Introduction
Colorectal cancer (CRC) is the third most common
tumor worldwide only after lung and breast cancer. From a global
perspective, the incidence of CRC among malignant tumors ranks
third in men, and second in women (1). In recent years, this incidence is
rising. Therefore, in-depth research on the pathogenesis of CRC is
of important realistic significance in improving the diagnosis and
treatment efficacy of CRC and evaluation of the prognosis.
In the last century, investigations of CRC have
focused on the tumor cells themselves, and the gene and epigenetic
changes causing the occurrence and development of tumors have been
widely accepted. However, increasing evidence suggests that the
tumor microenvironment plays an important role in tumorigenesis,
tumor infiltration and metastasis (2,3).
Cancer-associated fibroblasts (CAFs) in CRC tissues are the major
cells of the tumor microenvironment and are closely related to the
occurrence and development of CRC (4). CRC cells produce transforming growth
factor (TGF)-β, epidermal growth factor (EGF), platelet-derived
growth factor (PDGF) and other vital cytokines through paracrine
mechanisms to induce activation of CAFs (5,6).
Activated CAFs can upregulate matrix metalloproteinases (MMPs) and
plasminogen activator inhibitor (PAI)-1, ultimately playing a role
in breaking through the basilar membrane, the invasion and
metastasis of tumor cells (7,8).
According to experiments, CRC cells co-cultured with CAFs obviously
became spindle-shaped to increase the metastasis capacity.
Meanwhile, CAFs secrete MMPs to specifically degrade the
extracellular matrix (ECM), preparing for cancer cell migration
(9). Through 3-dimensional
co-culture of tumor cells and CAFs, Brentnall (10) found that CAFs can prompt cancer
cells to form pseudopodia and help them break through the basilar
membrane. All these findings indicate that CAFs can directly
increase the invasion ability of tumor cells and promote
metastasis, and CAFs are directly involved in the developmental
process of CRC.
The interaction between CAFs and tumor cells
involves many signaling pathways, such as TGF-β, PDGF, SDF-1/CXCR4
and hepatocyte growth factor (HGF), of which TGF-β has a critical
role (11). TGF-β can first induce
fibroblast activation, and these activated fibroblasts promote the
malignant transformation of epithelial cells (12). For example, according to a study by
Kuperwasser et al (13),
mammary stromal fibroblasts in mice overexpressing TGF-β and/or HGF
may induce human mammary epithelial cell cancerization, further
developing into ductal breast cancer. Activated fibroblasts have
been confirmed to promote tumor cell growth, while normal
fibroblasts inhibit tumor cell growth. This is achieved mainly by
secreting various factors or changing ECM components and
accelerating epithelial cell cancerization (14), yet the specific mechanism remains
unclear.
Studies have shown that TGF-β can induce CAFs to
express protein gene product (PGP9.5). In addition, PGP9.5 is a
ubiquitin hydrolase, also known as ubiquitin C terminal
hydrolase-L1 (UCH-L1). It regulates cellular processes in the cell
hydrolysis pathway including cell cycle division and cell death
(15). Current relevant research
indicates a high expression of PGP9.5 in many tumors including CRC,
medullary thyroid carcinoma, pancreatic, esophageal and bladder
cancer (15,16). In a previous study, in tumor
tissues, PGP9.5 induced an increase in cyclin ubiquitination
resulting in uncontrolled growth of undifferentiated cells, which
is one of the key factors leading to oncogene activation (17). Through analysis of patients with
ulcerative colitis, PGP9.5 was demonstrated to have a key function
in the malignant transformation process from ulcerative colitis to
carcinoma (18). In CRC, UCH-L1
hypomethylation was found to be closely associated with lymphatic
metastasis, and in CRC patients with lymphatic metastasis, the
methylation status of UCH-L1 was significantly reduced (19).
As found in the present study, TGF-β induced PGP9.5
expression in CAFs to influence CRC, which may be an important
mechanism of the interaction between CAFs and tumor cells.
Accordingly, the present study investigated the possible mechanisms
of this interaction, thus contributing to a better understanding of
the tumor growth environment, and the findings may aid in the
identification of new targets for cancer therapy.
Materials and methods
Cell culture
CAFs were obtained from surgically resected CRC
tissues at The First Affiliated Hospital of Dalian Medical
University, Dalian, China. The present study was approved by the
Ethics Committee of the National Cancer Center, China. The tissue
was minced into 2- to 3-mm fragments and seeded into Dulbecco's
modified Eagles medium (DMEM) containing 10% fetal bovine serum
(FBS) (both from Gibco-BRL, Carlsbad, CA, USA) and
penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) (20). Specifically, the procedure, which is
based on the selective growth advantage of fibroblasts under the
culture conditions used, allowed for a 100% pure fibroblast
population, as confirmed by positive staining for α-SMA (Abcam,
Shanghai, China). The fibroblast cultures were used until passage
10.
Human CRC cell lines LoVo and HCT116 were obtained
from the American Type Culture Collection (www.ATCC.org) and cultured in high-glucose DMEM
containing 10% FBS. The cells were maintained at 37°C in an
atmosphere of humidified air with 5% CO2 in a cell
culture incubator.
Immunofluorescence
CAFs were digested by 0.25% EDTA-trypsin
(Invitrogen) and seeded at a density of 1×106 cells/well
in 6-well plates treated with poly-L-lysine coverslips, and then
TGF-β1 (8 ng/ml; R&D Systems, Minneapolis, MN, USA) was added.
After 48 h the cells were fixed with 4% paraformaldehyde and then
incubated in 1 ml primary antibody (α-SMA) at 4°C overnight. After
the addition of secondary antibody, the cells were washed with
phosphate-buffered saline (PBS), treated with alcohol and xylene
for gradient and transparence and were mounted with 10% glycerol
phosphate. They were then placed under a fluorescence microscope
camera for immunofluorescence detection.
Overexpression vector construction and
transfection
RNA was extracted from LoVo cells using TRIzol
(Invitrogen) according to the manufacturers instructions, followed
by reverse transcription PCR to amplify the coding region of
TGF-β1. In addition, the primers were TGF-β1-F
(5′-cccAAGCTTgccaccATGCCGCCCTCCGGGCTG-3′) and TGF-β1-R
(5′-ccggaattcTCAGCTGCACTTGCAGGAGCGCA-3′). Then, the products were
digested with HindIII and EcoRI (Takara, Dalian,
China), cloned into pcDNA3.1 vectors, sequenced and verified. The
cells were seed into a 6-well plate at 1×105 cells/ml
and incubated for 24 h. Transfection was carried out using
Lipofectamine 2000 (Invitrogen) according to the manufacturers
instructions, when the cell confluence reached ~70%. The
concentrations of plasmid and siRNAs for transfection were 4
µg/well and 50 nM/well. The sequence of the sense oligo for the
siRNA directed against TGF-β1 was, 5′-GGG CUA CCA UGC CAA CUU
CTT-3′; for Smad2, 5′-GUC CCA UGA AAA GAC UUA ATT-3′; and for
Smad3, 5′-GGA GAA AUG GUG CGA GAA GTT-3′. After transfection,
TGF-β1 and LY2109761 (10 µmol/l; Calbiochem, China) were added or
not respectively, and then PD98059 (ERK1/2 inhibitor), LY294002
(PI3K inhibitor), SB203580 (p38 inhibitor) and SP600125 (JNK
inhibitor) (all purchased from Calbiochem) were added respectively
at a final concentration of 20 µM.
Western blot analysis
Total cellular proteins were extracted by incubating
cells in lysis buffer (1X PBS, 1% NP-40, 0.1% SDS, 5 mM EDTA, 0.5%
sodium deoxycholate and 1 mM sodium with protease inhibitors). The
protein concentrations in the cell lysates were determined by
bicinchoninic acid assay (Pierce, Rockford, IL, USA). SDS-PAGE was
carried out on 8% glycine gels (Bio-Rad, Hercules, CA, USA) loading
equal amount of proteins/lane. After electrophoresis, the separated
proteins were transferred to a nitrocellulose membrane (Pierce) and
blocked with 5% non-fat milk in Tris-buffered saline with Tween-20
(TBST) buffer for 1 h. After that, the membranes were incubated
with PGP9.5, Smad2, Smad3 and β-actin antibodies (Cell Signaling
Technology, USA) at 1:1000 dilutions in 5% non-fat milk overnight
at 4°C, and then anti-rabbit IgG monoclonal antibody conjugated
with horseradish peroxidase (Cell Signaling Technology, Danvers,
MA, USA) at 1:2,000 dilution for 1 h at room temperature. Protein
bands were detected using the West Femto system (Pierce).
Cell proliferation
Cell proliferation was estimated in 96-well plates
using a colorimetric immunoassay, based on the measurement of BrdU
incorporation during DNA synthesis (BrdU ELISA kit; Roche
Diagnostics, Mannheim, Germany). PGP9.5 siRNA was transfected and
TGF-β1 was added or TGF-β1 and UCH-L1 (PGP9.5) inhibitor (5 µM;
Calbiochem) were added at the same time. After cells were cultured
for 48 h, the medium was removed and cells were labeled with BrdU
(10 mM 5-bromo-2′-deoxyuridine) for 3 h at 37°C. Cells were fixed,
and incubated with peroxidase-conjugated anti-BrdU antibody for 90
min at room temperature. Then, the peroxidase substrate
3,3′,5,5′-tetramethylbenzidine was added, and BrdU incorporation
was quantitated by differences in absorbance at wavelength 370
minus 492 nm. Cell proliferation was expressed as the mean
percentage of the control values (set at 100%).
Cell cycle analysis
Cells were pelleted (400 × g for 5 min at 4°C),
washed twice with cold PBS, resuspended in 500 µl of cold PBS, and
then fixed for 1 h at 4°C by adding 500 µl of fixation solution (2%
w/v paraformaldehyde in PBS, pH 7.2). The fixed cells were
pelleted, washed with cold PBS, resuspended in 1 ml of 70% ethanol
added dropwise to the pellet while vortexing and then incubated
overnight at 4°C. The next day, cells were pelleted and resuspended
in 1 ml propidium iodide solution (40 µg/ml with 100 µg/ml RNase A)
for 30 min at 37°C in the dark and analyzed on a FACScan flow
cytometer (BD Biosciences, San Jose, CA, USA).
Transwell Matrigel invasion assay
Invasion of cells was evaluated by Transwell
Matrigel invasion assay (BD, China). Briefly, 200 µl of cells after
transfection (1×106 cells/ml) and 600 µl of the complete
medium was added to the upper and lower compartments of the
chamber, respectively. After an incubation of 48 h, cells that
migrated to the lower side of the filter were fixed with 4%
paraformaldehyde for 15 min at room temperature, washed with PBS,
stained with crystal violet, and then observed under an inverted
microscope (Olympus CKX41).
Statistical analyses
Experiments were carried out at least in triplicate
and the results are expressed as mean ± SD. Statistical analysis
was performed using SPSS statistical program version 17 (SPSS,
Inc., Chicago, IL, USA). Differences with a probability value
(P)<0.01 were considered statistically significant.
Results
TGF-β1 induces CAF activation while
promoting PGP9.5 expression simultaneously
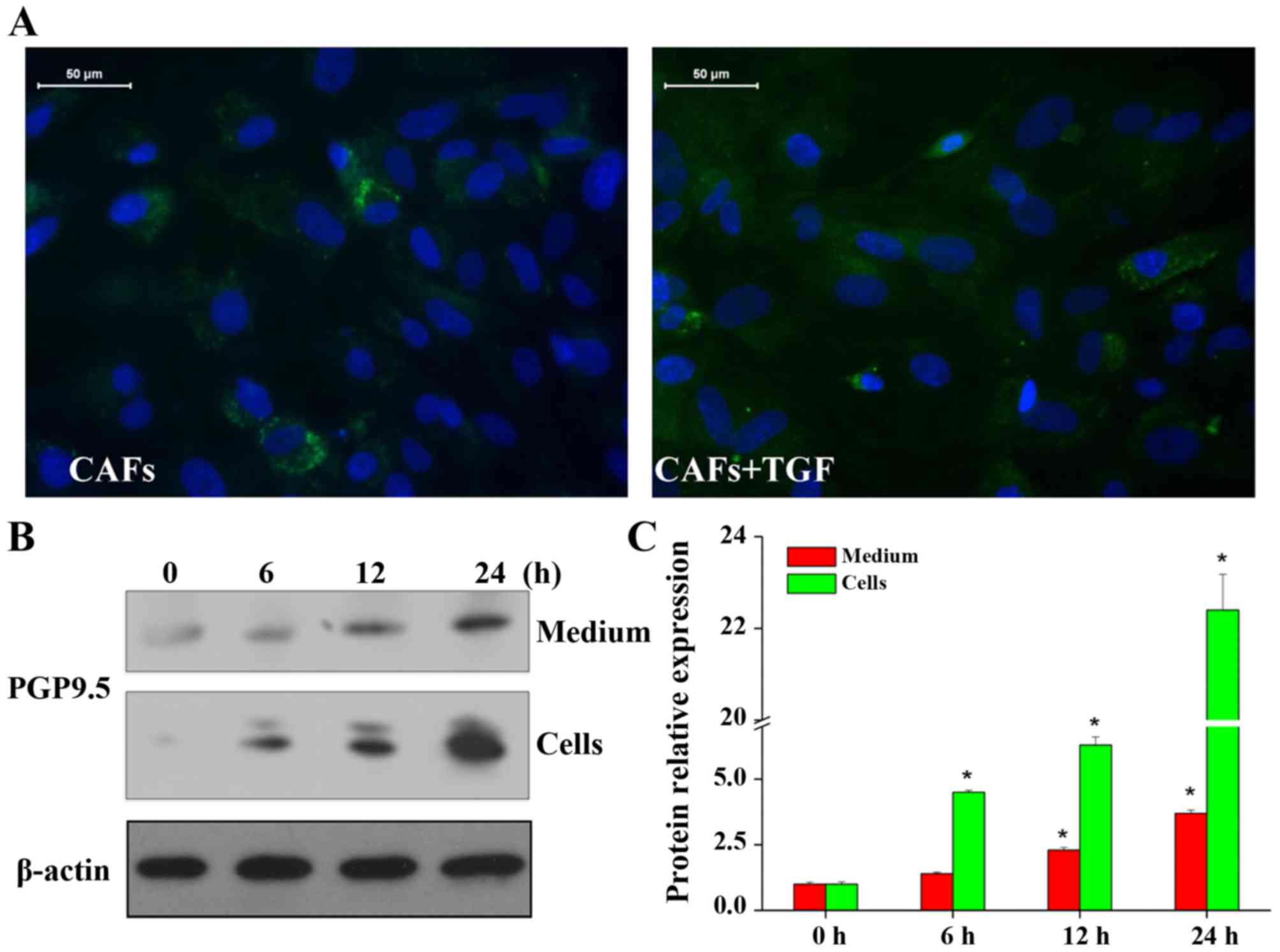
CAFs were isolated from surgically resected CRC
tissues, and an immunofluorescence assay was used to determine the
expression of α-SMA. The results showed that α-SMA expression was
significantly increased when TGF-β1 was added (Fig. 1A). An increase in α-SMA expression
is indicative of fibroblast activation. This showed that TGF-β1
promoted activation of CAFs in CRC. PGP9.5 expression was also
detected (Fig. 1B and C). With the
prolonged duration of TGF-β1 stimulation, the expression of PGP9.5
was gradually increased and the difference was significant.
However, PGP9.5 in the cells and media showed a consistent
increased expression trend. According to further validation of the
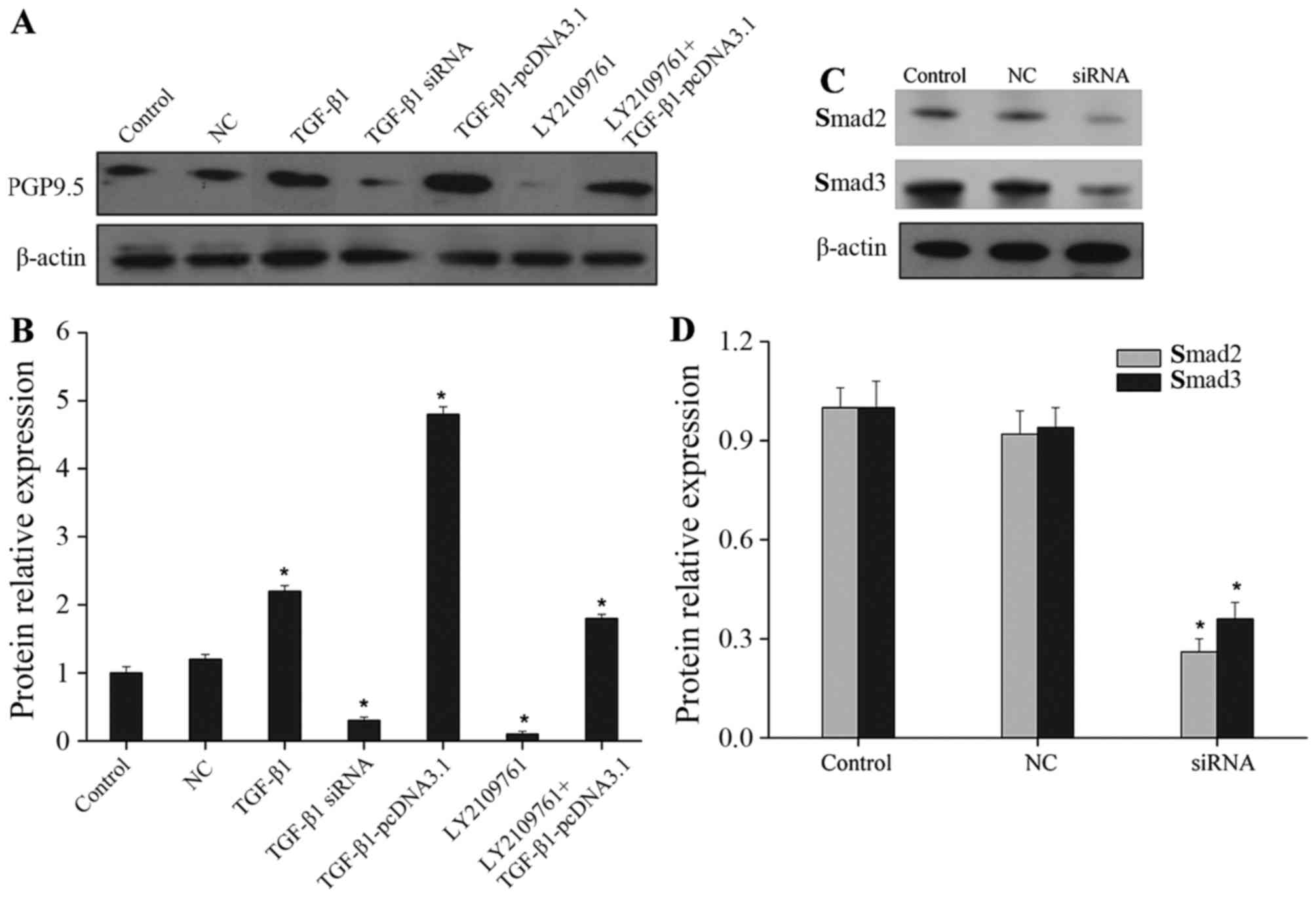
above mentioned results, PGP9.5 expression was obviously reduced
after adding siRNA specifically targeting TGF-β1 or its inhibitor
LY2109761. When the TGF-β1 overexpression vector was transfected,
the effect was similar to that after directly adding TGF-β1, both
of which showed a significant increase in PGP9.5 expression;
moreover, when the TGF-β1 overexpression vector was transfected and
the TGF-β1 inhibitor was added simultaneously, PGP9.5 expression
was markedly reduced when compared with the TGF-β1 overexpression
vector transfection group (Fig. 2A and
B). In light of the above data, TGF-β1 induced activation of
CAFs in CRC and promoted PGP9.5 expression.
TGF-β1 promotes PGP9.5 expression
through Smad, ERK1/2 and PI3K pathways
Research was continued to investigate the pathway by
which TGF-β1 promotes PGP9.5 expression in CAFs. The classical
TGF-β signaling pathway is mediated by the Smad protein family
mainly including Smad2/3 (21). The
inhibitory effects of Smad2 and Smad3 siRNAs were first examined by
western blotting (Fig. 2C and D).
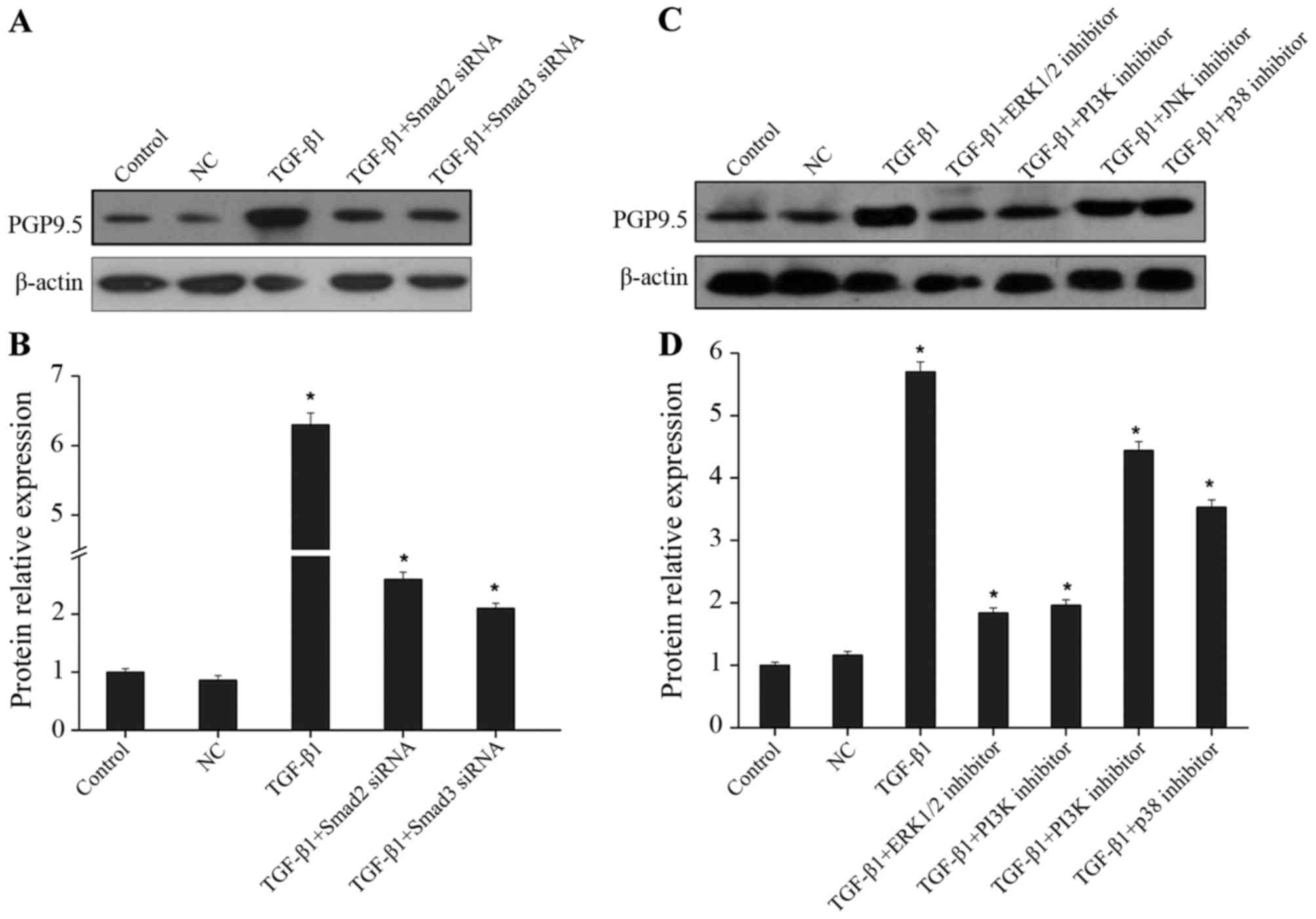
The effect of the Smad pathway on promotion of PGP9.5 expression by
TGF-β1 was then determined. As shown in Fig. 3A and B, after the specific siRNA was
used to downregulate Smad2 and Smad3 expression, the promoting
effect of TGF-β1 on PGP9.5 expression was obviously decreased,
suggesting that TGF-β1 regulated PGP9.5 expression through the Smad
pathway. In addition, TGF-β was previously found to activate
multiple intracellular signaling pathways, such as ERK1/2,
PI3K/Akt, JNK and p38, to regulate the biological behaviors of
cells (21). After TGF-β1 and the
specific inhibitors of the above mentioned pathways were added
simultaneously, the expression of PGP9.5 was detected. The results
indicated that after blocking the ERK1/2 and PI3K pathways, the
promoting effect of TGF-β1 on PGP9.5 expression was significantly
suppressed; after blocking the JNK and p38 pathways, PGP9.5
expression displayed a less significant decrease than that of the
TGF-β1 group (Fig. 3C and D). Thus,
in CRC CAFs, TGF-β1 promoted PGP9.5 expression mainly through the
Smad, ERK1/2 and PI3K pathways.
TGF-β1 promotes CAF-induced PGP9.5
expression to increase the proliferation of CRC cells
TGF-β1 can induce CAFs to express PGP9.5 which has
been proven to play an important role in the occurrence and
development of CRC (17–19). Further research was conducted to
validate whether one of the mechanisms for promoting cancer cell
growth by CAFs functioned through TGF-β1/PGP9.5. CAFs and CRC cells
were cultured on the upper and lower chambers of Transwell plates,
respectively, thereby establishing a co-culture model. When TGF-β1
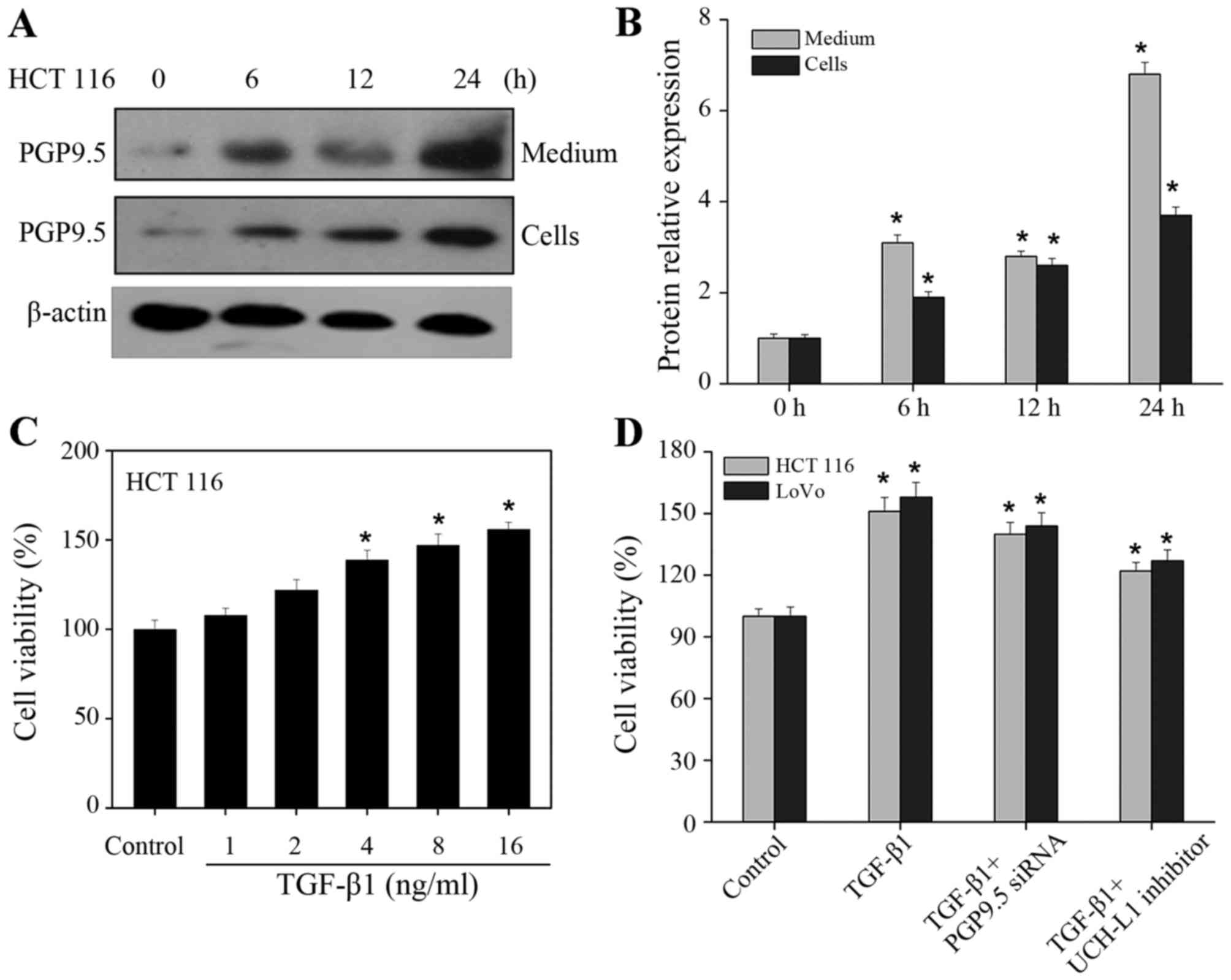
was added to the CAFs, western blotting was used to determine
PGP9.5 expression in the lower layer of CRC cells. According to the
results, with the extension of the duration of TGF-β1 induction,
PGP9.5 expression was significantly enhanced in the media of the
lower layer of CRC cells (Fig. 4A and
B). Then BrdU assay showed a gradual and significant increase
in the proliferation of CRC cells as the concentration of TGF-β1
was increased (Fig. 4C). When
TGF-β1 was added into CAFs and the PGP9.5 inhibitor was added to
the CRC cells concomitantly, the proliferation was increased
compared with the control group, but the proliferation rate was
obviously reduced compared with TGF-β1 group (Fig. 4D). This suggests that PGP9.5
expressed by CAFs under the induction effect of TGF-β1 can promote
the proliferation of CRC cells. In contrast, when PGP9.5 was
inhibited, the promoting effect of TGF-β1-activated CAFs on CRC
cells was obviously suppressed.
TGF-β1 promotes CAF-induced PGP9.5
expression to suppress CRC cell apoptosis
TGF-β1 not only induced CAFs to express PGP9.5, thus
promoting the proliferation of CRC cells, but may also affect other
biological behaviors of the cells. Therefore, flow cytometry was
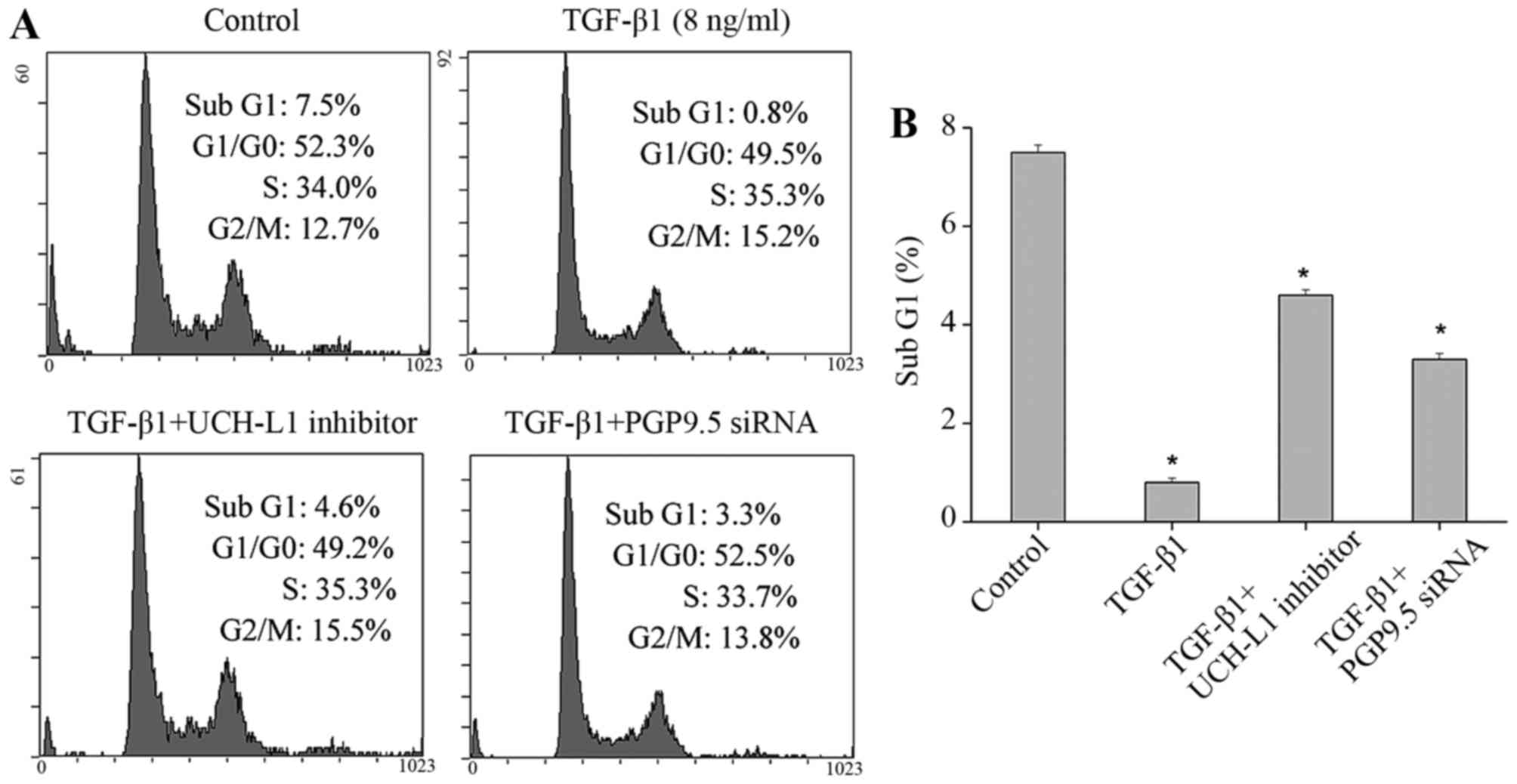
used to assess the cell cycle and apoptosis. The results indicated
that when TGF-β1 was added into the CAFs, the proportion of the
apoptosis peak (Sub G1 peak) of CRC cells was significantly reduced
(Fig. 5). When the TGF-β1 inhibitor
was added to the CRC cells, the proportion of the Sub G1 peak was
also decreased, but was higher than that of the TGF-β1 alone group
(Fig. 5). From the above data, we
concluded that addition of the TGF-β1 inhibitor alone into tumor
cells cannot completely block the effect of TGF-β1-activated CAFs
on tumor cells. Similarly, after the PGP9.5 inhibitor was added to
the CRC cells, the proportion of Sub G1 peak also declined compared
with the control group, but was still higher than that of the
TGF-β1 alone group (Fig. 5). This
suggested that PGP9.5 plays a role in the inhibitory process of
TGF-β1-activated CAFs against CRC cell apoptosis; however, the
effect of TGF-β1-activated CAFs against tumor cells was not
completely blocked by inhibiting the function of PGP9.5 and may
also secrete other factors to play a role.
TGF-β1 promotes CAF-induced PGP9.5
expression to promote the invasion of CRC cells
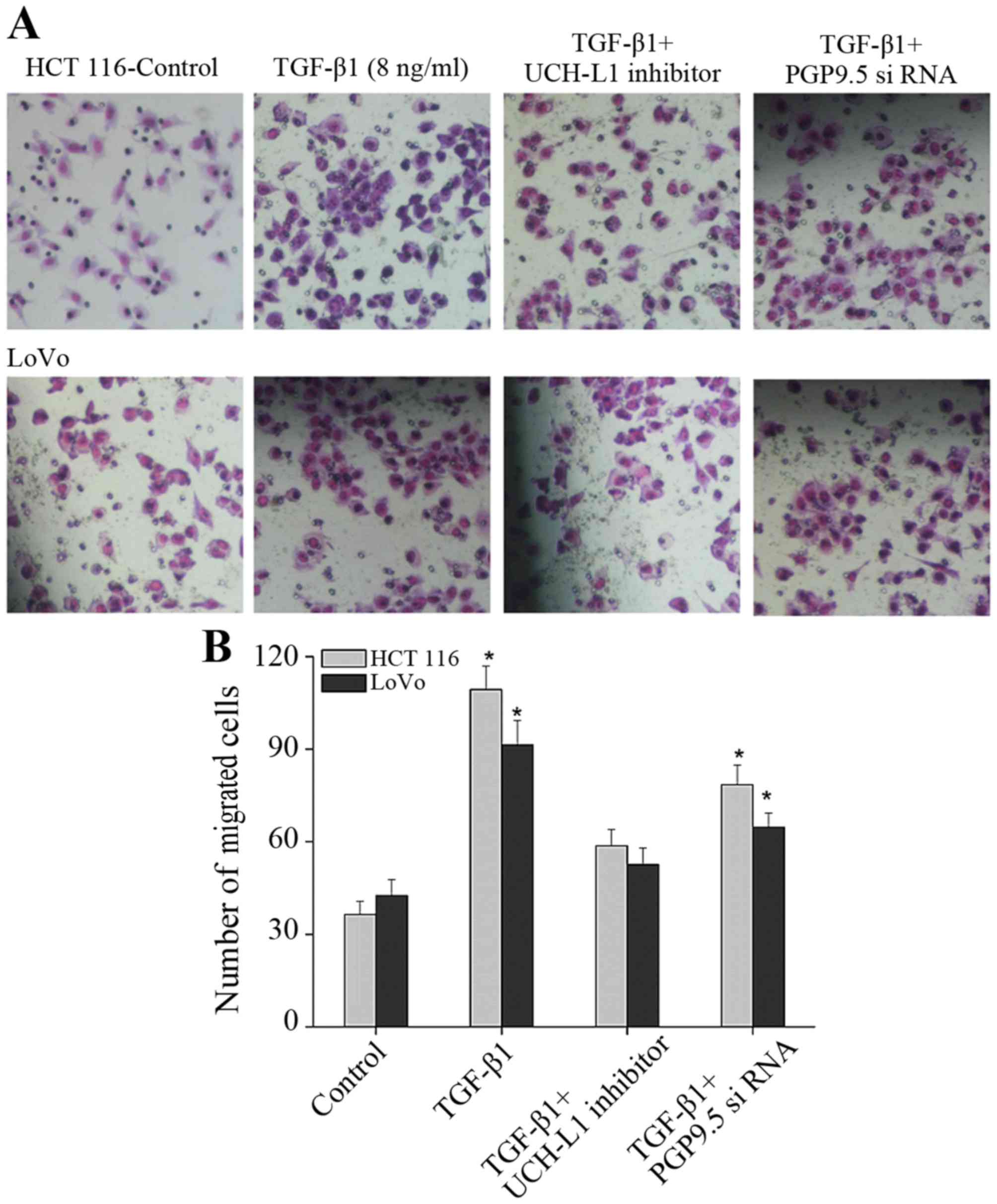
One significant effect of activated CAFs on tumor
cells is to promote invasion and metastasis. Thus, Transwell assay
was applied to detect the effect of PGP9.5 expressed by CAFs under
the induction effect of TGF-β1. After TGF-β1 was added into CAFs,
the invasion ability of CRC cells was significantly increased
(Fig. 6). When CAFs and the UCH-LI
(PGP9.5) inhibitor was added into CAFs and CRC cells, respectively,
however, the invasion ability of the cells increased compared with
the control group, but was obviously suppressed when compared with
the TGF-β1 alone group (Fig. 6).
Thus, after CAFs were activated by TGF-β1, the invasion of tumor
cells was significantly promoted, in which PGP9.5 has a key
function as inhibition of the PGP9.5 effect blocked the promotion
of TGF-β1-activiated CAFs on the invasion of CRC cells to some
extent.
Discussion
Using cancer-associated fibroblasts (CAFs) for
diagnosis and antitumor treatment for colorectal cancer (CRC) has
just begun. The in-depth research of CAFs may help to elucidate the
mechanism of tumorigenesis and tumor progression, providing new
specific markers for early diagnosis of tumors and new targets for
tumor treatment (2,3). Currently, although there has been some
progress in investigating the relationship between CAFs and CRC
development, there are still many issues which remain unsolved.
CRC cells secrete growth factors [e.g., transforming
growth factor (TGF)-β and IGF], promoting the acccumulation and
activation of fibroblasts involved in cancer cell proliferation,
metastasis and other pathophysiological processes (6). Sappino et al (22) reported that CRC cells release TGF-β
to induce mesenchymal reaction and prompt CAF activation, while
CAFs secreted various growth factors, collagens and proteases,
which affect the tumor cells themselves in turn. The present study
further demonstrated that TGF-β1 can promote activation of CAFs,
and the activiation marker α-SMA expression increased markedly. In
normal colon epithelial cells and early colon cancer, TGF-β mainly
inhibits cell proliferation, while in advanced colon cancer, it
promotes the invasion and metastasis of tumor cells by inducing
epithelial-mesenchymal transition (EMT). There have been many
studies to support the promotion of TGF-β-activated CAFs on the
growth of tumor cells (23).
The present study further found that TGF-β1 can
promote CAFs to overexpress PGP9.5 through the classical Smad,
ERK1/2 and PI3K pathways, in which JNK and p38 pathways were not
obvious. The relationship between PGP9.5 and tumors has been
reported in recent years (15,16). A
large number of clinical studies have shown that a high level of
PGP9.5 in breast cancer, CRC, pancreatic cancer, and other tumors
is closely associated with a lower survival rate (17,24,25).
The carcinogenesis of PGP9.5 has been reported in non-small cell
lung cancer and colon cancer (25,26).
According to previous studies, PGP9.5 enhanced the activity of CDKs
in tumor cells (27), promoted cell
survival by activating the Akt signaling pathway (28), and accelerated the metastasis of
tumor cells by activating the Wnt signaling pathway (29). Although PGP9.5 has been reported to
be a cancer-suppressor gene (30),
the findings concerning its role in CRC are consistent at present.
PGP9.5 was shown to play an important role in the progression of
CRC, which may be an independent prognostic factor (19,24,25).
Mizukami et al (19)
indicated that PGP9.5 was less frequently methylated in metastatic
CRC, suggesting that PGP9.5 hypomethylation may play an important
role in reexpression of the PGP9.5 gene in CRC. Thereby, routine
test of its expression in CRC contributes to evaluation of
diagnosis and prognosis of CRC. Akishima-Fukasawa et al
(25) also found that PGP9.5
expression in cultured normal fibroblasts was increased by TGF-β
stimulation, indicating the phenotypic alteration to activated
fibroblast. Thus, PGP9.5 overexpression induced by CAFs under the
induction effect of TGF-β1 may play a key role in CRC.
The further research results indicated that the
proliferation and invasion of CRC cells were promoted when CAFs
were activated by TGF-β1 while their apoptosis was inhibited. This
was also achieved by promoting PGP9.5 expression. Nevertheless, the
effect of TGF-β1-activiated CAFs against tumor cells was not
completely blocked when the function of PGP9.5 was inhibited. Based
on the above data, PGP9.5 played an important role in the process
of CAF activation by TGF-β1 and promotion of cancer cell growth,
but TGF-β1-activated CAFs may secrete other factors that also play
a role. Hawinkels et al (8)
also found that the interaction of tumor cells with resident
fibroblasts results in hyperactivated TGF-β1 signaling and
subsequent transdifferentiation of the fibroblasts into
α-SMA-positive CAFs. In turn this leads to cumulative production of
TGF-β and proteinases within the tumor microenvironment, creating a
cancer-promoting feedback loop. Gonzalez-Zubeldia et al
(31) showed that TGFβ1 promoted
co-travelling of CRC cells and CAFs to the liver to enhance
metastasis. Activation of the TGF-β pathway in cancer cells did not
influence tumor growth, but cancer cell-derived-TGF-β ligands
affected stromal cells in a paracrine manner, leading to fibroblast
activation and enhanced tumor growth (32). Functional evaluations in coculture
experiments showed that TGF-β enhanced the aggressiveness of
ovarian cancer cells by upregulating VCAN in CAFs (33). CAFs were measurably different from
normal fibroblasts in response to TGF-β1, suggesting that TGF-β
stimulates changes in CAFs that foster tumor invasion (11). Therefore, TGF-β1 can promote CAFs to
express a variety of factors such as PGP9.5, thus acting on cancer
cells and promoting the growth of cancer cells.
These findings of CAFs help to elucidate the
mechanism of tumorigenesis and tumor progression, suggesting new
specific markers for early diagnosis of tumors and new targets for
tumor treatment. Nevertheless, further in-depth research is needed
to investigate the mechanisms.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Joyce JA and Pollard JW:
Microenvironmental regulation of metastasis. Nat Rev Cancer.
9:239–252. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Coussens LM: Accessories to
the crime: Functions of cells recruited to the tumor
microenvironment. Cancer Cell. 21:309–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tommelein J, Verset L, Boterberg T,
Demetter P, Bracke M and De Wever O: Cancer-associated fibroblasts
connect metastasis-promoting communication in colorectal cancer.
Front Oncol. 5:632015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Denys H, Derycke L, Hendrix A, Westbroek
W, Gheldof A, Narine K, Pauwels P, Gespach C, Bracke M and De Wever
O: Differential impact of TGF-beta and EGF on fibroblast
differentiation and invasion reciprocally promotes colon cancer
cell invasion. Cancer Lett. 266:263–274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mishra P, Banerjee D and Ben-Baruch A:
Chemokines at the crossroads of tumor-fibroblast interactions that
promote malignancy. J Leukoc Biol. 89:31–39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calon A, Espinet E, Palomo-Ponce S,
Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung
P, Zhang XH, et al: Dependency of colorectal cancer on a
TGF-β-driven program in stromal cells for metastasis initiation.
Cancer Cell. 22:571–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hawinkels LJ, Paauwe M, Verspaget HW,
Wiercinska E, van der Zon JM, van der Ploeg K, Koelink PJ, Lindeman
JH, Mesker W, ten Dijke P, et al: Interaction with colon cancer
cells hyperactivates TGF-β signaling in cancer-associated
fibroblasts. Oncogene. 33:97–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taguchi A, Kawana K, Tomio K, Yamashita A,
Isobe Y, Nagasaka K, Koga K, Inoue T, Nishida H, Kojima S, et al:
Matrix metalloproteinase (MMP)-9 in cancer-associated fibroblasts
(CAFs) is suppressed by omega-3 polyunsaturated fatty acids in
vitro and in vivo. PLoS One. 9:e896052014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brentnall TA: Arousal of cancer-associated
stromal fibroblasts: Palladin-activated fibroblasts promote tumor
invasion. Cell Adhes Migr. 6:488–494. 2012. View Article : Google Scholar
|
|
11
|
Casey TM, Eneman J, Crocker A, White J,
Tessitore J, Stanley M, Harlow S, Bunn JY, Weaver D, Muss H, et al:
Cancer associated fibroblasts stimulated by transforming growth
factor beta1 (TGF-beta 1) increase invasion rate of tumor cells: A
population study. Breast Cancer Res Treat. 110:39–49. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dawes LJ, Eldred JA, Anderson IK, Sleeman
M, Reddan JR, Duncan G and Wormstone IM: TGF beta-induced
contraction is not promoted by fibronectin-fibronectin receptor
interaction, or alpha SMA expression. Invest Ophthalmol Vis Sci.
49:650–661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuperwasser C, Chavarria T, Wu M, Magrane
G, Gray JW, Carey L, Richardson A and Weinberg RA: Reconstruction
of functionally normal and malignant human breast tissues in mice.
Proc Natl Acad Sci USA. 101:4966–4971. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ao M, Williams K, Bhowmick NA and Hayward
SW: Transforming growth factor-beta promotes invasion in
tumorigenic but not in nontumorigenic human prostatic epithelial
cells. Cancer Res. 66:8007–8016. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mandelker DL, Yamashita K, Tokumaru Y,
Mimori K, Howard DL, Tanaka Y, Carvalho AL, Jiang WW, Park HL, Kim
MS, et al: PGP9.5 promoter methylation is an independent prognostic
factor for esophageal squamous cell carcinoma. Cancer Res.
65:4963–4968. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YM, Lee JY, Kim MJ, Bae HI, Park JY,
Kim SG and Kim DS: Hypomethylation of the protein gene product 9.5
promoter region in gallbladder cancer and its relationship with
clinicopathological features. Cancer Sci. 97:1205–1210. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ohta T and Fukuda M: Ubiquitin and breast
cancer. Oncogene. 23:2079–2088. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Watanabe T, Kobunai T, Toda E, Kanazawa T,
Kazama Y, Tanaka J, Tanaka T, Yamamoto Y, Hata K, Kojima T, et al:
Gene expression signature and the prediction of ulcerative
colitis-associated colorectal cancer by DNA microarray. Clin Cancer
Res. 13:415–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mizukami H, Shirahata A, Goto T, Sakata M,
Saito M, Ishibashi K, Kigawa G, Nemoto H, Sanada Y and Hibi K:
PGP9.5 methylation as a marker for metastatic colorectal cancer.
Anticancer Res. 28:2697–2700. 2008.PubMed/NCBI
|
|
20
|
Nakagawa H, Liyanarachchi S, Davuluri RV,
Auer H, Martin EW Jr, de la Chapelle A and Frankel WL: Role of
cancer-associated stromal fibroblasts in metastatic colon cancer to
the liver and their expression profiles. Oncogene. 23:7366–7377.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Derynck R and Zhang YE: Smad-dependent and
Smadindependent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sappino AP, Schürch W and Gabbiani G:
Differentiation repertoire of fibroblastic cells: Expression of
cytoskeletal proteins as marker of phenotypic modulations. Lab
Invest. 63:144–161. 1990.PubMed/NCBI
|
|
23
|
Calon A, Tauriello DV and Batlle E:
TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer
Biol. 25:15–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yamazaki T, Hibi K, Takase T, Tezel E,
Nakayama H, Kasai Y, Ito K, Akiyama S, Nagasaka T and Nakao A:
PGP9.5 as a marker for invasive colorectal cancer. Clin Cancer Res.
8:192–195. 2002.PubMed/NCBI
|
|
25
|
Akishima-Fukasawa Y, Ino Y, Nakanishi Y,
Miura A, Moriya Y, Kondo T, Kanai Y and Hirohashi S: Significance
of PGP9.5 expression in cancer-associated fibroblasts for prognosis
of colorectal carcinoma. Am J Clin Pathol. 134:71–79. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim HJ, Kim YM, Lim S, Nam YK, Jeong J,
Kim HJ and Lee KJ: Ubiquitin C-terminal hydrolase-L1 is a key
regulator of tumor cell invasion and metastasis. Oncogene.
28:117–127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kabuta T, Mitsui T, Takahashi M, Fujiwara
Y, Kabuta C, Konya C, Tsuchiya Y, Hatanaka Y, Uchida K, Hohjoh H,
et al: Ubiquitin C-terminal hydrolase L1 (UCH-L1) acts as a novel
potentiator of cyclin-dependent kinases to enhance cell
proliferation independently of its hydrolase activity. J Biol Chem.
288:12615–12626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hussain S, Foreman O, Perkins SL, Witzig
TE, Miles RR, van Deursen J and Galardy PJ: The de-ubiquitinase
UCH-L1 is an oncogene that drives the development of lymphoma in
vivo by deregulating PHLPP1 and Akt signaling. Leukemia.
24:1641–1655. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bheda A, Yue W, Gullapalli A, Whitehurst
C, Liu R, Pagano JS and Shackelford J: Positive reciprocal
regulation of ubiquitin C-terminal hydrolase L1 and
beta-catenin/TCF signaling. PLoS One. 4:e59552009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu J, Tao Q, Cheung KF, Jin H, Poon FF,
Wang X, Li H, Cheng YY, Röcken C, Ebert MP, et al: Epigenetic
identification of ubiquitin carboxyl-terminal hydrolase L1 as a
functional tumor suppressor and biomarker for hepatocellular
carcinoma and other digestive tumors. Hepatology. 48:508–518. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gonzalez-Zubeldia I, Dotor J, Redrado M,
Bleau AM, Manrique I, de Aberasturi AL, Villalba M and Calvo A:
Co-migration of colon cancer cells and CAFs induced by
TGFβ1 enhances liver metastasis. Cell Tissue Res.
359:829–839. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guido C, Whitaker-Menezes D, Capparelli C,
Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Andò S,
Martinez-Outschoorn U, et al: Metabolic reprogramming of
cancer-associated fibroblasts by TGF-β drives tumor growth:
Connecting TGF-β signaling with ‘Warburg-like’ cancer metabolism
and L-lactate production. Cell Cycle. 11:3019–3035. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yeung TL, Leung CS, Wong KK, Samimi G,
Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ and Mok SC: TGF-β
modulates ovarian cancer invasion by upregulating CAF-derived
versican in the tumor microenvironment. Cancer Res. 73:5016–5028.
2013. View Article : Google Scholar : PubMed/NCBI
|