Introduction
Uncontrolled cell proliferation, disorganized tissue
growth and subsequent tumor formation may be the result of
dysregulation in cell growth and antigrowth signals and changes in
programmed cell death pathways. The aberrant expression of multiple
genes makes normal cells ignore growth controlling signals,
resulting in tumor formation. Tumors of the central nervous system
(CNS) are characterized by heterogeneity within the cell population
and represent a very serious medical problem (1,2). The
grading system of brain tumors according to World Health
Organization (WHO) assigns grades I–IV in order of increasing
anaplasia and malignancy. Brain tumors with a low growth rate have
a benign character (WHO grade I or II) while tumors with malignant
character (WHO grade III or IV) can infiltrate into surrounding
tissue (3). The incidence of
primary malignant brain and CNS cancer in developed countries is
5.8 per 100,000 in males and 4.4 per 100,000 in females (4).
Gliomas originate from dedifferentiated mature
neural cells that may transform into cancer stem cells (CSCs)
(2,5). For treatment to be successful, it is
crucial to eliminate CSCs, which represent a key factor in tumor
reduction (6,7). According to the WHO classification
system established in 2007, glioblastoma (GB) is the most
aggressive WHO grade IV astrocytoma (3) and is the most frequent histological
type of brain tumor, accounting for 60–70% of all gliomas (8,9). The
prognosis remains very poor, with most patients dying within one
year of diagnosis. GB affects patients of different ages and
develops through different genetic pathways (10). Despite the immense pace of research
to increase our understanding of the molecular basis of GB, the
clinical utility of key genes and markers remains limited (11).
The majority of GBs arise de novo from
astrocytes and are designated primary GB, and are found mostly in
older patients. On the contrary, secondary GBs develop via
malignant transformation of lower grade astrocytomas and are more
often observed in younger patients (4,5,8,12).
The mean age of primary GB patients is 55–62 years, with a median
survival of 4.7 months, while secondary GB patients have a mean age
40–45 years, with a significantly longer median survival of 7.8
months (10,13). Standard treatment for patients with
newly diagnosed GB consists of surgical resection, radiotherapy and
chemotherapy. Despite considerable progress in research, the
survival rate of GB patients has not improved as expected and
resistance to clinical therapy is a major obstacle to successful
treatment (7,14). Radiotherapy and chemotherapy work
predominantly by inducing apoptosis (15). The unfavorable prognosis of patients
diagnosed with GB can also be explained by our poor understanding
of GB molecular pathway alterations. The elucidation of
disturbances present in apoptotic pathways is one way to avoid GB
resistance to therapy (14,16). Dysregulation of physiological
apoptosis mechanisms plays an important role in the pathogenesis
and progression of gliomas.
Difficult access to human tumors and normal brain
tissue makes commercially available cell lines useful and popular
experimental models. Therefore, the characterization of their
features and response to a wide range of agents can provide
important information and knowledge. The majority of spontaneous
cell deaths in malignant gliomas are due to apoptosis (9,17,18).
Resistance to apoptosis is a common feature of tumor cells and
leads to drug resistance mediated by the Bcl-2 family of proteins
(19).
Activation of apoptosis can be triggered by
different pathways distinguished according to pathological
conditions in a specific tissue type. Understanding the mechanisms
involved in apoptotic signaling pathways in GB may contribute to
identifying target molecules for molecular therapies (20). The two main apoptotic pathway types
have been classified depending on the origin of the stimulus of
death: (i) the extrinsic receptor pathway and (ii) the intrinsic
mitochondrial pathway (21,22). The final step in both pathways,
which leads to programmed cell death, is the activation of effector
caspases (23). The extrinsic
apoptotic pathway is activated by the stimulation of tumor necrosis
factor (TNF)-α or Fas cell surface death receptors. The TNF family
and its corresponding receptors play important roles in cell death
or survival, proliferation and maturation. All members of the TNF
superfamily activate nuclear factor of kappa light polypeptide gene
enhancer in B-cells (NF-κB), which suppresses apoptosis, cell
survival and proliferation (24–26).
The regulation of cell growth and activation of NF-κB by the TNF
family is mediated by the sequential activation of a set of cell
signaling proteins, namely TNF receptor-associated factors,
Fas-associated death domain and FADD-like ICE, caspases, receptor
interacting proteins, NF-κB-inducing kinases and IκBα kinases
(27). The intrinsic mitochondrial
pathway is controlled by interactions between anti- and
pro-apoptotic B-cell lymphoma 2 (Bcl-2) members which share
homology via BH3 domains (28). The
Bcl-2 family of proteins can be divided into two subgroups:
anti-apoptotic proteins, namely Bcl-2, Bcl-xL, Bcl-w, Mcl-1
(myeloid cell leukemia 1), A1, and pro-apoptotic proteins. The
pro-apoptotic proteins can be further divided into two classes: i)
the Bax-like proteins (Bax, Bak, Bok) and ii) the BH3-only proteins
(Bid, Bad, Bim, Bik, Blk, Hrk, Noxa, PUMA) that are unrelated in
their sequence to each other or other Bcl-2 family members with the
exception of the BH3 domain (23,29,30).
While anti-apoptotic members stabilize mitochondrial membrane
potential and prevent the release of cytochrome c and
apoptosis-inducing factors, pro-apoptotic members activate
programmed apoptotic cell death 26,31–33.
Anti-apoptotic family members share up to four highly conserved
Bcl-2 homology domains, i.e., BH1, BH2, BH3 and BH4 (23,29,34).
The multidomain pro-apoptotic Bcl-2 family proteins Bax and Bak are
essential for mitochondrial apoptosis; their activity is controlled
by the BH3-only pro-apoptotic Bcl-2 family of proteins (28). BH3-only Bcl-2 family members are
critical for cell death initiation, can function as tumor
suppressors and their loss can induce apoptosis. Their activity is
tightly controlled by diverse transcriptional and
post-translational mechanisms. BH3-only proteins selectively bind
to the hydrophobic groove of anti-apoptotic Bcl-2 family members,
leading to Bax/Bak activation (31). Activation of Bax and Bak results in
the release of apoptogens from the mitochondrial intermembrane
space and activation of an amplifying cascade of caspase-mediated
proteolysis (35,36). The release of cytochrome c
into the cytoplasm with the involvement of apoptotic peptidase
activating factor 1 (APAF1) and deoxy-ATP results in the formation
of the apoptosome, which stimulates caspases, a family of
apoptosis-related cysteine proteases (37,38).
Caspases that initiate cell death and play critical roles in human
cell apoptosis are divided into two classes, (i) initiator
caspases, i.e., caspase-1, −2, −4, −5, −8, −9, −10, −12 and −13 and
(ii) the effector caspases, i.e., caspase-3, −6 and −7 (26,36).
One of the cardinal features of cancer is the
deregulation of apoptosis (39).
Most drugs, including cancer chemotherapeutic agents, chemicals or
irradiation, induce apoptosis by activating the intrinsic apoptotic
pathway. High levels of anti-apoptotic Bcl-2 family members are
associated with the resistance of many tumor types to clinically
used chemotherapy. Apoptosis inhibitors bind selectively to
specific types of anti-apoptotic proteins and are useful tools in
mechanistic studies (34,40). Over the last few years, several
small BH3 mimetic molecules have been synthesized that can inhibit
the anti-apoptotic Bcl-2 family of proteins and thus induce
apoptosis (41–44).
The most potent small molecule inhibitors are the
Bad-like BH3 mimetic ABT-737 and its orally active analog ABT-263,
which bind with very high nanomolar affinity to the hydrophobic
pocket of the Bcl-2, Bcl-xL and Bcl-w anti-apoptotic proteins
(41,43,45).
Bcl-2 is expressed in the nervous system, and its function in
neuronal tumors was first described by Reed et al (46). Increased expression of Bcl-xL has
been found in many solid tumors, including neuronal tumors
(18). ABT-737 releases
pro-apoptotic proteins that activate Bax and Bak and thus is able
to induce apoptosis or sensitize cancer cells to cytotoxic agents
in a range of tumor types (34,43).
The apoptosis inhibitor ABT-737 does not directly initiate the
apoptotic process, but enhances the effect of death signals by
selectively binding with a high affinity directly to the
hydrophobic areas in the structure of the anti-apoptotic proteins
Bcl-2, Bcl-xL, Bcl-w, leading to their inhibition (19,43).
ABT-737 could be an important anticancer agent as it
functions by regulating gene transcription and sensitizing
resistant tumor types to alternative therapies (47). It displays synergistic cytotoxicity
with chemotherapeutics and radiation (43). ABT-737 treatment regulates changes
in the transcription of genes involved in cellular senescence
(48). As one of the most specific
BH3-mimetic agents, ABT-737 can be useful, but also ineffective at
inducing apoptosis in solid tumors. It targets Bcl-2, Bcl-xL and
Bcl-w, but not Mcl-1, and thus tumor cell expression of Mcl-1 is
associated with resistance to ABT-737. The control of Mcl-1 has
become an area of intense investigation (32,49,50).
Resistance to apoptosis is in part mediated by the increased
expression of Mcl-1 or Bcl2A1, which are not capable of binding
this compound.
The anti-apoptotic protein Mcl-1 was identified as
an early response gene induced during the differentiation of human
myeloblastic leukemia cells (51)
and has been shown to be expressed at relatively high levels in a
wide range of hematological as well as solid malignancies (31). Mcl-1 is the most labile
anti-apoptotic Bcl-2 protein with a very short half-life.
Furthermore, Mcl-1 has been identified in several studies as being
the major resistance factor for ABT-737. Destabilization of Mcl-1
makes such cells sensitive to apoptosis-induction by ABT-737
(32,50,52).
MIM-1 is a novel molecule that selectively targets the BH3 binding
groove of Mcl-1, with Bak-dependent apoptotic activity (42). It has the opposite biochemical and
cellular activity profile of ABT-737 and may have limited cell-type
dependent potency. MIM-1 may serve as a prototype for the
development of the next generation of small molecules that
effectively reduce the apoptotic threshold in cancers specifically
driven by anti-apoptotic Mcl-1 protein expression (42). The inhibition of Mcl-1 leads to the
inhibition of cell proliferation and metastasis. Hence, it is a
promising target for tumor therapy (53).
In our previous studies, we focused on gene
expression profiling in different types of healthy and tumor
tissues (54,55) as well as GB tissue (56). Genomic characterization can rapidly
expand knowledge of the molecular basis of GB (57,58).
We hypothesized that deregulation of the apoptotic pathway in
astrocyte and GB cells after selective apoptosis inhibitor
treatment may contribute to understanding brain tumor cell
metabolism and the process of tumor formation.
Materials and methods
Cell lines and culture conditions
The human astrocyte cell line (HA) was purchased
from ScienCell Research Laboratories (Carlsbad, CA USA). Cells were
cultivated in 96% (v/v) Astrocyte Medium supplemented with 2% (v/v)
fetal bovine serum (FBS), 1% (v/v) Astrocyte Growth Supplement and
1% (v/v) penicillin/streptomycin solution (all from ScienCell
Research Laboratories) and incubated in a humidified atmosphere
with 5% CO2 at 37°C. The culture medium was renewed
every third day. The human GB cell line (T98G) was purchased from
the European Collection of Cell Cultures (Salisbury, UK). Cells
were cultivated in 89% (v/v) Dulbeccos modified Eagles medium (PAA,
Linz, Austria) supplemented with 10% (v/v) FBS (Gibco, USA), 1%
(v/v) penicillin/streptomycin solution (PAA) and incubated in a
humidified atmosphere with 5% CO2 at 37°C. The culture
medium was renewed every third day.
Drug treatment
The IC50 value, defined as the
concentration that reduces the global growth of cells by 50%, was
determined for the apoptosis inhibitors ABT-737 and MIM-1,
individually, for the HA and GB (T98G) cell lines. The apoptosis
inhibitor concentrations and treatment time periods were selected
experimentally according to preliminary experiments. The final
ABT-737 treatment was performed with 10-fold increasing
concentrations in the range of 0.001–100 µmol/l, and the final
MIM-1 treatment was performed with 4-fold increasing concentrations
in the range of 0.4–400 µmol/l, for 48 h.
Cell viability assay
The biochemical colorimetric
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrayolium bromide (MTT)
assay, based on the enzymatic conversion of MTT to a violet
formazan salt (59), was used to
assess the viability of the HA and T98G cells. Briefly, the cells
in culture medium were seeded (3.5×103 cells/well for
the HA cell line, 3.0×103 cells/well for T98G cell line)
in 96-well microtiter plates. On the third day, the medium was
changed to culture medium supplemented with the apoptosis inhibitor
ABT-737 (Abbott, USA) or MIM-1 (Calbiochem, Billerica, MA, USA) at
varied concentrations and incubation continued for another two
days. After the treatment with the apoptosis inhibitors, cells were
rinsed once with Dulbeccos phosphate buffer saline (DPBS) and
further incubated in medium supplemented with 0.5 mg/ml MTT in a
humidified atmosphere for 6 h. During a subsequent incubation for
16 h in medium containing SDS [5% (w/v)], the precipitated
formazan, the amount of which is proportional to the number of live
cells, was solubilized. The absorbance of the formazan-containing
solution was measured at 540 nm using an ELISA plate reader
(Bio-Rad PR2100; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The absorbance was also determined for the medium of the control
cells not exposed to the apoptosis inhibitors. The percentage of
cell viability was calculated relative to the untreated control
cells. The IC50 values were determined for both human
brain cell lines after individual apoptosis inhibitor
treatment.
RNA extraction and reverse
transcription
Total RNA was extracted from the HA and T98G cell
lines after 48 h of apoptosis inhibitor treatment (individually) at
the IC50 with TRI Reagent® (MRC, Cincinnati,
OH, USA) following the manufacturers protocol. The RNA quality of
each sample was checked using a NanoDrop® apparatus
(Thermo Fisher Scientific Inc., Wilmington, DE, USA) and by
MCE®-202 MultiNA (Shimadzu Corporation, Kyoto, Japan).
Five micrograms of total RNA were reverse transcribed in a total
volume of 14 µl using the Maxima First Strand cDNA Synthesis kit
(Thermo Scientific, USA) according to the manufacturers
protocol.
TaqMan gene expression array
The regulation of gene expression was studied using
TaqMan® Human Apoptosis Array (Applied Biosystems™, USA)
based on the qRT-PCR reaction. The predesigned 384-well
microfluidic card contained 93 human apoptotic genes and three
endogenous control genes: eukaryotic 18S rRNA (18S), β-actin (ACTB)
and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). A reaction
mixture with 100 ng cDNA template and an equal volume of the
TaqMan® Gene Expression Master Mix was loaded into each
line of the microfluidic card. The PCR mix (2×100 µl) was
distributed into the wells by two centrifugations at 450 × g for 1
min, sealed and loaded into the ViiA7 Real-Time PCR system (Applied
Biosystems Life Technologies, Foster City, CA, USA). The standard
amplification protocol consisted of a ramp of 50°C for 2 min and a
hot start of 94.5°C for 10 min, followed by 40 cycles of 15 sec at
95.0°C and 60 sec at 60.0°C. Samples were measured in duplicate and
the levels of the genes of interest were normalized to the three
endogenous controls (18S, ACTB, GAPDH), determined using the ΔΔCt
method. The expression data from the untreated cells were used as a
reference in the ∆∆Ct method calculation for each cell line
individually. The levels of the genes were considered to be
significant when their average fold change (FC) was ≤-2.0 or
≥2.0.
Results
Sensitivity of the cell lines to
apoptosis inhibitor treatment
To estimate the sensitivity of HA and T98G cells to
the apoptosis inhibitors ABT-737 and MIM-1, the colorimetric MTT
assay was used to detect cell viability and to determine the
IC50 value. The astrocyte and GB cell lines demonstrated
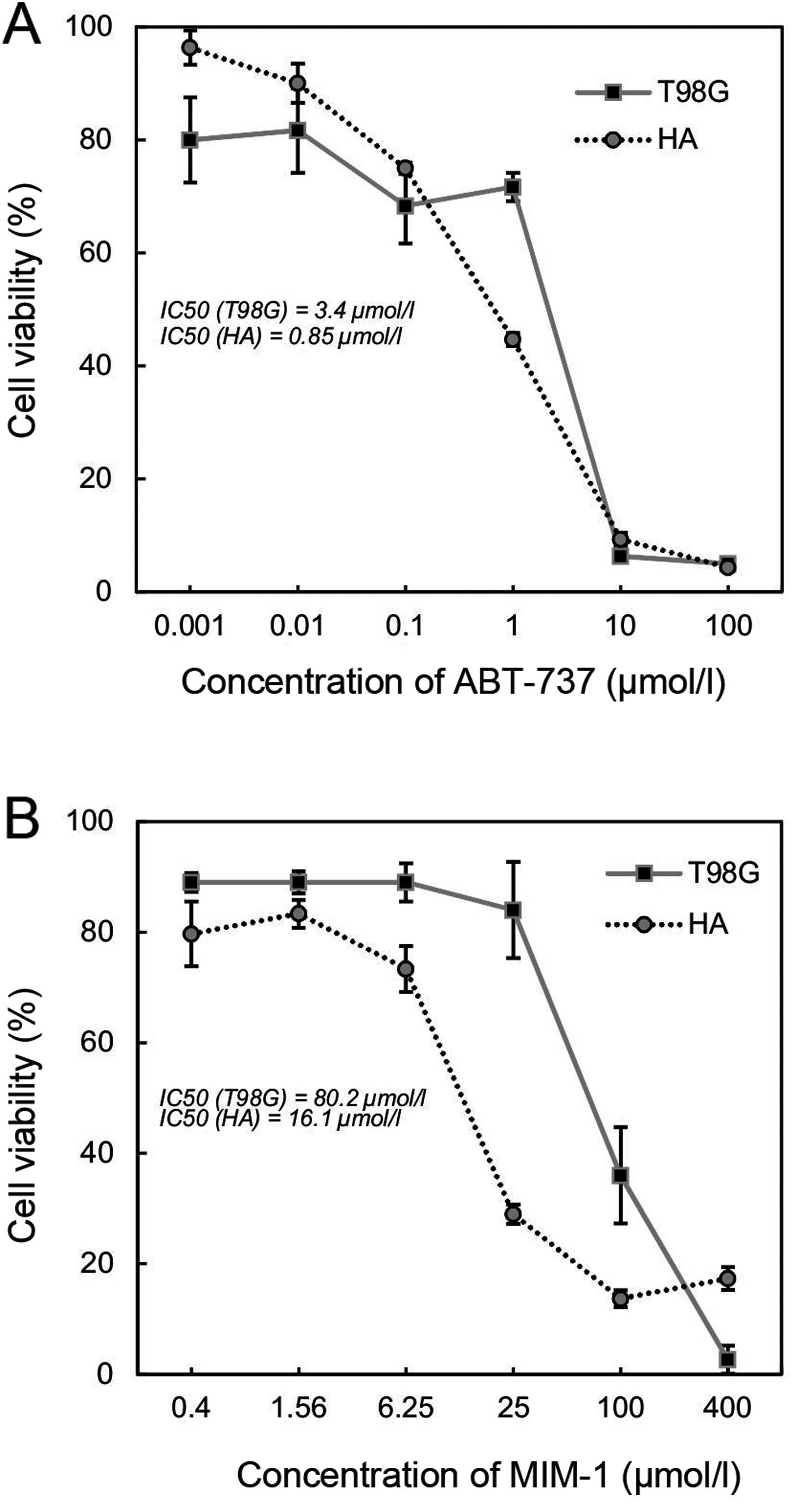
different sensitivities to the inhibitors after 48 h (Fig. 1). The IC50 value of the
astrocyte cell line HA was 4-fold lower (0.85 µmol/l) compared with
the IC50 of the GB cell line T98G (3.40 µmol/l) after
ABT-737 inhibitor treatment (Fig.
1A). The IC50 value of the HA cell line was almost
5-fold lower (16.10 µmol/l) compared with the IC50 of
the T98G cell line (80.20 µmol/l) after MIM-1 inhibitor treatment
(Fig. 1B). The viability of the GB
cells refers to their relative resistance to the apoptosis
inhibitors compared to the astrocyte cell line.
Effect of apoptosis inhibitors on the
expression of apoptosis-associated genes
The expression levels of 93 apoptosis-associated
genes were studied in the astrocyte and GB cell lines after 48 h of
apoptosis-inhibitor treatment (individually) at the IC50
using an intact cell line as a reference. The pre-designed
TaqMan® Human Apoptosis Array Micro Fluidic Cards
(Applied Biosystems Life Technologies) contain the most important
apoptosis signaling pathway-related genes and three internal
controls (18S, ACTB, GAPDH) for data normalization.
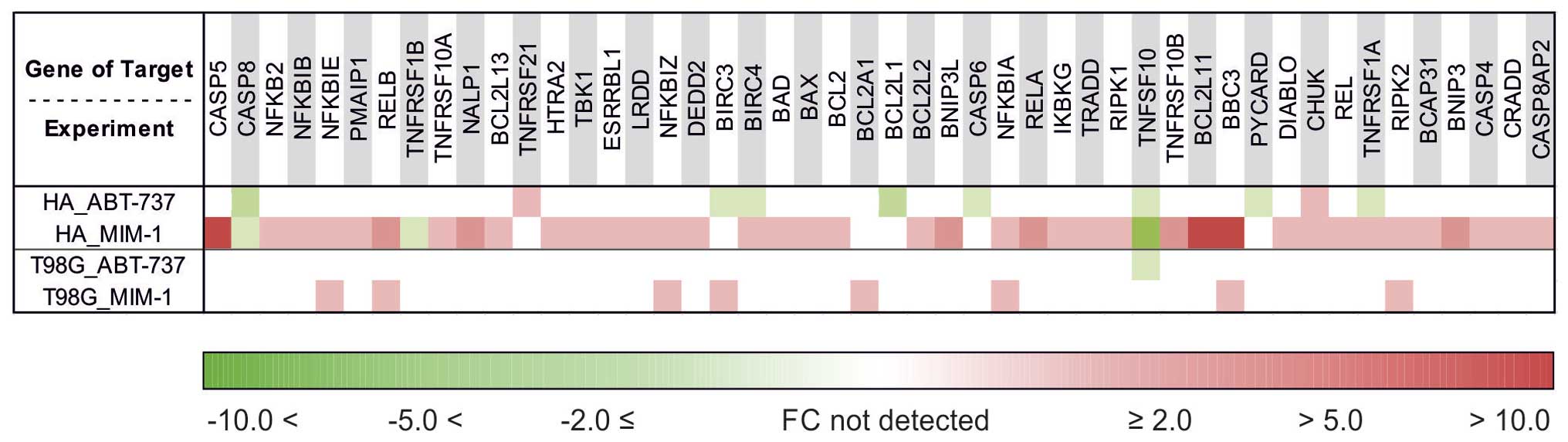
HA cell line and MIM-1 treatment
Bcl-2 family regulated pathway
The expression patterns of the apoptotic genes were
significantly different in the astrocyte cell line HA after
treatment with the apoptosis inhibitor MIM-1 compared to the
reference untreated HA cell line (Fig.
2). The significantly higher expression of Bcl-2 (FC, 4.43) and
Bcl-w (FC, 3.93) may serve as a protective factor against apoptosis
in astrocytes. Other upregulated genes included the apoptosis
facilitator Bim (FC, 11.77), which plays an important role in
neuronal apoptosis and can be induced by nerve growth factor, as
well as upregulation of the pro-apoptotic genes PUMA (FC, 13.12),
Bad (FC, 2.86), Bax (FC, 2.70), NOXA (FC, 2.59), BCL2L13 (FC,
4.42), BNIP3 (FC, 5.86) and BNIP3L (FC, 6.09). The most significant
genes associated with apoptosis, Bax and Bcl-2, were both
upregulated, but upregulation of Bcl-2 was markedly higher (FC of
4.43 compared to 2.70).
TNF receptor pathway
The astrocyte cell line HA demonstrated altered
regulation of the TNF receptor pathway after treatment with the
apoptosis inhibitor MIM-1 (Fig. 3).
The most pronounced changes were significant decreased expression
of the genes TNFSF10 (TRAIL) (FC, −17.02) and TNFRSF1B (FC, −4.22)
and increased expression of TNFRSF10B (FC, 7.46), LRDD (FC, 4.36)
and CASP8AP2 (FC, 3.50). Slight upregulation was detected in the
genes CRADD, RIPK1, RIPK2, TNFRSF1A, TNFRSF10A and TRADD.
Caspases
Our results showed pronounced significantly
increased expression of the inflammatory caspase CASP5 (FC, 16.14)
as well as upregulation of the associated caspase CASP4 (FC, 4.13),
but there did not seem to be an important causative role of CASP8
activation (FC, −3.88), which was downregulated (Fig. 3).
NF-κB signaling pathway
Genetic analysis revealed significant upregulation
of RELB (FC, 8.88), RELA (FC, 5.55), REL (FC, 3.39), NFKBIB (FC,
4.96) and TBK1 (FC, 4.78) and slight upregulation of CHUK, IKBKG,
NFKB2, NFKBIA, NFKBIE and NFKBIZ in the NF-κB signaling pathway
(Fig. 3).
Inhibitor of apoptosis (IAP) family
Baculoviral IAP repeat-containing (BIRC) proteins
are members of the IAP gene family which encode for proteins that
prevent apoptotic cell death (60–62).
Significantly increased expression of the gene BIRC4 (FC, 2.60) was
observed (Fig. 3).
Caspase activation and recruitment domain (CARD)
family
CARDs are interaction motifs found in a wide array
of proteins, typically those involved in apoptosis (63–66).
In the astrocyte cell line, MIM-1 treatment significantly
upregulated NALP1 expression (FC, 8.30) (Fig. 3).
Changes observed in the expression of other
genes
There was a significant increase in the expression
of HTRA2, DEDD2, DIABLO, BCAP31 and ESRRBL1 (Fig. 3).
HA cell line and ABT-737 treatment
The expression patterns of apoptotic genes were
significantly different for only 10 apoptosis-associated genes,
involved in different regulatory pathways, in the astrocyte cell
line HA after treatment with the apoptosis inhibitor ABT-737
compared to the reference, untreated HA cell line (Fig. 3). Significantly lower expression was
detected for the genes BCL2L1 (FC, −6.95), TNFRSF1A (FC, −2.68),
TNFSF10 (FC, −2.03), CASP8 (FC, −8.63), CASP6 (FC, −2.08), BIRC3
(FC, −2.50), BIRC4 (FC, −2.26) and PYCARD (FC, −2.17), while
expression of the genes TNFRSF21 (FC, 2.42) and CHUK (FC, 2.06) was
significantly higher.
Human GB cell line and MIM-1 treatment
The expression patterns of apoptotic genes were
significantly different for only eight apoptosis-associated genes
in the GB cell line T98G after treatment with the apoptosis
inhibitor MIM-1 compared to the reference, untreated T98G cell line
(Fig. 3). Genes with prominently
altered expression are involved in different regulated pathways,
most of them (n=4) in the NF-κB signaling pathway. Significantly
higher expression was observed for the genes BBC3 (FC, 2.04),
BCL2A1 (FC, 2.50), NFKBIA (FC, 2.26), NFKBIE (FC, 2.50), NFKBIZ
(FC, 4.79), RELB (FC, 2.51), RIPK2 (FC, 2.49) and BIRC3 (FC,
3.62).
Human GB cell line and ABT-737 treatment
A difference in in the expression of only one
apoptotic gene was observed in the panel of apoptosis-associated
genes in the GB cell line T98G after treatment with the apoptosis
inhibitor ABT-737 compared to intact T98G cells (Fig. 3). Significantly lower expression was
observed for the gene TNFSF10 (FC, −2.78), which is involved in the
TNF receptor regulated pathway.
Discussion
Brain tumors are very diverse in their biological
behavior and therefore are considered a major issue in modern
medicine. GB, WHO grade IV, arises from astrocytes de novo
as primary GB or by malignant transformation of lower grade
astrocytomas as secondary GB. Generally, GB is the most aggressive
brain tumor in adulthood and is often resistant to clinical
treatment. Searching for new markers and treatment targets is the
aim of current research studies (3,11).
Notably, human malignant gliomas exhibit high Bcl-2 protein levels,
and glioma cells with stem cell features are characterized by high
expression of Mcl-1 (67).
Therefore, targeting these proteins in human GB and astrocytes may
contribute to understanding the process of apoptotic cell death in
this aggressive brain tumor.
The aim of our study was to assess the sensitivity
of astrocyte and GB cells to apoptosis inhibitors (ABT-737 and
MIM-1) by the colorimetric MTT assay (Fig. 1) and to analyze apoptotic gene
expression changes after apoptosis inhibitor treatment (Figs. 2 and 3) using the commercially available set of
genes included in the TaqMan® Human Apoptosis Array
Microarray Card. Cell sensitivity to apoptosis inhibitors depends
on the expression levels of appropriately inhibited anti-apoptotic
proteins. Based on the cell viability results and IC50
values, GB cells express higher levels of Bcl-2, Bcl-xL, Bcl-w and
Mcl-1 proteins compared to astrocytes. The results of our study
demonstrate the relative resistance of the GB cell line T98G to
apoptosis inhibitors compared to the astrocyte cell line HA and
show that the expression patterns of several apoptotic genes are
significantly different between the cell lines after individual
apoptosis inhibitor treatment against the appropriate untreated
cell line as a reference (Fig. 2).
Our understanding all the biological attributes and molecular
phenotypes of astrocytes and GB cells is still limited and remains
one of the most challenging investigations worldwide. Sensitivity
determination in the HA and T98G cell lines to selective apoptosis
inhibitors and the characterization of gene expression signatures
can help to us understand the mechanism of GB malignant
transformation and increase our knowledge about GB physiology.
The development of Bcl-2 inhibitors with the ability
to specifically inhibit BH3-Bcl-2 family member protein-protein
interactions at low nanomolar concentrations with no toxicity is
potentially a significant development in cancer therapy. The great
challenge now is to explore how to best utilize these compounds in
different tumor types. The Bcl-2 family of protein inhibitors
represent a promising avenue of anticancer therapy, either to be
used as single agents, or they may be very beneficial in
combination with other modalities (32,41).
ABT-737 potentiates the cytotoxicity of the chemotherapeutic drugs
vincristine and etoposide, and the inhibition of proliferation and
the induction of apoptosis by ABT-737 are less efficient in glioma
stem cells than in non-stem cell-like glioma cells. The resistance
of glioma stem cells is associated with high Mcl-1 expression
levels (67). The combination of
ABT-737 and MIM-1 results in synergistic cytotoxicity (42). Therefore, the identification of cell
sensitivity and gene alterations in tumor and normal brain cells
after specific apoptotic pathway inhibition may lead to important
knowledge and help in the development of significant therapeutic
advances. In the present study, the astrocyte and GB cell lines
demonstrated different sensitivity to individual ABT-737 and MIM-1
inhibitor treatment, and allowed us to identify the IC50
values. According to our results (Fig.
1), the IC50 value of the astrocyte cell line HA was
4-fold lower compared with the IC50 of the GB cell line
T98G after ABT-737 treatment. Similarly, with MIM-1 inhibitor
treatment, the astrocyte cell line HA was more sensitive with an
IC50 value almost 5-fold lower compared with the
IC50 value of the GB cell line T98G. The viability of GB
cells was in reference to the relative resistance of the astrocyte
cell line to apoptosis inhibitors.
Many recent studies have analyzed and confirmed
changes in p53 gene expression in human tumor tissue or cell lines.
Higher expression of this gene is associated with the activation of
programmed apoptotic cell death (68–71).
One possible reason of differentially expressed apoptotic genes is
that studied cell lines have different p53 status (p53 is a tumor
suppressor protein that regulates the expression of a wide variety
of genes involved in apoptosis), because normal HAs are wild-type
and the T98G cell line is mutated. Therefore, alterations in
apoptosis pathways are considered to play a key role in tumor
formation and progression. Tumorigenesis can be driven by the
deregulation and perturbation of apoptotic cell death pathways and
represents one of the major reasons for the failure of conventional
anticancer treatment in the clinic (31,41,72).
GB cells show globally lower expression of caspase-3, compared to
the expression of the Bcl-2 protein, suggesting that anti-apoptotic
mechanisms in tumor cells are more active than pro-apoptotic
pathways (9). Highly invasive
cancer cells are protected from apoptosis by the upregulation of
various anti-apoptotic molecules, including Bcl-2. Human malignant
GB cells also present high levels of the Bcl-2, which may confer
resistance to apoptosis (9,67). Overexpression of Bcl-2 provides a
survival advantage to cancer cells in response to a wide range of
apoptotic stimuli through the inhibition of mitochondrial
cytochrome c release (26).
Inhibition of Mcl-1 is associated with synergistic upregulation of
Bim by the displacement of Bak from Mcl-1 (73). Moreover, blocking Mcl-1-mediated
suppression of Bid-induced Bax activation in vitro can
activate cell death pathways (42).
Our results showed that normal HAs showed upregulated expression of
Bim by >11-fold and Bax by >2-fold after Mcl-1 inhibition by
MIM-1. The potential function of Bcl-2 in neuronal cells was first
described by Reed et al (46). Bcl-2 is therefore considered to be a
predominant anti-apoptotic protein that preserves neuronal cell
survival. Consistent with these findings, our results showed
upregulation of Bcl-2 and Bcl-w in the normal HA cell line, which
may serve as an essential protective factor against apoptosis after
Mcl-1 inhibition by MIM-1.
BH3-only proteins are essential initiators of
apoptosis signaling, including Bcl-2-binding component 3 (BBC3) or
also a p53 upregulated modulator of apoptosis (PUMA), which is a
pro-apoptotic member of the BH3-only subgroup of the Bcl-2 family
(31). Biochemical studies have
shown that PUMA interacts with anti-apoptotic Bcl-2 family members
such as Bcl-xL, Bcl-2, Mcl-1, Bcl-w, BCL2A1 and inhibits their
interaction with the pro-apoptotic molecules, Bax and Bak (74). A subsequent study demonstrated that
PUMA was able to induce apoptosis of glioma cells and
overexpression of PUMA induces activation of caspases and
cytochrome c release (75).
We obtained results of PUMA upregulation after MIM-1 inhibitor
treatment; p53-wild type astrocyte cell line HA exhibited PUMA
expression several-fold higher compared to the GB p53-mutated
(T98G) cell line.
The protein Diablo acts as an IAP antagonist
(24), but its slight upregulation
after MIM-1 treatment in astrocytes did not cause changes in IAP
family expression (except for the upregulation of BIRC4). According
to our previous results (56), we
detected in human GB biopsy tissue a pronounced significant
increase in the gene coding neuronal apoptosis inhibitory protein
BIRC1 (NAIP). The protein BIRC1 prevents neuronal apoptosis induced
by a variety of signals and enhances neuronal survival under
pathological conditions (76–78).
We did not observe changes in BIRC1 gene expression after ABT-737
or MIM-1 inhibitor treatment in the GB cell line.
Caspase activity is regulated by certain members of
the IAP family, most potently by BIRC4 (XIAP), which is able to
bind to and inactivate members of the caspase family of cell death
proteases, i.e., caspase-9, −3 and −7 (30). BIRC4 functions also by binding to
the tumor necrosis factor receptor-associated factors TRAF1 and
TRAF2, which form a heterodimeric complex required for the
activation of NF-κB. Active NF-κB stimulates the expression of
genes that maintain cellular proliferation and protect cells from
conditions leading cells to apoptosis. According to our results,
slight upregulation of XIAP after MIM-1 treatment in astrocytes was
associated with deregulation of the NF-κB signaling pathway.
Previous studies have demonstrated that the extrinsic apoptotic
pathway is severely inhibited in high-grade gliomas 17,79–81. We
observed changes in multiple gene encoding proteins involved in the
extrinsic apoptotic pathway in astrocytes, but only several slight
expressional changes in genes involved in the extrinsic apoptotic
pathway in GB cells after selective intrinsic apoptotic pathway
inhibition. The extrinsic apoptotic pathway is triggered by the
binding of death ligands of the TNF family to their appropriate
death receptors on the cell surface. TNF family members are
responsible for the transmission of signals from extracellular
death ligands through death receptors to the cells apoptotic
machinery. High levels of Mcl-1 are often associated with
resistance to TRAIL (25), although
Mcl-1 is involved in the intrinsic mitochondrial pathway and TRAIL
plays a role in the extrinsic receptor pathway of apoptotic cell
death (27,31). Our results showed a significant
decrease, i.e., >17-fold, in the expression of TNFSF10 after
anti-apoptotic protein Mcl-1 inhibition, which confirms the
association between the effectiveness and activation of this
protein.
Multiple changes in genes encoding for proteins
associated in the TNF and NF-κB signaling pathways resulted in
CASP8 downregulation after treatment with the individual apoptosis
inhibitors in the astrocyte cell line. Low endogenous caspase-8
levels may be responsible for a decrease in the activation of the
effector caspase-3 and result in the resistance of cells to death
ligands (79,81). The frequent occurrence of low levels
of caspase-8 in glioma may complicate the future development of
therapies for patients with this malignancy. Our data indicate
alterations in various members of the TNF and NF-κB signaling
pathway in astrocytes after Mcl-1 inhibition. We observed cognate
results with the downregulation of caspase-8. The extrinsic
apoptotic pathway in HA and GB cells has not yet been
systematically studied, but previous reports have demonstrated
inhibition of the extrinsic pathway in high-grade gliomas (17,80,81).
Our experiments showed a significant downregulation in TNFSF10,
which may affect formation of death-inducing signaling complex
(DISC) (82), and subsequently
result in low levels of caspase-8 (81); in keeping with this, our experiments
showed significant downregulation of CASP8. These changes may
prevent apoptotic cell death via the activation of the extrinsic
apoptotic pathway in astrocytes after the inhibition of
anti-apoptotic proteins involved in the intrinsic apoptotic pathway
and ensure cell survival. On the contrary, our results showed an
increase in gene expression of inflammatory caspases-4 and −5 in
the astrocyte cell line after Mcl-1 inhibition, which could be
involved in immunoregulation (65).
The development of DNA microarray technologies over
the past decade has revolutionized translational cancer research.
Over the past decade, tremendous progress has been made in DNA
microarray-based gene expression profiling of gliomas. Such
progress in this research field may lead to identifying subsets of
genes uniquely responsive to specific adjuvant therapies and
provide individualized clinical care of glioma patients in the
future (57). Targeting apoptotic
pathways in selected cell lines offers a unique opportunity to
develop novel therapeutic strategies that may overcome tumor
resistance. We have shown in our study that HAs are more
susceptible to selective apoptosis inhibitors (individually) than
GB cells, in which apoptosis-associated gene expression is
relatively stable after ABT-737 or MIM-1 treatment. Improved
characterization of apoptotic gene expression differences in
astrocytes and GB cells may lead to a better understanding of
normal and cancerous brain cell metabolism and improve the
treatment of brain tumors. Monitoring of differentially expressed
human apoptosis-associated genes in cell lines might provide a
useful research tool to investigate experimental models of CNS
cells. Furthermore, cDNA microarrays may aid in establishing and
improving gene expression patterns capable of predicting individual
responses to therapy. The results of several recent studies have
shown a synergistic effect of apoptosis inhibitors with clinically
used cytostatics which enhanced their cytotoxicity (42,43,73,83).
The concept is that the combination of apoptotic Bcl-2 inhibitors
with a chemotherapeutic agent such as temozolomide, which is
routinely used in clinical practice, may provide a better clinical
effect. This combination may lead to apoptosis if the threshold to
undergo apoptosis is sufficiently low. Recognition of all the
biological attributes of GB growth inhibition still remains one of
the most challenging questions worldwide.
Acknowledgements
The authors are thankful to Dr Mahmoodova and Dr
Murin for donation and preparation of HA cell line for experiments.
We appreciate support by the Slovak Research and Development Agency
under contract no. APVV-0224-12 and by the project ‘Biomedical
Center Martin’ ITMS: no. 26220220187, which is co-financed from EU
sources.
References
|
1
|
Richterová R, Jurečeková J, Evinová A,
Kolarovszki B, Benčo M, De Riggo J, Sutovský J, Mahmood S, Račay P
and Dobrota D: Most frequent molecular and immunohistochemical
markers present in selected types of brain tumors. Gen Physiol
Biophys. 33:259–279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ricard D, Idbaih A, Ducray F, Lahutte M,
Hoang-Xuan K and Delattre JY: Primary brain tumours in adults.
Lancet. 379:1984–1996. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ahmed R, Oborski MJ, Hwang M, Lieberman FS
and Mountz JM: Malignant gliomas: Current perspectives in
diagnosis, treatment, and early response assessment using advanced
quantitative imaging methods. Cancer Manag Res. 6:149–170.
2014.PubMed/NCBI
|
|
5
|
Persano L, Rampazzo E, Basso G and Viola
G: Glioblastoma cancer stem cells: Role of the microenvironment and
therapeutic targeting. Biochem Pharmacol. 85:612–622. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pointer KB, Clark PA, Zorniak M, Alrfaei
BM and Kuo JS: Glioblastoma cancer stem cells: Biomarker and
therapeutic advances. Neurochem Int. 71:1–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schonberg DL, Lubelski D, Miller TE and
Rich JN: Brain tumor stem cells: Molecular characteristics and
their impact on therapy. Mol Aspects Med. 39:82–101. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jovčevska I, Kočevar N and Komel R: Glioma
and glioblastoma - how much do we (not) know? Mol Clin Oncol.
1:935–941. 2013.PubMed/NCBI
|
|
9
|
Tirapelli LF, Bolini PH, Tirapelli DP,
Peria FM, Becker AN, Saggioro FP and Carlotti CG Jr: Caspase-3 and
Bcl-2 expression in glioblastoma: An immunohistochemical study. Arq
Neuropsiquiatr. 68:603–607. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohgaki H and Kleihues P: Genetic
alterations and signaling pathways in the evolution of gliomas.
Cancer Sci. 100:2235–2241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Karsy M, Neil JA, Guan J, Mahan MA, Colman
H and Jensen RL: A practical review of prognostic correlations of
molecular biomarkers in glioblastoma. Neurosurg Focus. 38:E42015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bralten LBC and French PJ: Genetic
alterations in glioma. Cancers (Basel). 3:1129–1140. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kleihues P, Louis DN, Scheithauer BW,
Rorke LB, Reifenberger G, Burger PC and Cavenee WK: The WHO
classification of tumors of the nervous system. J Neuropathol Exp
Neurol. 61:215–229. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barazzuol L, Jena R, Burnet NG, Jeynes JC,
Merchant MJ, Kirkby KJ and Kirkby NF: In vitro evaluation of
combined temozolomide and radiotherapy using X rays and high-linear
energy transfer radiation for glioblastoma. Radiat Res.
177:651–662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Westhoff MA, Brühl O, Nonnenmacher L,
Karpel-Massler G and Debatin KM: Killing me softly-future
challenges in apoptosis research. Int J Mol Sci. 15:3746–3767.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Redmond KM, Wilson TR, Johnston PG and
Longley DB: Resistance mechanisms to cancer chemotherapy. Front
Biosci. 13:5138–5154. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Riffkin CD, Gray AZ, Hawkins CJ, Chow CW
and Ashley DM: Ex vivo pediatric brain tumors express Fas (CD95)
and FasL (CD95L) and are resistant to apoptosis induction. Neuro
Oncol. 3:229–240. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krajewski S, Krajewska M, Ehrmann J,
Sikorska M, Lach B, Chatten J and Reed JC: Immunohistochemical
analysis of Bcl-2, Bcl-X, Mcl-1, and Bax in tumors of central and
peripheral nervous system origin. Am J Pathol. 150:805–814.
1997.PubMed/NCBI
|
|
19
|
Klymenko T, Brandenburg M, Morrow C, Dive
C and Makin G: The novel Bcl-2 inhibitor ABT-737 is more effective
in hypoxia and is able to reverse hypoxia-induced drug resistance
in neuroblastoma cells. Mol Cancer Ther. 10:2373–2383. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kögel D, Fulda S and Mittelbronn M:
Therapeutic exploitation of apoptosis and autophagy for
glioblastoma. Anticancer Agents Med Chem. 10:438–449. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Portt L, Norman G, Clapp C, Greenwood M
and Greenwood MT: Anti-apoptosis and cell survival: A review.
Biochim Biophys Acta. 1813:238–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thomas S, Quinn BA, Das SK, Dash R, Emdad
L, Dasgupta S, Wang XY, Dent P, Reed JC, Pellecchia M, et al:
Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther
Targets. 17:61–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tchoghandjian A, Jennewein C, Eckhardt I,
Momma S, Figarella- Branger D and Fulda S: Smac mimetic promotes
glioblastoma cancer stem-like cell differentiation by activating
NF-κB. Cell Death Differ. 21:735–747. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sayers TJ: Targeting the extrinsic
apoptosis signaling pathway for cancer therapy. Cancer Immunol
Immunother. 60:1173–1180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wong RSY: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gaur U and Aggarwal BB: Regulation of
proliferation, survival and apoptosis by members of the TNF
superfamily. Biochem Pharmacol. 66:1403–1408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hervouet E, Cheray M, Vallette FM and
Cartron PF: DNA methylation and apoptosis resistance in cancer
cells. Cells. 2:545–573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Labi V, Grespi F, Baumgartner F and
Villunger A: Targeting the Bcl-2-regulated apoptosis pathway by BH3
mimetics: A breakthrough in anticancer therapy? Cell Death Differ.
15:977–987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Llambi F and Green DR: Apoptosis and
oncogenesis: Give and take in the BCL-2 family. Curr Opin Genet
Dev. 21:12–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim H, Tu HC, Ren D, Takeuchi O, Jeffers
JR, Zambetti GP, Hsieh JJ and Cheng EH: Stepwise activation of BAX
and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis.
Mol Cell. 36:487–499. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vogler M, Dinsdale D, Dyer MJ and Cohen
GM: Bcl-2 inhibitors: Small molecules with a big impact on cancer
therapy. Cell Death Differ. 16:360–367. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Galluzzi L, Maiuri MC, Vitale I, Zischka
H, Castedo M, Zitvogel L and Kroemer G: Cell death modalities:
Classification and pathophysiological implications. Cell Death
Differ. 14:1237–1243. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Degterev A, Boyce M and Yuan J: A decade
of caspases. Oncogene. 22:8543–8567. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yuan S and Akey CW: Apoptosome structure,
assembly, and procaspase activation. Structure. 21:501–515. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bender A, Opel D, Naumann I, Kappler R,
Friedman L, von Schweinitz D, Debatin KM and Fulda S: PI3K
inhibitors prime neuroblastoma cells for chemotherapy by shifting
the balance towards pro-apoptotic Bcl-2 proteins and enhanced
mitochondrial apoptosis. Oncogene. 30:494–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vogler M: Targeting BCL2 proteins for the
treatment of solid tumours. Adv Med. 2014:9436482014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cohen NA, Stewart ML, Gavathiotis E,
Tepper JL, Bruekner SR, Koss B, Opferman JT and Walensky LD: A
competitive stapled peptide screen identifies a selective small
molecule that overcomes MCL-1-dependent leukemia cell survival.
Chem Biol. 19:1175–1186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oltersdorf T, Elmore SW, Shoemaker AR,
Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges
J, Hajduk PJ, et al: An inhibitor of Bcl-2 family proteins induces
regression of solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Degterev A, Lugovskoy A, Cardone M, Mulley
B, Wagner G, Mitchison T and Yuan J: Identification of
small-molecule inhibitors of interaction between the BH3 domain and
Bcl-xL. Nat Cell Biol. 3:173–182. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tse C, Shoemaker AR, Adickes J, Anderson
MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et
al: ABT-263: A potent and orally bioavailable Bcl-2 family
inhibitor. Cancer Res. 68:3421–3428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Reed JC, Meister L, Tanaka S, Cuddy M, Yum
S, Geyer C and Pleasure D: Differential expression of bcl2
protooncogene in neuroblastoma and other human tumor cell lines of
neural origin. Cancer Res. 51:6529–6538. 1991.PubMed/NCBI
|
|
47
|
Song JH, Kandasamy K, Zemskova M, Lin YW
and Kraft AS: The BH3 mimetic ABT-737 induces cancer cell
senescence. Cancer Res. 71:506–515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cichowski K and Hahn WC: Unexpected pieces
to the senescence puzzle. Cell. 133:958–961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen S, Dai Y, Harada H, Dent P and Grant
S: Mcl-1 down-regulation potentiates ABT-737 lethality by
cooperatively inducing Bak activation and Bax translocation. Cancer
Res. 67:782–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
van Delft MF, Wei AH, Mason KD, Vandenberg
CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et
al: The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and
efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized.
Cancer Cell. 10:389–399. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kozopas KM, Yang T, Buchan HL, Zhou P and
Craig RW: MCL1, a gene expressed in programmed myeloid cell
differentiation, has sequence similarity to BCL2. Proc Natl Acad
Sci USA. 90:3516–3520. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Varin E, Denoyelle C, Brotin E,
Meryet-Figuière M, Giffard F, Abeilard E, Goux D, Gauduchon P,
Icard P and Poulain L: Downregulation of Bcl-xL and
Mcl-1 is sufficient to induce cell death in mesothelioma cells
highly refractory to conventional chemotherapy. Carcinogenesis.
31:984–993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li RY, Chen LC, Zhang HY, Du WZ, Feng Y,
Wang HB, Wen JQ, Liu X, Li XF, Sun Y, et al: MiR-139 inhibits Mcl-1
expression and potentiates TMZ-induced apoptosis in glioma. CNS
Neurosci Ther. 19:477–483. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zubor P, Hatok J, Moricova P, Kapustova I,
Kajo K, Mendelova A, Sivonova MK and Danko J: Gene expression
profiling of histologically normal breast tissue in females with
human epidermal growth factor receptor 2 positive breast cancer.
Mol Med Rep. 11:1421–1427. 2015.PubMed/NCBI
|
|
55
|
Zubor P, Hatok J, Galo S, Dokus K,
Klobusiakova D, Danko J and Racay P: Anti-apoptotic and
pro-apoptotic gene expression evaluated from eutopic endometrium in
the proliferative phase of the menstrual cycle among women with
endometriosis and healthy controls. Eur J Obstet Gynecol Reprod
Biol. 145:172–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Blahovcova E, Richterova R, Kolarovszki B,
Dobrota D, Racay P and Hatok J: Apoptosis-related gene expression
in tumor tissue samples obtained from patients diagnosed with
glioblastoma multiforme. Int J Mol Med. 36:1677–1684.
2015.PubMed/NCBI
|
|
57
|
Vitucci M, Hayes DN and Miller CR: Gene
expression profiling of gliomas: Merging genomic and
histopathological classification for personalised therapy. Br J
Cancer. 104:545–553. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shiozaki EN and Shi Y: Caspases, IAPs and
Smac/DIABLO: Mechanisms from structural biology. Trends Biochem
Sci. 29:486–494. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Verhagen AM, Coulson EJ and Vaux DL:
Inhibitor of apoptosis proteins and their relatives: IAPs and other
BIRPs. Genome Biol. 2:REVIEWS30092001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wen X, Lin ZQ, Liu B and Wei YQ:
Caspase-mediated programmed cell death pathways as potential
therapeutic targets in cancer. Cell Prolif. 45:217–224. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, et
al: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kurokawa M and Kornbluth S: Caspases and
kinases in a death grip. Cell. 138:838–854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tagscherer KE, Fassl A, Campos B, Farhadi
M, Kraemer A, Böck BC, Macher-Goeppinger S, Radlwimmer B, Wiestler
OD, Herold-Mende C, et al: Apoptosis-based treatment of
glioblastomas with ABT-737, a novel small molecule inhibitor of
Bcl-2 family proteins. Oncogene. 27:6646–6656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wei B, Wang L, Du C, Hu G, Wang L, Jin Y
and Kong D: Identification of differentially expressed genes
regulated by transcription factors in glioblastomas by
bioinformatics analysis. Mol Med Rep. 11:2548–2554. 2015.PubMed/NCBI
|
|
69
|
Stancheva G, Goranova T, Laleva M,
Kamenova M, Mitkova A, Velinov N, Poptodorov G, Mitev V, Kaneva R
and Gabrovsky N: IDH1/IDH2 but not TP53 mutations predict prognosis
in Bulgarian glioblastoma patients. BioMed Res Int.
2014:6547272014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
England B, Huang T and Karsy M: Current
understanding of the role and targeting of tumor suppressor p53 in
glioblastoma multiforme. Tumour Biol. 34:2063–2074. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li J: Di Ch, Mattox AK, Wu L and Adamson
DC: The future role of personalized medicine in the treatment of
glioblastoma multiforme. Pharm Genomics Pers Med. 3:111–127.
2010.
|
|
72
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Song T, Chai G, Liu Y, Xie M, Chen Q, Yu
X, Sheng H and Zhang Z: Mechanism of synergy of BH3 mimetics and
paclitaxel in chronic myeloid leukemia cells: Mcl-1 inhibition. Eur
J Pharm Sci. 70:64–71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Nakano K and Vousden KH: PUMA, a novel
proapoptotic gene, is induced by p53. Mol Cell. 7:683–694. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ito H, Kanzawa T, Miyoshi T, Hirohata S,
Kyo S, Iwamaru A, Aoki H, Kondo Y and Kondo S: Therapeutic efficacy
of PUMA for malignant glioma cells regardless of p53 status. Hum
Gene Ther. 16:685–698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Maier JK, Lahoua Z, Gendron NH, Fetni R,
Johnston A, Davoodi J, Rasper D, Roy S, Slack RS, Nicholson DW, et
al: The neuronal apoptosis inhibitory protein is a direct inhibitor
of caspases 3 and 7. J Neurosci. 22:2035–2043. 2002.PubMed/NCBI
|
|
77
|
Holcik M, Thompson CS, Yaraghi Z, Lefebvre
CA, MacKenzie AE and Korneluk RG: The hippocampal neurons of
neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice
display increased vulnerability to kainic acid-induced injury. Proc
Natl Acad Sci USA. 97:2286–2290. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mercer EA, Korhonen L, Skoglösa Y, Olsson
PA, Kukkonen JP and Lindholm D: NAIP interacts with hippocalcin and
protects neurons against calcium-induced cell death through
caspase-3-dependent and -independent pathways. EMBO J.
19:3597–3607. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Saggioro FP, Neder L, Stávale JN,
Paixão-Becker AN, Malheiros SM, Soares FA, Pittella JE, Matias CC,
Colli BO, Carlotti CG Jr, et al: Fas, FasL, and cleaved caspases 8
and 3 in glioblastomas: A tissue microarray-based study. Pathol Res
Pract. 210:267–273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Siegelin MD, Gaiser T and Siegelin Y: The
XIAP inhibitor Embelin enhances TRAIL-mediated apoptosis in
malignant glioma cells by down-regulation of the short isoform of
FLIP. Neurochem Int. 55:423–430. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ashley DM, Riffkin CD, Muscat AM, Knight
MJ, Kaye AH, Novak U and Hawkins CJ: Caspase 8 is absent or low in
many ex vivo gliomas. Cancer. 104:1487–1496. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Peter ME and Krammer PH: The
CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26–35. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Konopleva M, Contractor R, Tsao T, Samudio
I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, et al:
Mechanisms of apoptosis sensitivity and resistance to the BH3
mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 10:375–388.
2006. View Article : Google Scholar : PubMed/NCBI
|