LAMB2, MAX, RUNX1,
MAPK8, MMP2, FGF2, FGF5, FGF14, LEF1, TCF7, WNT5A, FZD6, RALA,
GLI2, MMP1 |
| KEGG_CELL_CYCLE | 12 | 3.78E-04 | RB1, ABL1, CDK6,
SKP2, CDC25B, MCM2, MCM3, MCM4, CDC20, ESPL1, E2F4, RBL2 |
| KEGG_ENDOCYTOSIS | 14 | 3.78E-04 | CBL, CBLB, PIP5K1C,
PIP5K1A, AP2M1, AP2A1, EEA1, SMURF2, NEDD4, PARD6B, ACAP3, LDLRAP1,
LDLR, VPS36 |
|
KEGG_CYTOKINE_CYTOKINE_
RECEPTOR_INTERAERACTION | 17 | 3.78E-04 | IL1A, IL6R, IL6ST,
IL11, LEPR, PRLR, TNFRSF10D, TNFRSF10C, CCL2, CXCL2, CXCL3, BMPR2,
TNFRSF9, TNFRSF21, TNFRSF11B, TNFSF14, CD70 |
|
KEGG_PHOSPHATIDYLINOSITOL_
SIGNALING_SY_SYSTEM | 9 | 4.51E-04 | PIK3R1, PIP5K1C,
PIP5K1A, ITPR1, SYNJ2, INPP5A, OCRL, INPP4B, DGKE |
|
BIOCARTA_ARF_PATHWAY | 5 | 4.79E-04 | RB1, ABL1, PIK3R1,
RAC1, POLR1A |
|
KEGG_SMALL_CELL_LUNG_CANCER | 9 | 7.38E-04 | RB1, CDK6, SKP2,
PIK3R1, AKT3, ITGA6, FN1, LAMB2, MAX |
|
KEGG_CHRONIC_MYELOID_LEUKEMIA | 8 | 1.66E-03 | RB1, ABL1, CDK6, CBL,
CBLB, PIK3R1, AKT3, RUNX1 |
|
KEGG_FOCAL_ADHESION | 13 | 1.93E-03 | PIK3R1, RAC1, AKT3,
ITGA6, FN1, LAMB2, MAPK8, PIP5K1C, ELK1, THBS1, ARHGAP5, COL1A1,
COL5A2 |
|
KEGG_GNRH_SIGNALING_PATHWAY | 9 | 2.05E-03 | MAPK8, MMP2, ITPR1,
ELK1, MAP2K4, ADCY8, ADCY7, ADCY6, GNA11 |
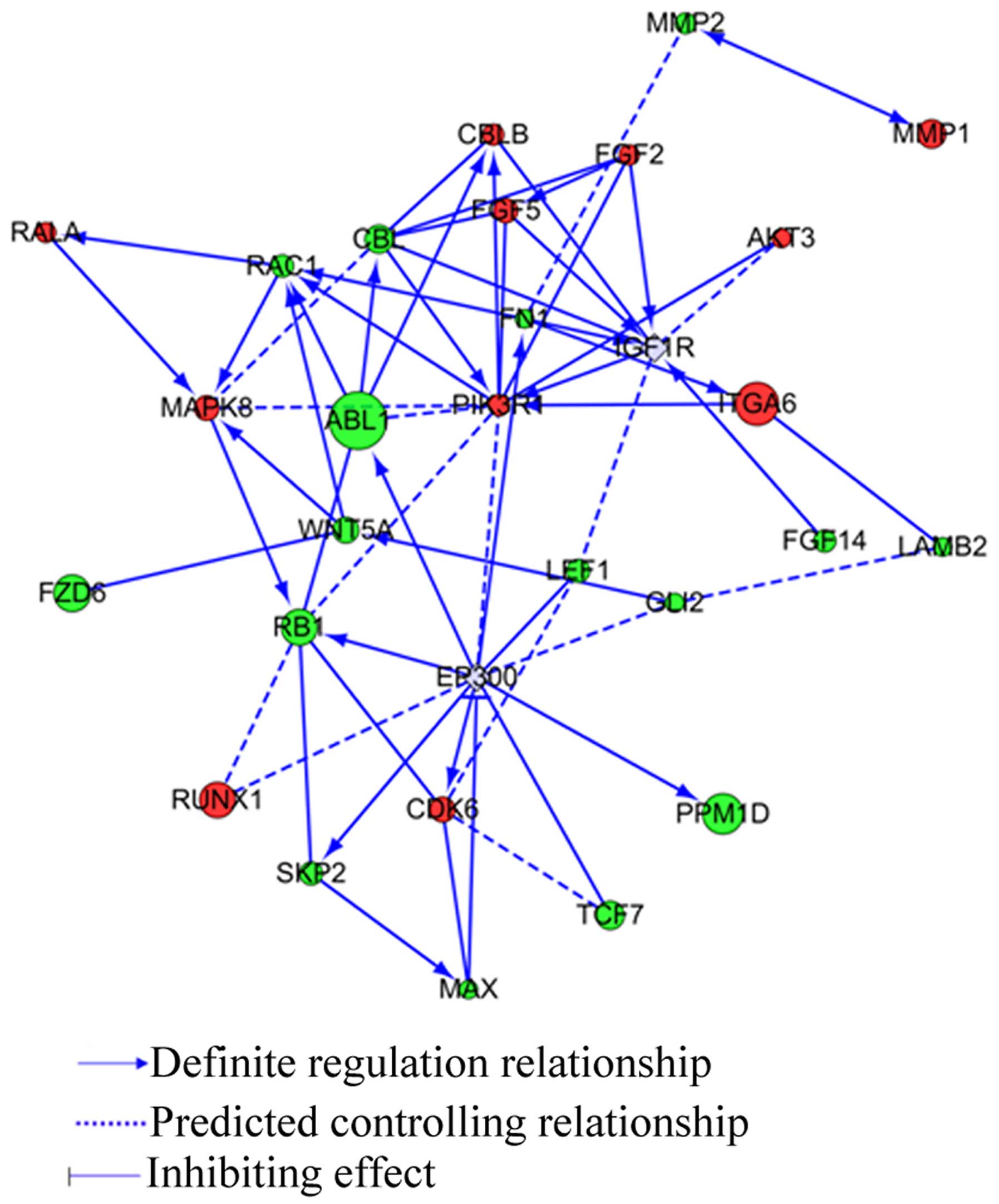
To further analyze the PPM1D effects when U-87MG
cells were administered with TMZ, the most significant differential
expression pathway in cancer in KEGG pathway was confirmed in gene
network construction. The relationship between PPM1D and the 26
differential genes is shown in Fig.
6. IGFR1R, PIK3R1, MAPK8, and EP300 are core genes in the
network.
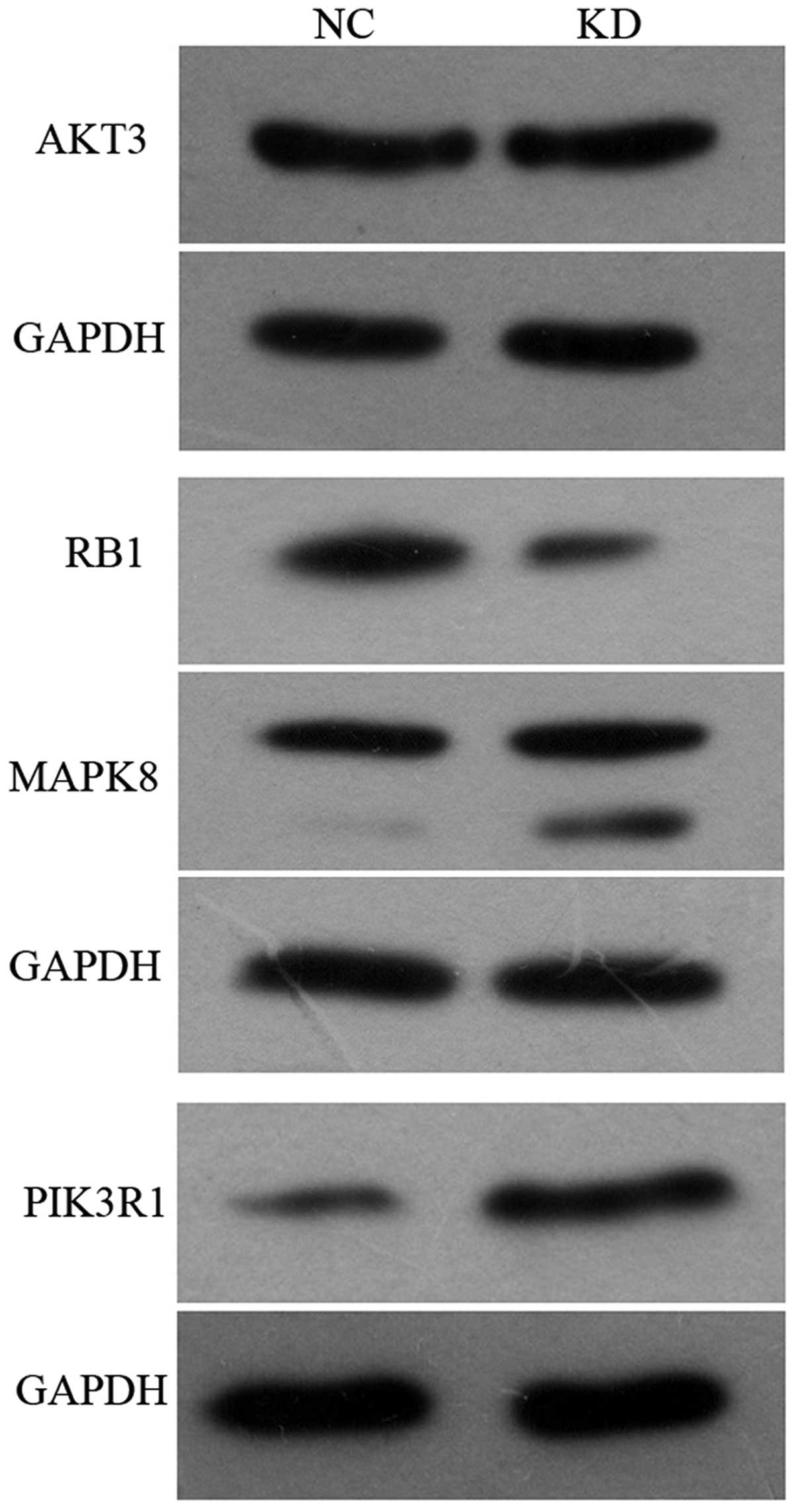
Western blotting
Several target genes deemed biologically interesting
because of their differential expression levels in glioma cells in
pathways in cancer were validated by western blot analysis. As
shown in Fig. 7, MAPK8 and PIK3R1
protein expression levels in KD group were increased by 64.1% and
261.6%, respectively, relative to the expression level in NC group.
It was consistent with that detected in gene expression profiling.
RB1 protein expression was decreased by 61.5% compared with NC
group. The expression of AKT3 was not significantly different.
Discussion
Malignant gliomas account for close to 50% of all
CNS tumors. The median survival of patients with GBM remains close
to 1 year from the time of diagnosis in spite of surgical resection
followed by radiotherapy and chemotherapy (1). The average five year survival is less
than 3%, leading to the fact that GBM is the most lethal form of
brain tumor (19). Such poor
outcome has led to the exploration of a wide variety of novel
therapies, and some of them have been incorporated as a standard of
care for patients with GBM. TMZ is approved for use as first-line
treatment for patients with primary and recurrent high-grade
gliomas. It has been shown that TMZ combined with radiotherapy can
improve the 2-year survival rate from 10.4% with radiotherapy alone
to 26.5% in patients with GBM and radiotherapy (1). Whereas the efficacy of TMZ is
encouraging, additional prolongation of survival remains a
challenge. GBM shows chemoresistance shortly after the initiation
of treatment. Additionally, recent studies suggest that 60–75% of
patients with GBM derive no benefit from treatment with TMZ
(2). Thus, exploring alternative
therapeutic strategies is required.
TMZ is an alkylating agent that binds to DNA and
interferes with replication, resulting in the insertion of DNA
strand breaks and, ultimately, cell death (3). p53 facilitates favorable antitumor
drug response through a variety of key cellular functions,
including cell cycle arrest, senescence, and apoptosis (4). p53 status in addition to MGMT plays a
role in chemotherapy resistance to TMZ. p53 wild-type glioma cells
are significantly more sensitive than p53-mutated glioma cells to
apoptosis induced by TMZ (18). GBM
patients with low mutant p53 expression have higher
progression-free survival time and may have longer life expectancy
in comparison with the high mutant p53 expression group (6). Initially identified as a p53-regulated
allele located on 17q23-24, PPM1D is a member of the protein
phosphatase 2C (PP2C) family and expressed in a p53-dependent
manner (7). PPM1D can negatively
regulate key DNA damage response proteins such as p53, ATM, and
p38MAPK and control cell cycle checkpoints in response to DNA
damage (20). PPM1D is frequently
amplified and overexpressed in many cancers, including gliomas,
breast cancers, neuroblastomas, ovarian clear cell adenocarcinomas,
and medulloblastomas (8–13). PPM1D is recognized as a novel
oncogene and is widely believed to be a promising therapeutic
target for cancers (21,22). In addition, we found that PPM1D may
serve as an oncogene important to astrocytoma progression,
especially in astrocytomas with wild-type p53 (8). However, whether PPM1D plays a role in
TMZ-induced DNA damage through regulating p53 functions has not
been clarified yet.
In the current study, we demonstrated that PPM1D
silencing can increase the antiproliferative activity of TMZ in
glioma cells. Cell cycle and apoptosis analyses showed that the
decreased proliferative activity was partly due to increased cell
apoptosis. It has been reported that U87-MG cells with p53
wild-type are sensitive to apoptosis induced by TMZ (18). However, it was shown that although
TMZ-treated cells underwent cell cycle arrest, the apoptotic cells
were few and did not significantly increase throughout the 10 days
after TMZ treatment (17). In
addition, it was reported that there was no significant difference
in the rate of apoptosis 72 h after treatment with either DMSO
control or 0.1 mmol/l TMZ, though TMZ reduced cell viability and
caused cell cycle arrest (16). TMZ
induced autophagic, but not apoptotic processes in glioma cells.
our results established that apoptotic cells were few when TMZ was
administrated alone. However, PPM1D silencing could significantly
induce cell apoptosis in TMZ-treated cells. Cell cycle arrest is
one of the important mechanisms through which TMZ exerts antitumor
effects. In this study, we found that PPM1D silencing combined with
TMZ could induce significantly increased G2/M cells and
decreased G1 cells. Glioma cells with PPM1D silencing
are more sensitive to TMZ induced apoptosis and cell cycle is
arrested in G2/M. Therefore, our results established
that PPM1D controls cell cycle checkpoints in response to TMZ-
induced DNA damage.
To investigate the mechanisms of changes in cell
biology when PPM1D gene silencing U-87MG cells were treated with
TMZ, cells were assessed with microarray-based gene expression
profile. Our results demonstrated that many genes attribute to the
different changes when PPM1D silencing glioma cells were treated
with TMZ. Pathway in cancer was the most significant pathway. Our
results proved that PPM1D is associated with glioma development,
which is consistent with the fact that PPM1D is recognized as a
novel oncogene in many cancers (21,22).
PPM1D was initially identified as a p53-regulated allele and can
negatively regulate key DNA damage response proteins such as p53,
ATM, and p38MAPK (20). The most
significant network arose around the IGFR1R, PIK3R1, MAPK8 and
EP300, when glioma cells were treated with combination of PPM1D
silencing and TMZ. Accumulating evidence has shown that GBM
frequently displays hyperactivation of the AKT pathway (23,24)
and endogenous AKT kinase activity can be activated in response to
clinically relevant concentrations of TMZ (25,26).
Over 80% of GBMs have an acquired alteration in the RTK/PI3K/AKT
pathway (27). It has been detected
that PPM1D knocked out mesenchymal stem cells may be attributed to
increases in the pAKT/AKT ratio and PI3K/AKT signaling axis
(28).
Our microarray-based gene expression profile and
western blot results detected that PIK3R1 is upregulated and RB1
protein expression was decreased when PPM1D silencing glioma cells
were treated with TMZ. However, AKT3 protein expression levels were
not different between the two groups. It was proved that Rb was a
substrate of PI3K/Akt signaling pathway (29). Activation of PI3K/AKT pathway
contributed to promote Rb phosphorylation and degradation (30). It has been previously shown that
inhibition of PI3K/Akt/mToR-mediated signaling in glioblastoma cell
lines strongly amplifies cell death induced by radiotherapy and a
wide range of chemotherapeutics. As a regulatory subunit of
phosphoinositide-3-kinase (PI3K), PIK3R1 target the PI3K/AKT
pathway. However, our results showed different tendency of change
of PIK3R1. Thus, PPM1D may not regulate PI3K/AKT signaling axis
through PIK3R1 (31).
It has been demonstrated that downregulated
expression of COX-2, Akt1 and PIK3R1 could upregulate p53 and exert
proliferation and invasion inhibition effects on U251 cells
(32). We infer it should be partly
due to the different status of p53. The exact mechanism still
remains to be explored.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81201995), the Natural
Science Foundation of Guangdong Province, China (grant no.
2015A030313532) and the Science and Technology Planning Project of
Guangdong Province, China (grant no. 2014A020212678).
References
|
1
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: European Organisation for Research and Treatment of
Cancer Brain Tumor and Radiotherapy Groups; National Cancer
Institute of Canada Clinical Trials Group: Radiotherapy plus
concomitant and adjuvant temozolomide for glioblastoma. N Engl J
Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chamberlain MC: Temozolomide: Therapeutic
limitations in the treatment of adult high-grade gliomas. Expert
Rev Neurother. 10:1537–1544. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marchesi F, Turriziani M, Tortorelli G,
Avvisati G, Torino F and De Vecchis L: Triazene compounds:
Mechanism of action and related DNA repair systems. Pharmacol Res.
56:275–287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miao W, Liu X, Wang H, Fan Y, Lian S, Yang
X, Wang X, Guo G, Li Q and Wang S: p53 upregulated modulator of
apoptosis sensitizes drug-resistant U251 glioblastoma stem cells to
temozolomide through enhanced apoptosis. Mol Med Rep. 11:4165–4173.
2015.PubMed/NCBI
|
|
5
|
Martinez-Rivera M and Siddik ZH:
Resistance and gain-of-resistance phenotypes in cancers harboring
wild-type p53. Biochem Pharmacol. 83:1049–1062. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li S, Jiang T, Li G and Wang Z: Impact of
p53 status to response of temozolomide in low MGMT expression
glioblastomas: Preliminary results. Neurol Res. 30:567–570. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fiscella M, Zhang H, Fan S, Sakaguchi K,
Shen S, Mercer WE, Vande Woude GF, O'Connor PM and Appella E: Wip1,
a novel human protein phosphatase that is induced in response to
ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci
USA. 94:6048–6053. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang P, Rao J, Yang H, Zhao H and Yang L:
Wip1 overexpression correlated with TP53/p14(ARF) pathway
disruption in human astrocytomas. J Surg oncol. 104:679–684. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bulavin DV, Demidov ON, Saito S,
Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G,
Nebreda AR, Anderson CW, et al: Amplification of PPM1D in human
tumors abrogates p53 tumor-suppressor activity. Nat Genet.
31:210–215. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Yang Y, Peng Y, Austin RJ, van
Eyndhoven WG, Nguyen KC, Gabriele T, McCurrach ME, Marks JR, Hoey
T, et al: Oncogenic properties of PPM1D located within a breast
cancer amplification epicenter at 17q23. Nat Genet. 31:133–134.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saito-Ohara F, Imoto I, Inoue J, Hosoi H,
Nakagawara A, Sugimoto T and Inazawa J: PPM1D is a potential target
for 17q gain in neuroblastoma. Cancer Res. 63:1876–1883.
2003.PubMed/NCBI
|
|
12
|
Liang C, Guo E, Lu S, Wang S, Kang C,
Chang L, Liu L, Zhang G, Wu Z, Zhao Z, et al: Over-expression of
wild-type p53-induced phosphatase 1 confers poor prognosis of
patients with gliomas. Brain Res. 1444:65–75. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hirasawa A, Saito-Ohara F, Inoue J, Aoki
D, Susumu N, Yokoyama T, Nozawa S, Inazawa J and Imoto I:
Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas
with poor prognosis and identification of PPM1D and APPBP2 as
likely amplification targets. Clin Cancer Res. 9:1995–2004.
2003.PubMed/NCBI
|
|
14
|
Wang P, Rao J, Yang H, Zhao H and Yang L:
PPM1D silencing by lentiviral-mediated RNA interference inhibits
proliferation and invasion of human glioma cells. J Huazhong Univ
Sci Technolog Med Sci. 31:94–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Spiro T, Liu L and Gerson S: New cytotoxic
agents for the treatment of metastatic malignant melanoma:
Temozolomide and related alkylating agents in combination with
guanine analogues to abrogate drug resistance. Forum (Genova).
10:274–285. 2000.
|
|
16
|
Shen W, Hu JA and Zheng JS: Mechanism of
temozolomideinduced antitumour effects on glioma cells. J Int Med
Res. 42:164–172. 2014. View Article : Google Scholar
|
|
17
|
Hirose Y, Berger MS and Pieper RO: p53
effects both the duration of G2/M arrest and the fate of
temozolomide-treated human glioblastoma cells. Cancer Res.
61:1957–1963. 2001.PubMed/NCBI
|
|
18
|
Batista LF, Roos WP, Kaina B and Menck CF:
p53 mutant human glioma cells are sensitive to UV-C-induced
apoptosis due to impaired cyclobutane pyrimidine dimer removal. Mol
Cancer Res. 7:237–246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tivnan A, Zakaria Z, O'leary C, Kögel D,
Pokorny JL, Sarkaria JN and Prehn JH: Inhibition of multidrug
resistance protein 1 (MRP1) improves chemotherapy drug response in
primary and recurrent glioblastoma multiforme. Front Neurosci.
9:2182015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu YH, Zhang CW, Lu L, Demidov ON, Sun L,
Yang L, Bulavin DV and Xiao ZC: Wipl regulates the generation of
new neural cells in the adult olfactory bulb through p53-dependent
cell cycle control. Stem Cells. 27:1433–1442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang YL, Liu X, Gao SY, Feng H, Jiang YP,
Wang SS, Yang J, Jiang J, Ma XR, Tang YJ, et al: WIP1 stimulates
migration and invasion of salivary adenoid cystic carcinoma by
inducing MMP-9 and VEGF-C. Oncotarget. 6:9031–9044. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gilmartin AG, Faitg TH, Richter M, Groy A,
Seefeld MA, Darcy MG, Peng X, Federowicz K, Yang J, Zhang SY, et
al: Allosteric Wip1 phosphatase inhibition through flap-subdomain
interaction. Nat Chem Biol. 10:181–187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hirose Y, Katayama M, Mirzoeva OK, Berger
MS and Pieper RO: Akt activation suppresses Chk2-mediated,
methylating agent-induced G2 arrest and protects from
temozolomide-induced mitotic catastrophe and cellular senescence.
Cancer Res. 65:4861–4869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Molina JR, Hayashi Y, Stephens C and
Georgescu MM: Invasive glioblastoma cells acquire stemness and
increased Akt activation. Neoplasia. 12:453–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Caporali S, Levati L, Starace G, Ragone G,
Bonmassar E, Alvino E and D'Atri S: AKT is activated in an
ataxia-telangiectasia and Rad3-related-dependent manner in response
to temozolomide and confers protection against drug-induced cell
growth inhibition. Mol Pharmacol. 74:173–183. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
De Salvo M, Maresca G, D'agnano I,
Marchese R, Stigliano A, Gagliassi R, Brunetti E, Raza GH, De Paula
U and Bucci B: Temozolomide induced c-Myc-mediated apoptosis via
Akt signalling in MGMT expressing glioblastoma cells. Int J Radiat
Biol. 87:518–533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Signore M, Pelacchi F, di Martino S, Runci
D, Biffoni M, Giannetti S, Morgante L, De Majo M, Petricoin EF,
Stancato L, et al: Combined PDK1 and CHK1 inhibition is required to
kill glioblastoma stem-like cells in vitro and in vivo. Cell Death
Dis. 5:e12232014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang Y, Liu L, Sheng M, Xiong K, Huang L,
Gao Q, Wei J, Wu T, Yang S, Liu H, et al: Wip1 knockout inhibits
the proliferation and enhances the migration of bone marrow
mesenchymal stem cells. Exp Cell Res. 334:310–322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao N, Zhang Z, Jiang BH and Shi X: Role
of PI3K/AKT/mTOR signaling in the cell cycle progression of human
prostate cancer. Biochem Biophys Res Commun. 310:1124–1132. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bao JM, He MY, Liu YW, Lu YJ, Hong YQ, Luo
HH, Ren ZL, Zhao SC and Jiang Y: AGE/RAGE/Akt pathway contributes
to prostate cancer cell proliferation by promoting Rb
phosphorylation and degradation. Am J Cancer Res. 5:1741–1750.
2015.PubMed/NCBI
|
|
31
|
Westhoff MA, Faham N, Marx D, Nonnenmacher
L, Jennewein C, Enzenmüller S, Gonzalez P, Fulda S and Debatin KM:
Sequential dosing in chemosensitization: Targeting the
PI3K/Akt/mTOR pathway in neuroblastoma. PLoS one. 8:e831282013.
View Article : Google Scholar
|
|
32
|
Fu Y, Zhang Q, Kang C, Zhang K, Zhang J,
Pu P, Wang G and Wang T: Inhibitory effects of adenovirus mediated
COX-2, Akt1 and PIK3R1 shRNA on the growth of malignant tumor cells
in vitro and in vivo. Int J oncol. 35:583–591. 2009.PubMed/NCBI
|