Introduction
Gastrointestinal stromal tumors (GISTs), the most
common mesenchymal neoplasms of the gastrointestinal tract
(1), are characterized by
gain-of-function mutations of the KIT or PDGFRA genes
(2,3). They most likely originate from the
interstitial cells of Cajal (ICC) or Cajal-like precursor cells
whose immunohistochemical marker is CD117 (4,5). In
recent years, the receptor tyrosine kinase (RTK) inhibitor imatinib
mesylate (Gleevec; Novartis Pharmaceuticals Corp., East Hanover,
NJ, USA), which targets the KIT and PDGFRA oncoproteins, has
frequently been used to treat patients with advanced GISTs or has
been used as an adjuvant agent (6).
Although up to 70% of GIST patients respond to imatinib well at
first, ~15% of patients may develop primary resistance, and most
responding patients will eventually develop secondary resistance
and have disease progression (7).
Thus, novel therapeutic targets and effective antineoplastic
strategies are still needed.
Primary resistance is mainly caused by KIT
exon 9 mutations and the lack of activating KIT mutations,
resulting in resistance to imatinib. The PDGFRA mutation
D842V on exon 18 also contributes to primary resistance in GISTs
(8). Acquired secondary mutations
in the kinase domains of KIT or PDGFRA will lead to
the development of imatinib resistance in most patients (9). Moreover, it has been reported that
stem cell factor (SCF), a KIT ligand, may contribute to the
acquired imatinib resistance (10).
Although an increasing number of new-generation RTK inhibitors,
such as sunitinib, nilotinib, vatalanib and masatinib, have
demonstrated their antineoplastic activities in GISTs (11), the therapeutic effect varies
depending on the pattern of genetic alterations Therefore, it is
imperative that novel molecular targets and selective agents be
developed and investigated to revolutionize antitumor
strategies.
CREB-binding protein (CBP) and p300 (collectively
referred to as CBP/p300) are transcriptional co-activators that
integrate and maintain various gene regulator pathways and protein
acetylation events with intrinsic histone acetyltransferase (HAT)
activity (12). Over the last few
years, their biological and pathological functions have been under
extensive investigation. CBP/p300 play pivotal roles in
differentiation, apoptosis and the cell cycle (13,14).
However, there is a distinct relationship between CBP/p300 activity
and tumorigenesis and malignancy. CBP/p300 inactivation will not
only inhibit the growth of prostate cancer cells and melanoma cells
(15,16) but will also induce apoptosis and
cell cycle arrest in leukaemia cells (17). Interestingly, it has been
illustrated that the co-activators CBP and p300 interact with an
oncoprotein ETS translocation variant 1 (ETV1) (18), a distinctive transcription factor
that is overexpressed in most prostate cancers (19). In addition, p300 directly acetylates
ETV1 and thereby enhances its stability, DNA-binding capacity, and
transcriptional activity in vitro (20). Recent studies have demonstrated that
ETV1 is a specific survival factor that cooperates with KIT in
GISTs, and our previous findings showed that ETV1 was highly
upregulated within tumor tissues in conjunction with KIT expression
(21,22). Therefore, CBP/p300 may play a vital
role in tumorigenesis and progression of GISTs by regulating the
functions of ETV1 and KIT-dependent pathways, serving as promising
targets for antineoplastic therapy.
Recently, a small molecule inhibitor targeting
CBP/p300, C646, has gained much attention for its anticancer
potential. C646 administration inhibits histone acetylation and
cell growth with relatively high selectivity and potency,
suggesting its application as a prospective anticancer agent
(23). Although the antitumor
activity of C646 has been investigated in other tumors (15,17),
its effects on GISTs have not yet been assessed. Here, we explored
the therapeutic relevance of CBP/p300 inhibition in GISTs and
further investigated the possible mechanisms underlying its
antineoplastic activity in vitro.
Materials and methods
Cell culture and reagents
The GIST882 cell line carrying the homozygous KIT
exon 13 K642E (KIT-exon13) mutation was a generous gift from Dr
Jonathan Fletcher (Dana-Farber Cancer Institute, Boston, MA, USA).
GIST882 cells were grown in RPMI-1640 medium (Corning Inc.,
Corning, NY, USA) with 15% fetal bovine serum (FBS), 100 U/ml
penicillin and 0.1 mg/ml streptomycin. The GIST cells were
maintained in a humidified incubator at 37°C in a 5% CO2
atmosphere. The CBP/p300 inhibitor, C646, was purchased from
Selleck Chemicals LLC (Shanghai, China).
Transfection of short interfering RNA
(siRNA)
To determine the role of CBP/p300 in GIST
proliferation and apoptosis, GIST882 cells were cultured in 6- and
96-well plates until 70–80% confluence, respectively. siRNA
transfections were performed with Lipofectamine RNAiMAX reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) in
antibiotics-free medium according to the manufacturer's
instructions. GIST882 cells were transfected with 10 nmol/l siRNA.
The control siRNA and siRNA against CBP and p300 were purchased
from Santa Cruz Biotechnology, Inc. (Dallas, Texas, USA).
Cell proliferation assay
GIST882 cells (1.5×104/well) were seeded
in 96-well plates and further cultured for 48 h before C646 and
siRNA were added. After administration of C646 (for 24 and 48 h) or
transfection with siRNA (for 24, 48 and 72 h), 10 µl of Cell
Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan)
solution was added to each well, followed by incubation for 2 h at
37°C. The optical density (OD) was measured at 450 nm with a
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The mean OD values from 4-wells for each treatment were used as the
index of cell viability. All experimental points were replicated in
three plates. Cell viability was calculated using the following
formula: OD value of each set/OD value of control group (DMSO
control and blank control, respectively) × 100.
Caspase activity assay
GIST882 cells (1.5×104/well) were
cultured in a 96-well plate for 48 h before being transfected with
siRNA or treated with C646. After transfection with siRNA (for 24,
48 and 72 h) or administration of C646 (for 24 and 48 h),
caspase-Glo 3/7 reagent (Promega, Madison, WI, USA) was added to
each well at a ratio of 1:1 following the manufacturer's
instructions. The luminescence intensity of each sample was
measured in a plate-reading luminometer (Promega). Caspase activity
was expressed as a percentage of the control level [blank and
dimethyl sulfoxide (DMSO) controls, respectively]. Luminescence
intensity was measured in quadruplicate, and all experimental
points were replicated in three plates.
Cell apoptosis assay
Apoptosis induction was detected using an Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
assay kit (Invitrogen Life Technologies). GIST882 cells
(7×105/well) were plated on 6-well plates and further
cultured for 48 h before being treated with C646. After incubation
for 24, 48 or 72 h, the cells were harvested by trypsin without
EDTA and centrifuged at 200 × g for 5 min. The cell pellets were
resuspended in 100 µl of 1X binding buffer. Next, 5
µl of Annexin V-FITC and 1 µl of PI were added and
mixed well. After incubation for 15 min in the dark, 400 µl
of 1X binding buffer was added to each sample. The stained cells
were evaluated by flow cytometry (NovoCyte; ACEA Biosciences, Inc.,
San Diego, CA, USA), and the data were analysed using FlowJo 7.6.2
software (Tree Star, Inc., Ashland, OR, USA). All experimental sets
were conducted in triplicate.
Cell cycle assay
Cell cycle analysis was performed using a Cell Cycle
kit purchased from Multi Sciences (Lianke) Biotech Co., Ltd.
(Hangzhou, China). GIST882 cells (7×105/well) were
seeded in 6-well plates and further cultured for 48 h before being
treated with C646. After administration of C646 for 24, 48 or 72 h,
the cells were trypsinized and collected by centrifugation at 200 ×
g for 5 min. The cells were then fixed with 75% ethanol at −20°C
overnight and rehydrated with ice-cold PBS for 15 min before being
stained with 1 ml of DNA staining solution. The stained cells were
then evaluated by flow cytometry as described above, and the data
were analysed using ModFit LT V3.3.11 software (Verity Software
House, Inc., Topsham, ME, USA). All experimental sets were
conducted in triplicate.
Quantitative real-time polymerase chain
reaction (PCR)
According to the transfection protocol, GIST882
cells were divided into four groups: blank, negative control (NC),
sip300 and siCBP. The total RNA of each sample was extracted using
TRIzol reagent (Invitrogen Life Technologies) following the
manufacturer's instructions. Total RNA (2 µg) was
reverse-transcribed using the PrimeScript™ RT reagent kit with gDNA
Eraser (Takara Biotechnology, Co., Ltd., Dalian, China). DNA
products were amplified with SYBR® Premix Ex Taq™ (Tli
RNaseH Plus) (Takara Biotechnology, Co., Ltd.). The primer
sequences were as follows: forward, 5′-CCTGAG TAGGGGCAACAAGA-3′ and
reverse, 5′-GTGTCTCCACA TGGTGCTTG-3′ for human p300;
forward, 5′-CTGCACAC GACATGACT-3′ and reverse, 5′-GAAGTGGCATTCTG
TTG-3′ for human CBP; and forward, 5′-AGAAGGCTGG
GGCTCATTTG-3′ and reverse, 5′-AGGGGCCATCCACA GTCTTC-3′ for human
glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Real-time
PCR was performed using a 7500 Real-Time PCR system (Applied
Biosystems, Foster City, CA, USA). The thermocycling conditions
were 10 min at 95°C as the initial denaturation step, followed by
40 cycles at 95°C for 30 sec and 60°C for 34 sec. Gene expression
levels were measured using the threshold cycle (Ct) value, and
relative fold-expression changes were normalized to GAPDH
amplification. The 2−ΔΔCT method was used to measure the
relative changes in gene expression.
Western blotting
Total proteins were harvested from GIST882 cells
using cell lysis buffer (Cell Signaling Technology, Inc., Beverly,
MA, USA) according to the manufacturer's instructions. Equal
amounts of protein were applied to 8–12% gels and were subjected to
SDS-PAGE. The samples were then transferred to polyvinylidene
difluoride (PVDF) membranes using a semidry transfer system
(Bio-Rad Laboratories, Inc.). Membranes were blocked for 2 h at
room temperature with 5% dry milk in Tris-buffered saline with
Tween®-20 (TBST) and were incubated overnight at 4°C
with specific polyclonal rabbit anti-human antibodies corresponding
to c-KIT (1:2,000), phospho-c-KIT (Tyr703) (1:2,000), CBP
(1:1,000), extracellular signal-regulated kinase (ERK)1/2
(1:2,000), phospho-ERK1/2 (Thr202/Tyr204 for ERK1 and Thr183/Tyr185
for ERK2 (1:2,000), c-Jun NH2-terminal kinase (JNK)
(1:1,000), phospho-JNK (Thr183/Tyr185) (1:1,000), Bax (1:2,000),
Bcl-2 (1:1,000), and GAPDH (1:5,000) (Cell Signaling Technology,
Inc.), along with rabbit anti-human antibodies against p300
(1:1,000) (Santa Cruz Biotechnology, Inc.) and ETV1 (1:1,000)
(Abcam, Cambridge, UK). Membranes were washed with TBST and
incubated with 1:5,000 anti-rabbit horseradish peroxidase
(HRP)-conjugated secondary antibody (Abcam) for 1 h at room
temperature and then washed again. Immune complexes were detected
with an enhanced chemiluminescence reagent (Millipore, Billerica
MA, USA), acquired in the linear range of the scanner and analysed
using Quantity One software (Bio-Rad Laboratories, Inc.).
Statistical analysis
Results are expressed as the means ± standard
deviations. Multiple comparisons between groups were performed
using Tukey's range test. Statistically significant differences are
indicated by P<0.05. All analyses were performed using GraphPad
Prism 6 (GraphPad Software, Inc., San Diego, CA, USA).
Results
CBP inhibition suppresses GIST882 cell
proliferation
To determine whether CBP/p300 can serve as
antineoplastic targets for GISTs, specific siRNAs against
CBP and p300 were transfected into GIST882 cells. As
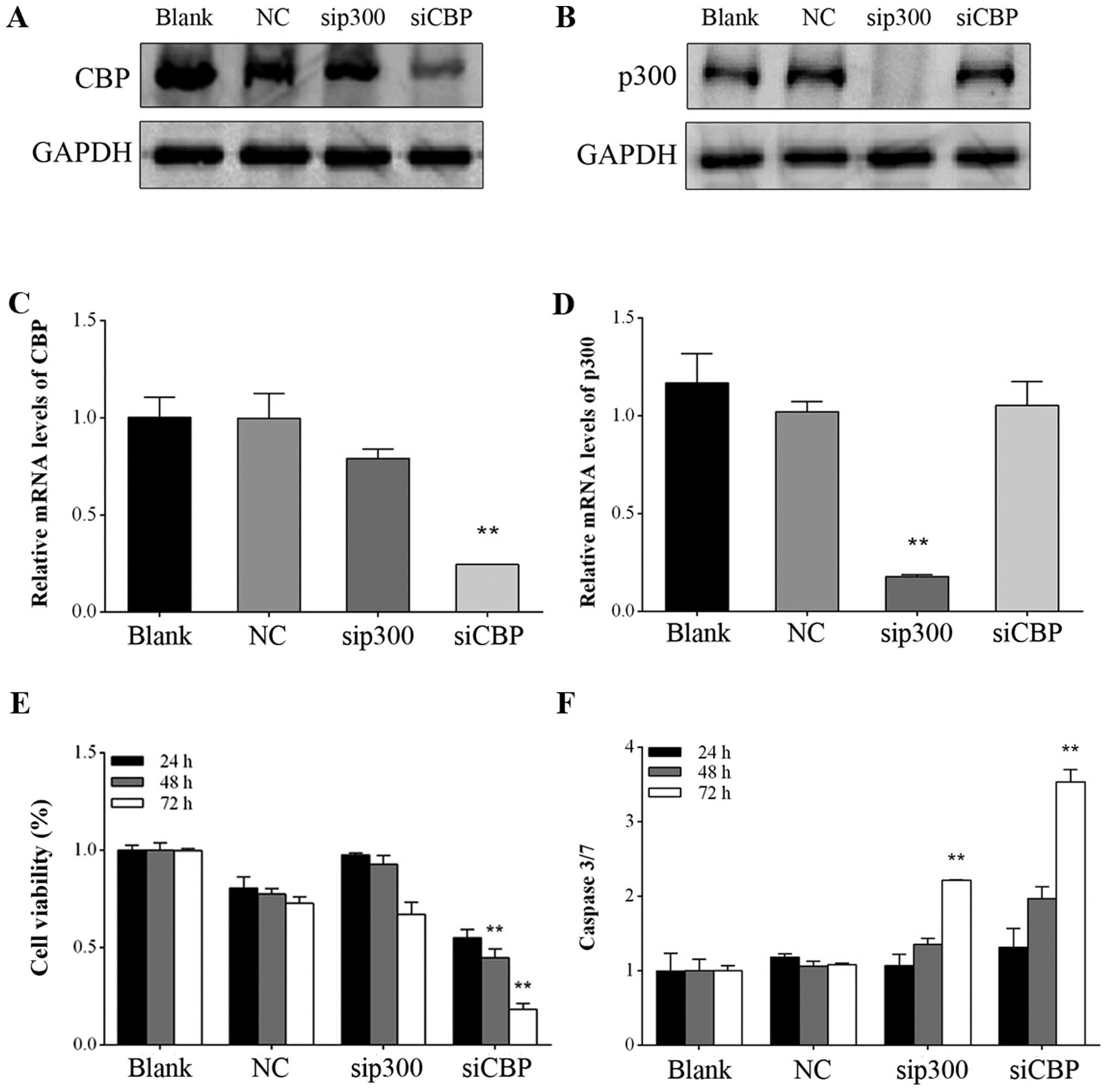
shown in Fig. 1A and B, siRNA
transfection for 72 h attenuated CBP and p300 protein expression,
consistent with their decreased mRNA expression levels (Fig. 1C and D). CBP downregulation led to
decreased cell viability, whereas a similar effect was not observed
upon p300 depletion in GIST882 cells (Fig. 1E), indicating that CBP and p300
potentially play different roles in GIST proliferation in spite of
their high homology.
CBP/p300 downregulation enhances
caspase-3/7 activity in GIST882 cells
In the present study, caspase-3/7 activity was
measured to evaluate apoptosis induction following CBP/p300
downregulation. As shown in Fig.
1F, consistent with the antiproliferative effects induced by
CBP silencing, caspase-3/7 activity in GIST882 cells was
markedly enhanced 3.5-fold compared with the negative control after
transfection for 72 h (P<0.01). Notably, inhibition of
p300 by siRNA also increased caspase-3/7 activity 2.2-fold,
which was much higher than that in the negative control group after
transfection for 72 h (P<0.01).
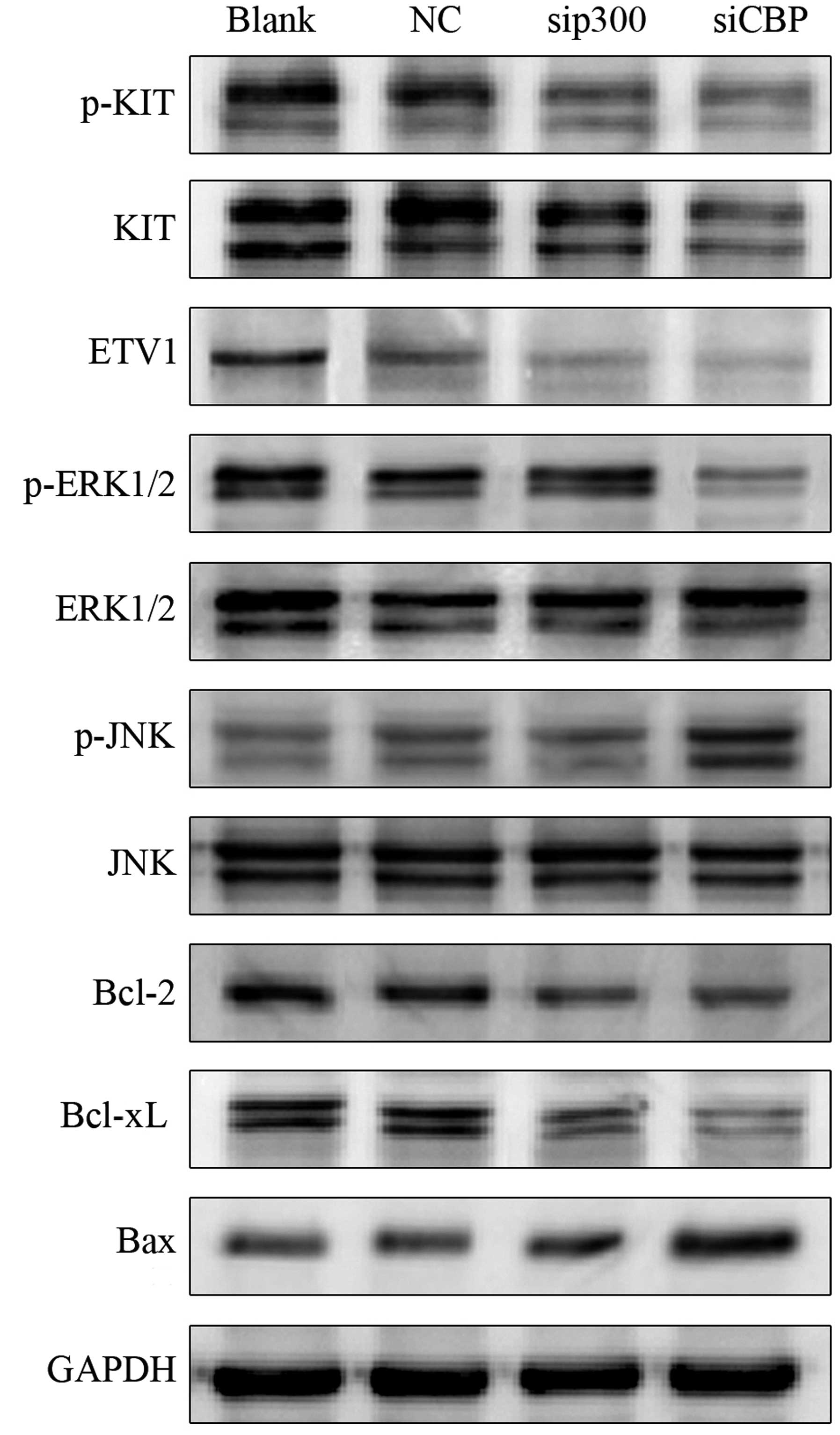
CBP/p300 silencing results in inhibition
of ETV1 and KIT-dependent pathways and activation of apoptotic
pathways
To elucidate the molecular mechanisms involved in
the antineoplastic effects on GIST882 cells, we detected ETV1
protein levels and KIT-dependent pathway regulation following siRNA
transfection for 72 h. As shown in Fig.
2, both CBP and p300 inhibition caused
considerable ETV1 degradation compared with the negative control.
Phosphorylation of KIT decreased considerably as CBP and p300 were
downregulated. In addition, silencing of CBP, rather than
p300, strongly attenuated the phosphorylation of ERK1/2 and
activated JNK in GIST882 cells; the total ERK1/2 and JNK expression
levels scarcely changed within the groups. In this study, the
expression of several mitochondrial-related apoptotic proteins,
including Bcl-2, Bcl-xL and Bax, was also evaluated to shed light
on the possible mechanisms of apoptosis induced by CBP/p300
inhibition. Our results demonstrated that the expression of
anti-apoptotic Bcl-2 and Bcl-xL decreased after siRNA transfection,
whereas pro-apoptotic Bax expression increased considerably.
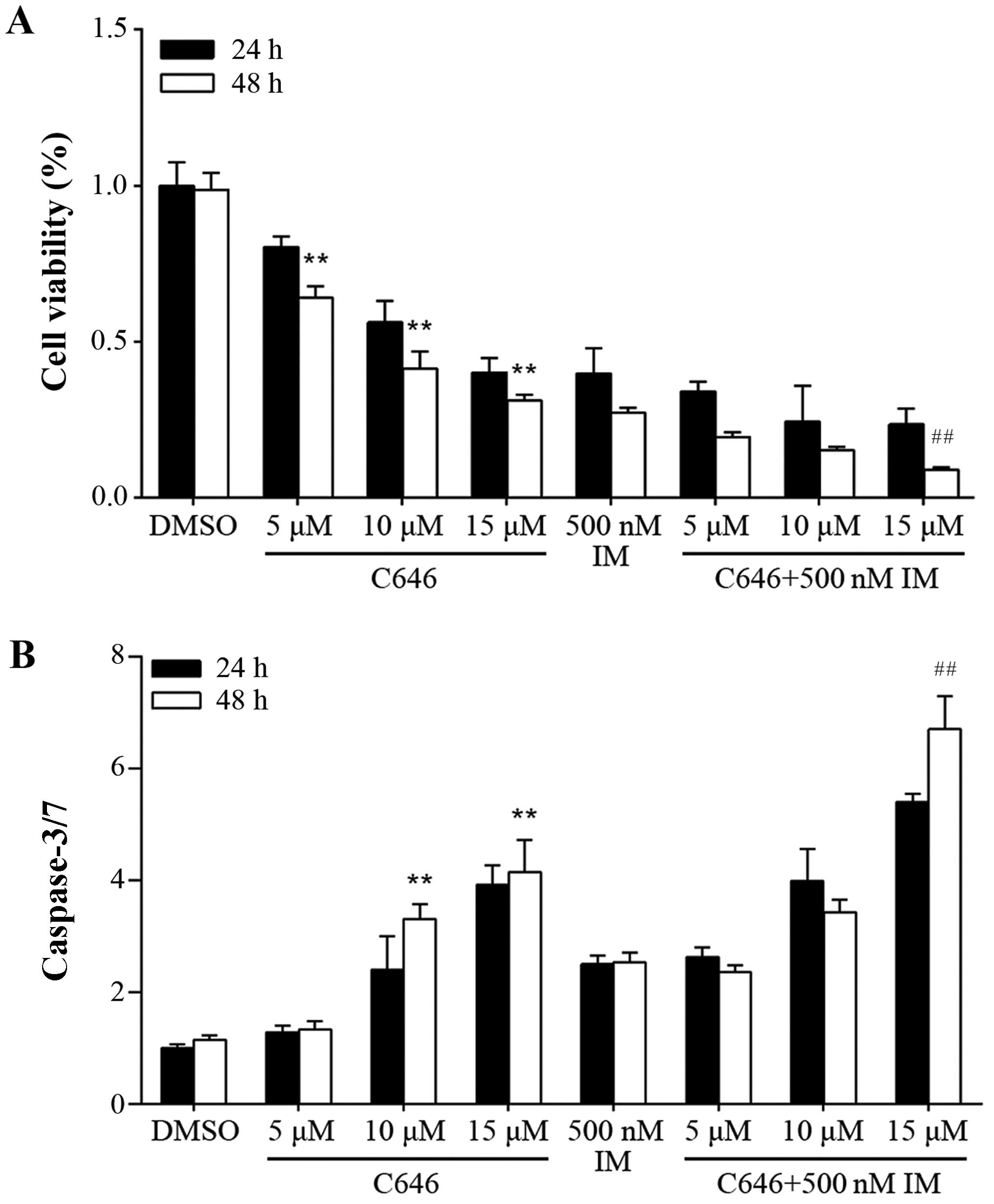
C646 inhibits cell proliferation and
activates caspase-3/7 in GIST882 cells
As mentioned above, CBP/p300 downregulation exerted
antiproliferative effects on GIST882 cells via inhibition of cell
proliferation and caspase-3/7 activation. Therefore, we next
investigated whether C646, a selective CBP/p300 inhibitor, was able
to exert similar effects. Its potential antineoplastic activity was
validated by treating GIST882 cells with an increasing
concentration of C646 (5–15 µmol/l) for 24 and 48 h,
respectively. Moreover, imatinib (500 nmol/l) alone or in
combination with C646 at the same doses (5–15 µmol/l) were
used to assess the possible additive effect. As shown in Fig. 3A, C646 administration had strong
inhibitory effects on cell proliferation in the GIST822 cells. The
cell viability of GIST882 cells significantly decreased after
treatment with imatinib combined with C646 (15 µmol/l) for
48 h compared with the group treated with 15 µmol/l C646
alone (P<0.01). To detect caspase-3/7 activity, C646 (1–15
µmol/l) alone or in combination with imatinib (500 nmol/l)
was administered to GIST cells for 24 and 48 h, respectively. As
shown in Fig. 3B, exposure to 15
µmol/l of C646 led to a 4.1-fold upregulation in caspase-3/7
activity after 48 h compared with DMSO control group (P<0.01).
Administration of imatinib (500 nmol/l) alone for 48 h resulted in
a 2.5-fold increase in caspase-3/7 activity compared with DMSO
control group (P<0.01). In the combination studies, caspase-3/7
activity significantly increased after treatment with imatinib
combined with C646 (15 µmol/l) for 48 h compared with the
group treated with 15 µmol/l C646 alone (P<0.01).
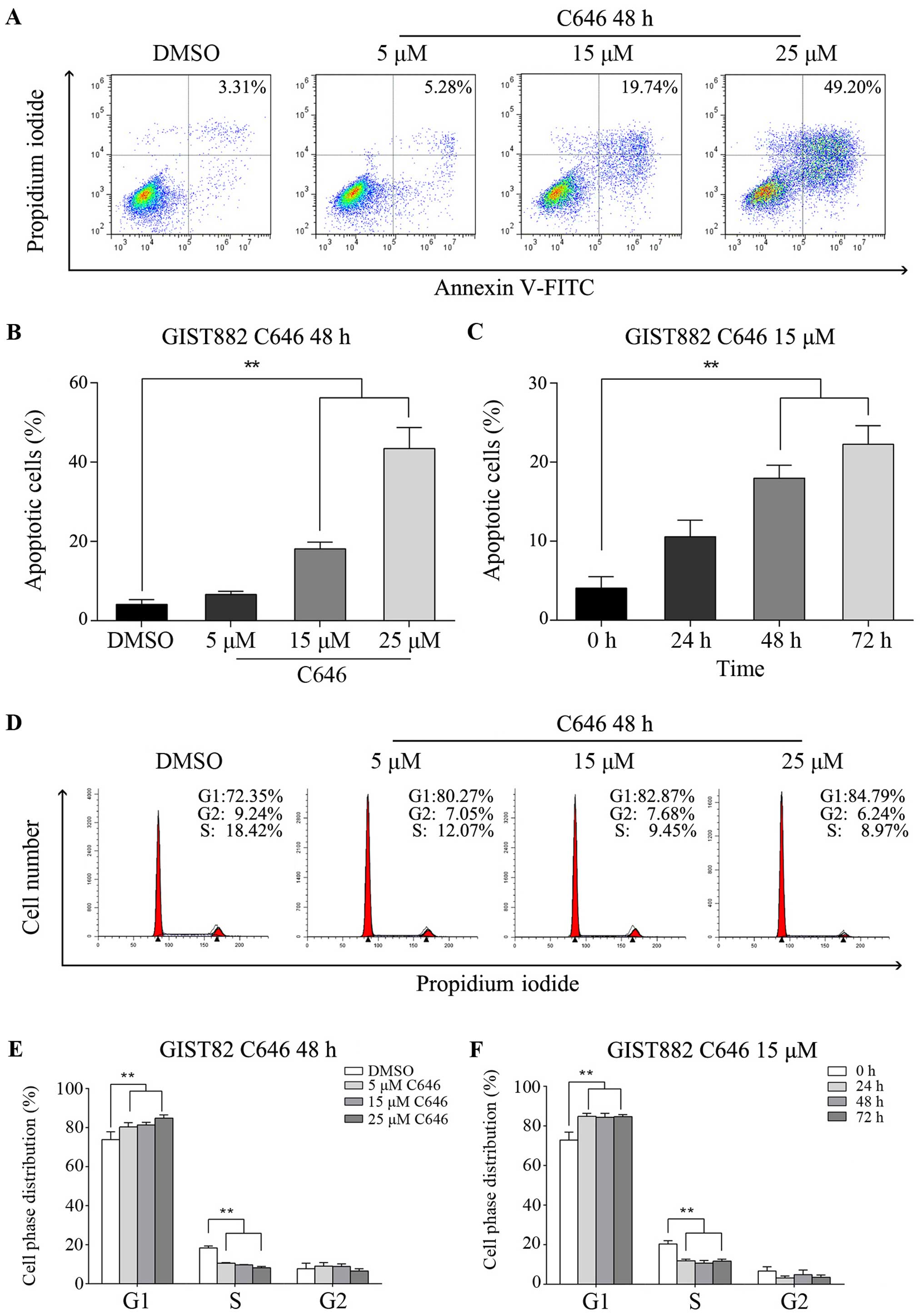
C646 exerts its antineoplastic effect via
apoptosis induction and cell cycle arrest in GIST882 cells
To further assess the apoptosis induced by C646,
Annexin V-FITC/PI staining and flow cytometry were performed. C646
administration induced significant apoptosis in GIST cells compared
with DMSO control group (Fig. 4A).
As shown in Fig. 4B, apoptotic cell
death was induced in a dose-dependent manner, and 15 µmol/l
C646 caused significant apoptosis in GIST cells compared with DMSO
group (P<0.01). In addition, the apoptotic rate increased
accordingly over time in GIST882 cells exposed to 15 µmol/l
C646 (Fig. 4C). The effect of C646
on the cell cycle was examined via PI staining and flow cytometry.
Our results indicated that C646 treatment led to an increase in
GIST cells in the G1-phase, whereas the S-phase population declined
considerably (Fig. 4D). Notably,
C646 (15 µmol/l) immediately induced cell cycle arrest at
the G1-phase in GIST882 cells within 24 h; this inhibitory effect
did not follow a dose- or time-dependent pattern (Fig. 4E and F).
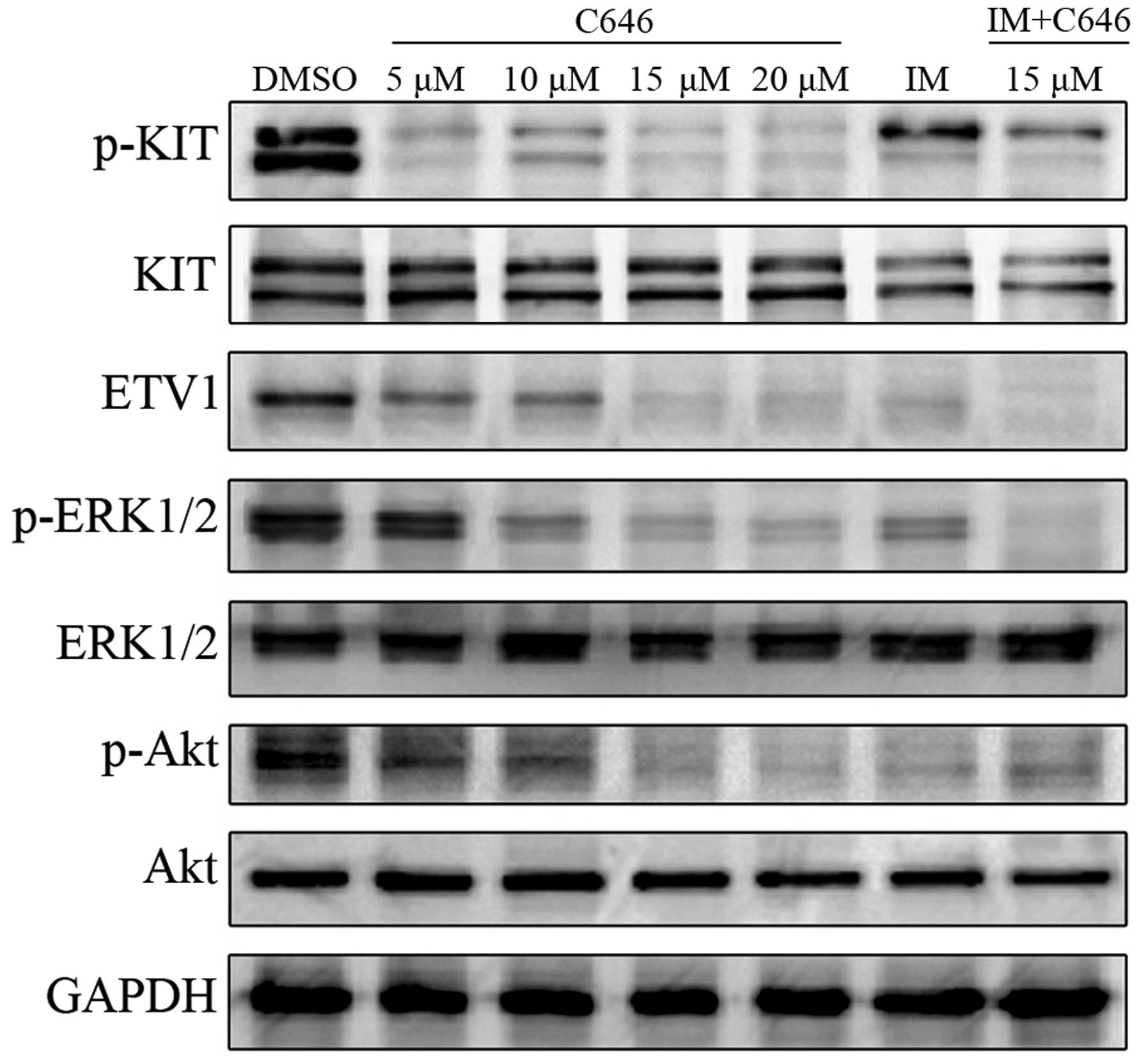
C646 attenuates ETV1 expression and
inactivates KIT-dependent pathways
To address the molecular mechanisms underlying the
antineoplastic effects induced by C646 in GIST882 cells, the
expression levels of ETV1 and functional changes in the KIT pathway
were explored. The GIST cells were treated with C646 (5–20
µmol/l) and imatinib (500 nmol/l) alone and in combination
with C646 (15 µmol/l) and were further incubated for 24 h.
As shown in Fig. 5, C646
administration strongly decreased ETV1 expression in a
dose-dependent manner. Considerable inhibition of KIT
phosphorylation was observed following C646 treatment. Moreover,
the phosphorylation levels of Akt and ERK1/2 were attenuated
accordingly after exposure of the cells to C646 for 24 h. In
contrast, C646 had little effect on the expression levels of total
Akt and ERK1/2. In the combination study, synergistic inhibitory
effects on KIT and ERK1/2 activation were observed in GIST882 cells
after treatment with comparatively low concentrations of imatinib
(500 nmol/l) and C646 (15 µmol/l). Taken together, C646 may
exert its antineoplastic effects through inhibition of ETV1
expression and inactivation of the KIT-dependent pathway, leading
to suppressed phosphorylation of Akt and ERK1/2.
Discussion
Recent advances in the knowledge of GIST
pathogenesis have contributed to the rapid developments of
therapeutic agents targeting abnormalities in KIT and
PDGFRA. Although the advent of potent RTK inhibitors, such
as imatinib and sunitinib, has greatly improved median overall
survival in inoperable and metastatic cases (24–26),
most patients eventually develop resistance due to various
resistance mechanisms (27,28), representing a clinical challenge to
antitumor therapy. As secondary mutations have appeared in the
majority of the patients who develop resistance (9), present antineoplastic strategies are
aimed at improving the efficiency and selectivity of the anticancer
therapy in spite of these mutations. Based on this goal,
posttranslational modifications involving oncology and therapy have
been intensively studied in GISTs over the past few decades. It has
been suggested that microRNA-221/222 may serve as a new therapeutic
strategy for treating TKI-resistant GIST (29). Here, we explore the functions of the
HAT proteins CBP and p300, which are involved in GIST
tumorigenesis.
Being a part of the vital molecules in
posttranslational modifications, the acetyltransferases CBP/p300
play crucial roles in oncogenesis and progression. CBP/p300 are
known to have tumor-promoting effects as they have been shown to
accelerate the progression of colon and prostate cancers (30,31).
However, mutations of the CBP and p300 genes or
dysfunction of the proteins will promote cell proliferation and
consequently increase the incidence of haematological malignancies,
suggesting a tumor-suppressor role of CBP/p300 in carcinogenesis
(32,33). Thus, the function of CBP/p300 varies
accordingly under different circumstances. The present study
demonstrated that blockage of CBP and p300 by
specific siRNAs induced cell growth suppression in GITS882 cells.
Interestingly, whereas transcriptional inhibition of CBP
exhibited a strong antiproliferative activity, p300
inhibition did not exert a similar inhibitory effect on cell
proliferation, implying that CBP rather than p300 played a dominant
role in the regulation of cell proliferation. These results support
the idea that despite their high degree of homology, CBP and p300
may have non-overlapping functions and accordingly may play
different roles (34). Nonetheless,
silencing of CBP and p300 significantly enhanced
caspase-3/7 activity, indicating that apoptosis induction did not
completely parallel growth inhibition. Our results revealed that
depletion of CBP/p300 resulted in decreased ETV1 expression levels
along with KIT inactivation in GIST882 cells. Previous studies have
reported that CBP/p300 directly interacted with ETV1 and enhanced
its DNA-binding and transactivation through acetylation (20). Additionally, upregulated ETV1
expression was detected in ICC, which are precursors of GISTs
(35). Therefore, CBP/p300 may
represent a specific target for GISTs due to its role in regulating
ETV1, the key transcriptional factor that is highly expressed in
both GISTs and its precursor cells. These findings prompt us to
hypothesize that CBP/p300 may stabilize ETV1 via direct acetylation
and activate KIT-dependent pathways, contributing to cell
proliferation and survival in GISTs.
The data reported herein indicate that CBP plays a
key role in the regulation of ERK1/2 and JNK activities, whereas
p300 does not act in a similar way. It is widely acknowledged that
ERK1/2, a subtype of mitogen-activated protein kinases (MAPKs),
regulates cell proliferation, differentiation and migration through
KIT-dependent pathways (36). JNK,
another subtype of MAPKs that is expressed but not phosphorylated
in GISTs in vitro (37),
participates in the death receptor signaling pathway required for
apoptosis induction in various cancers (38–40).
Therefore, CBP is required for tumor growth through ERK1/2
phosphorylation and apoptosis regulation via inactivation of JNK in
GIST882 cells, whereas p300 does not appear to contribute to these
activities. However, both CBP and p300 silencing led to the
upregulation of proapoptotic Bax and the downregulation of
anti-apoptotic Bcl-2 and Bcl-xL, members of the Bcl-2 family that
are involved in the mitochondrial cell death pathway (41), suggesting that CBP/p300 affect
apoptosis through the regulation of Bcl-2 family proteins. Thus,
CBP may regulate apoptosis via both the death receptor signaling
pathway and the mitochondrial cell death pathway in GIST cells. In
contrast, p300 affects apoptotic cell death through the
mitochondrial cell death pathway and has little effect on the death
receptor signaling pathway.
To date, a variety of histone deacetylase (HDAC)
inhibitors have been shown to induce apoptosis in tumor cells and
have been introduced into clinical trials (42). Although pharmacological inhibition
of deacetylation by HDAC inhibitors can exert antiproliferative and
proapoptotic effects on several GIST cell lines (43), not much is known regarding
acetyltransferase inhibitors. To this end, the present study is the
first to show strong antiproliferative and proapoptotic effects in
imatinib-sensitive GIST cells by C646, a competitive inhibitor of
HATs. The GIST882 cells appear to be highly sensitive to the HAT
inhibitor based on the observation that GIST cell proliferation is
reduced substantially following exposure to C646. Treatment with
C646 resulted in marked apoptotic cell death, demonstrating potent
antineoplastic activity. In addition to proapoptotic activity, C646
treatment also induced considerable cell cycle arrest at an early
time-point (24 h), showing the high efficacy of its antigrowth
effect. Moreover, C646 administration inhibited KIT phosphorylation
and deactivated ERK1/2, disrupting the downstream cascade for tumor
proliferation and survival. Overall, C646-induced antineoplastic
effects follow a similar pattern as that of CBP downregulation.
Therefore, it is conceivable that C646 is able to exert these
antitumor effects mainly through inhibition of CBP activity. Our
data further showed that imatinib and C646 combination therapy
would exert synergistic inhibitory effects on GIST cells,
suggesting a potential usage of C646 in GIST therapy especially for
imatinib-resistant cases. This effect may be partly attributed to
the inactivation of KIT and ERK1/2. Additionally, C646 exhibits a
strong inhibitory effect on the phosphorylation of Akt, another
downstream component of KIT and a critical molecule in GIST
tumorigenesis (44). Taken
together, our study indicates that C646 may be a promising
antineoplastic reagent for GIST therapy.
However, this study has some limitations. Further
investigations of the expression and function of CBP/p300 in
clinical GIST samples or animal models are required to clarify its
role in tumorigenesis and progression in vivo. Moreover,
although an immediate synergism with imatinib was acquired
following C646 administration in GIST882 cells, these effects
require further examination in other GIST cell lines that are
sensitive and resistant to imatinib. The efficacy and safety of
C646 as an antineoplastic agent for GISTs have not yet been fully
characterized and remain to be validated both in vitro and
in vivo.
In conclusion, our results demonstrate that
CBP silencing will inhibit cell proliferation and promote
apoptosis induction, whereas p300 inhibition has little
effect on cell growth, posing some advantages of CBP as a more
effective target for GIST therapy. The molecular mechanisms
underlying this antineoplastic effect may be attributed to ETV1
downregulation and inactivation of KIT-dependent pathways. CBP
regulates apoptosis through both the death receptor pathway and the
mitochondrial cell death pathway in GIST882 cells, whereas p300 has
little effect on the death receptor pathway. Furthermore, the HAT
inhibitor C646 attenuates ETV1 expression and inactivates
KIT-dependent pathways, triggering antiproliferative effects in
GIST cells and inducing apoptosis or cell cycle arrest.
Consequently, the present study provides mechanistic insight and
supports the notion that CBP/p300 inactivation by HAT inhibitors
may serve as a prospective approach for antitumor therapeutics
against GISTs.
Acknowledgments
This study was supported by the Scientific Research
Foundation of the National Health and Family Planning Commission of
the People's Republic of China (WKJ-ZJ-1614). We are very grateful
to the State Key Laboratory for Diagnosis and Treatment of
Infectious Diseases of the First Affiliated Hospital of Zhejiang
University and the Department of Oncology of Hangzhou First
People's Hospital for providing excellent technical assistance.
References
|
1
|
Rubin BP, Heinrich MC and Corless CL:
Gastrointestinal stromal tumour. Lancet. 369:1731–1741. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Corless CL, Barnett CM and Heinrich MC:
Gastrointestinal stromal tumours: origin and molecular oncology.
Nat Rev Cancer. 11:865–878. 2011.PubMed/NCBI
|
|
5
|
D'Amato G, Steinert DM, McAuliffe JC and
Trent JC: Update on the biology and therapy of gastrointestinal
stromal tumors. Cancer Control. 12:44–56. 2005.PubMed/NCBI
|
|
6
|
Joensuu H, Hohenberger P and Corless CL:
Gastrointestinal stromal tumour. Lancet. 382:973–983. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Joensuu H and DeMatteo RP: The management
of gastrointestinal stromal tumors: a model for targeted and
multidisciplinary therapy of malignancy. Annu Rev Med. 63:247–258.
2012. View Article : Google Scholar :
|
|
8
|
Heinrich MC, Corless CL, Blanke CD,
Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M,
Fletcher CD, Sandau K, et al: Molecular correlates of imatinib
resistance in gastrointestinal stromal tumors. J Clin Oncol.
24:4764–4774. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lim KH, Huang MJ, Chen LT, Wang TE, Liu
CL, Chang CS, Liu MC, Hsieh RK and Tzen CY: Molecular analysis of
secondary kinase mutations in imatinib-resistant gastrointestinal
stromal tumors. Med Oncol. 25:207–213. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hou XW, Bai CG, Liu XH, Qiu C, Huang L, Xu
JJ and Ma DL: Expression of stem cell factor in gastrointestinal
stromal tumors: implications for proliferation and imatinib
resistance. Oncol Lett. 5:552–558. 2013.PubMed/NCBI
|
|
11
|
Rutkowski P, Symonides M, Zdzienicki M and
Siedlecki JA: Developments in targeted therapy of advanced
gastrointestinal stromal tumors. Recent Patents Anticancer Drug
Discov. 3:88–99. 2008. View Article : Google Scholar
|
|
12
|
Kalkhoven E: CBP and p300: HATs for
different occasions. Biochem Pharmacol. 68:1145–1155. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giordano A and Avantaggiati ML: p300 and
CBP: partners for life and death. J Cell Physiol. 181:218–230.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goodman RH and Smolik S: CBP/p300 in cell
growth, transformation, and development. Genes Dev. 14:1553–1577.
2000.PubMed/NCBI
|
|
15
|
Santer FR, Höschele PP, Oh SJ, Erb HH,
Bouchal J, Cavarretta IT, Parson W, Meyers DJ, Cole PA and Culig Z:
Inhibition of the acetyltransferases p300 and CBP reveals a
targetable function for p300 in the survival and invasion pathways
of prostate cancer cell lines. Mol Cancer Ther. 10:1644–1655. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bandyopadhyay D, Okan NA, Bales E,
Nascimento L, Cole PA and Medrano EE: Down-regulation of p300/CBP
histone acetyltransferase activates a senescence checkpoint in
human melanocytes. Cancer Res. 62:6231–6239. 2002.PubMed/NCBI
|
|
17
|
Gao XN, Lin J, Ning QY, Gao L, Yao YS,
Zhou JH, Li YH, Wang LL and Yu L: A histone acetyltransferase p300
inhibitor C646 induces cell cycle arrest and apoptosis selectively
in AML1-ETO-positive AML cells. PLoS One. 8:e554812013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Papoutsopoulou S and Janknecht R:
Phosphorylation of ETS transcription factor ER81 in a complex with
its coactivators CREB-binding protein and p300. Mol Cell Biol.
20:7300–7310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baena E, Shao Z, Linn DE, Glass K, Hamblen
MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, et al: ETV1
directs androgen metabolism and confers aggressive prostate cancer
in targeted mice and patients. Genes Dev. 27:683–698. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goel A and Janknecht R:
Acetylation-mediated transcriptional activation of the ETS protein
ER81 by p300, P/CAF, and HER2/Neu. Mol Cell Biol. 23:6243–6254.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chi P, Chen Y, Zhang L, Guo X, Wongvipat
J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, et al: ETV1 is
a lineage survival factor that cooperates with KIT in
gastrointestinal stromal tumours. Nature. 467:849–853. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Gu ML, Zhou XX, Ma H, Yao HP and
Ji F: Altered expression of ETV1 and its contribution to
tumorigenic phenotypes in gastrointestinal stromal tumors. Oncol
Rep. 32:927–934. 2014.PubMed/NCBI
|
|
23
|
Bowers EM, Yan G, Mukherjee C, Orry A,
Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, et
al: Virtual ligand screening of the p300/CBP histone
acetyltransferase: identification of a selective small molecule
inhibitor. Chem Biol. 17:471–482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cohen MH, Farrell A, Justice R and Pazdur
R: Approval summary: imatinib mesylate in the treatment of
metastatic and/or unresectable malignant gastrointestinal stromal
tumors. Oncologist. 14:174–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Judson I and Demetri G: Advances in the
treatment of gastrointestinal stromal tumours. Ann Oncol. 18(Suppl
10): x20–x24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Demetri GD, van Oosterom AT, Garrett CR,
Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich
MC, Morgan JA, et al: Efficacy and safety of sunitinib in patients
with advanced gastrointestinal stromal tumour after failure of
imatinib: a randomised controlled trial. Lancet. 368:1329–1338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JH, Kim Y, Choi JW and Kim YS:
Correlation of imatinib resistance with the mutational status of
KIT and PDGFRA genes in gastrointestinal stromal tumors: a
meta-analysis. J Gastrointestin Liver Dis. 22:413–418.
2013.PubMed/NCBI
|
|
28
|
Tarn C, Rink L, Merkel E, Flieder D,
Pathak H, Koumbi D, Testa JR, Eisenberg B, von Mehren M and Godwin
AK: Insulin-like growth factor 1 receptor is a potential
therapeutic target for gastrointestinal stromal tumors. Proc Natl
Acad Sci USA. 105:8387–8392. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koelz M, Lense J, Wrba F, Scheffler M,
Dienes HP and Odenthal M: Down-regulation of miR-221 and miR-222
correlates with pronounced Kit expression in gastrointestinal
stromal tumors. Int J Oncol. 38:503–511. 2011. View Article : Google Scholar
|
|
30
|
Ionov Y, Matsui S and Cowell JK: A role
for p300/CREB binding protein genes in promoting cancer progression
in colon cancer cell lines with microsatellite instability. Proc
Natl Acad Sci USA. 101:1273–1278. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ianculescu I, Wu DY, Siegmund KD and
Stallcup MR: Selective roles for cAMP response element-binding
protein binding protein and p300 protein as coregulators for
androgen-regulated gene expression in advanced prostate cancer
cells. J Biol Chem. 287:4000–4013. 2012. View Article : Google Scholar :
|
|
32
|
Bayly R, Chuen L, Currie RA, Hyndman BD,
Casselman R, Blobel GA and LeBrun DP: E2A-PBX1 interacts directly
with the KIX domain of CBP/p300 in the induction of proliferation
in primary hematopoietic cells. J Biol Chem. 279:55362–55371. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kung AL, Rebel VI, Bronson RT, Ch'ng LE,
Sieff CA, Livingston DM and Yao TP: Gene dose-dependent control of
hematopoiesis and hematologic tumor suppression by CBP. Genes Dev.
14:272–277. 2000.PubMed/NCBI
|
|
34
|
Kawasaki H, Eckner R, Yao TP, Taira K,
Chiu R, Livingston DM and Yokoyama KK: Distinct roles of the
co-activators p300 and CBP in retinoic-acid-induced F9-cell
differentiation. Nature. 393:284–289. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rubin BP: Bioinformatic mining of gene
expression datasets identifies ETV1 as a critical regulator of
oncogenesis in gastrointestinal stromal tumors. Cancer Cell.
18:407–408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Duensing A, Medeiros F, McConarty B,
Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD and
Fletcher JA: Mechanisms of oncogenic KIT signal transduction in
primary gastrointestinal stromal tumors (GISTs). Oncogene.
23:3999–4006. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang D, Lu J, Liu Y, Meng Q, Xie J, Wang Z
and Teng L: Liquiritigenin induces tumor cell death through
mitogen-activated protein kinase- (MPAKs-) mediated pathway in
hepatocellular carcinoma cells. Biomed Res Int.
2014:9653162014.PubMed/NCBI
|
|
39
|
Yoon JY, Cho HS, Lee JJ, Lee HJ, Jun SY,
Lee JH, Song HH, Choi S, Saloura V, Park CG, et al: Novel TRAIL
sensitizer Taraxacum officinale F.H. Wigg enhances TRAIL-induced
apoptosis in Huh7 cells. Mol Carcinog. 55:387–396. 2016. View Article : Google Scholar
|
|
40
|
Kuo KL, Ho IL, Shi CS, Wu JT, Lin WC, Tsai
YC, Chang HC, Chou CT, Hsu CH, Hsieh JT, et al: MLN4924, a novel
protein neddylation inhibitor, suppresses proliferation and
migration of human urothelial carcinoma: in vitro and in vivo
studies. Cancer Lett. 363:127–136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Krestnikova N, Stulpinas A, Imbrasaite A,
Sinkeviciute G and Kalvelyte AV: JNK implication in adipocyte-like
cell death induced by chemotherapeutic drug cisplatin. J Toxicol
Sci. 40:21–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mühlenberg T, Zhang Y, Wagner AJ,
Grabellus F, Bradner J, Taeger G, Lang H, Taguchi T, Schuler M,
Fletcher JA, et al: Inhibitors of deacetylases suppress oncogenic
KIT signaling, acetylate HSP90, and induce apoptosis in
gastrointestinal stromal tumors. Cancer Res. 69:6941–6950. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Patel S: Exploring novel therapeutic
targets in GIST: focus on the PI3K/Akt/mTOR pathway. Curr Oncol
Rep. 15:386–395. 2013. View Article : Google Scholar : PubMed/NCBI
|