Introduction
Epidermal growth factor receptor (EGFR) is a
tyrosine kinase (TK) receptor that is activated by binding with
ligands such as EGF and other growth factors. Activation of EGFR
leads to the triggering of several downstream pathways including
RAS/MAPK, PI3K/Akt and JAK/STAT pathways that regulate cell
proliferation, survival, adhesion, migration, and differentiation
(1). Overexpression of EGFR and
dysregulation of EGFR-mediated signaling pathways have been
observed in tumors from patients with various cancers, especially
with non-small cell lung cancer (NSCLC), contributing to
tumorigenesis and leading to a poor prognosis (2). Thus, EGFR is a promising molecular
target as a therapeutic agent for patients with NSCLC and several
anti-EGFR drugs have been developed for this purpose.
Lung cancer is the second most common cancer and
remains the leading cause of cancer-related mortality worldwide.
Notably, the majority of lung cancers (over 85% of patients) are
categorized as NSCLC (3).
Traditionally, lung cancer has been treated with surgery,
radiation, and chemotherapy (4).
Although chemotherapy has been the standard of care for patients
with NSCLC, current clinical efforts are directed at molecular
target drugs to improve outcome and reduce toxicity. Gefitinib,
which primarily functions as an EGFR tyrosine kinase inhibitor
(TKI), was approved in Japan for the treatment of NCLC in 2002 and
in the USA in 2003. In 2004, the presence of somatic mutations in
the TK domain of EGFR was identified in patients with NSCLC
responding to gefitinib (5,6). These mutations, consisting of in-frame
deletions in EGFR exon 19 and the L858R substitution in exon
21, were associated with both in vitro sensitivity to
gefitinib and therapeutic efficacy and are commonly referred to as
‘activating mutations’ as the mutant products are constitutively
activated and oncogenic (1,7). Together, these mutations constitute
80–90% of all EGFR mutations in NSCLC. In addition, mutations
involving G719 and L861 are also associated with gefitinib
sensitivity, but their incidence is much lower (7). Thus, for patients with known EGFR
activating mutations, treatment with an EGFR TKI (gefitinib,
erlotinib or afatinib) represents current standard first-line
therapy (8,9).
However, the clinical efficacy of gefitinib and
erlotinib is ultimately limited by the development of acquired drug
resistance such as by mutation of the gatekeeper T790 residue
(T790M), which is the most frequent of acquired resistance
mutations occurring in ~60% of patients after treatment with EGFR
TKIs (1,7,9).
Therefore, several EGFR TKIs have been developed for overcoming
this acquired resistance to gefitinib and erlotinib. The so-called
‘second-generation’ of EGFR TKIs, which irreversibly and covalently
bind with the catalytic site of the EGFR TK domain and widely
inhibit TK receptors of the ErbB family (of which EGFR is a
member), have been examined in clinical trials (1,9).
However, despite promising preclinical evidence of activity against
EGFR-mutated cell lines harboring the T790M mutation (10–12),
the second-generation inhibitors (afatinib, neratinib and
dacomitinib) did not demonstrate significant activity in patients
harboring the T790M mutation (13–15).
Consequently, to overcome the limitations of the second-generation
inhibitors, a novel class of mutant-selective ‘third-generation’
inhibitors has been developed. Among these, rociletinib (16,17)
and osimertinib (AZD9291) (18,19),
which irreversibly and covalently inhibit the T790M resistance
mutation as well as the activating mutations (exon 19 deletions and
L858R), showed activities against T790M-positive NSCLC in clinical
trials.
An efficient cell-based assay system for the
identification of clinically efficacious EGFR mutant-selective
inhibitors is required. Although the cell-based assays with human
EGFR-mutated cell lines have been already reported (20–22),
the activity against currently utilized EGFR-mutated cell lines
harboring the T790M mutation is inconsistent with activity of the
agents in patients harboring the T790M mutation. In addition,
although the assay systems with EGFR mutant-overexpressing murine
cell lines for EGFR TKIs have been reported (23–25),
the assay systems with a human cell line have been not reported
yet. Thus, we have developed a novel cell-based assay with a human
non-tumorigenic epithelial cell line for the evaluation of
anti-EGFR drug efficacy against EGFR mutation. Wild-type, T790M
mutant, and L858R mutant EGFR genes were introduced into
human non-tumorigenic immortalized breast epithelial MCF 10A cells
that exhibit EGF-dependent growth using a retrovirus system to
effect overexpression. To predict the construct validity of our
system, the activity of EGFR TKIs including first, second and
third-generation agents was evaluated utilizing these EGFR
mutant-expressing cells in comparison to currently utilized
isogenic lines.
Materials and methods
Compounds
The 21 EGFR TKIs of the first, second and
third-generation were used in this study (Table I). The stock solutions (10 mM) of
the compounds were prepared in dimethyl sulfoxide (DMSO) and stored
at −80°C until use. The stock solutions were arrayed in 384-well
plates and serially diluted 3 times to yield a concentration range
from 10 mM to 52 nM. The purity and integrity of all compound
solutions were measured using ultra performance liquid
chromatography-mass spectrometry (Waters, Milford, MA, USA) as
follows: a Waters CORTECS C18 column (1.6 µm, i.d. 2.1×50 mm) was
developed with an aqueous acetonitrile containing a 0.1% formic
acid linear gradient system (5–90% MeCN, 1.6 min; flow rate, 1
ml/min), verifying the UV adsorption and mass of the major UV peaks
(Table I).
 | Table I.EGFR TKIs used in this study. |
Table I.
EGFR TKIs used in this study.
| Compound | Generation | Supplier | Purity (%) |
|---|
| Erlotinib | First | Carbosynth | 100 |
| Gefitinib | First | Chemscene | 100 |
| AEE-788 | First | Active Biochem | 100 |
| AG-1478 | First | Selleck | 100 |
| Icotinib | First | Selleck | 100 |
| WHI-P154 | First | Selleck | 100 |
| PD 153035 | First | Selleck | 100 |
| Afatinib | Second | Selleck | 100 |
| Lapatinib | Second | LC
Laboratories | 100 |
| AC 480 | Second | Selleck | 100 |
| Dacomitinib | Second | Selleck | 100 |
| Pelitinib | Second | Selleck | 100 |
| Varlitinib | Second | Selleck |
97.7 |
| AZD 8931 | Second | Selleck | 100 |
| AST-1306 | Second | Selleck |
95.2 |
| WZ3146 | Third | Selleck | 100 |
| WZ4002 | Third | Selleck | 100 |
| WZ8040 | Third | Selleck | 100 |
| Osimertinib
(AZD9291) | Third | MedChem | 100 |
| Rociletinib | Third | MedChem | 100 |
| CUDC-101 |
Multitargeta | Selleck | 100 |
Construction of retroviral plasmids
containing the mutant genes
A wild-type EGFR cDNA clone was obtained from
our human proteome expression resource library (HuPEX) (26). EGFR mutations were introduced
into the wild-type cDNA using the QuickChange Lightning Multi
Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara,
CA, USA). pRetro-GW-IH, which was produced from pRetroX (Takara
Bio, Shiga, Japan), was used as the retroviral vector. Wild-type
and mutant EGFR constructs were transferred to the pRetro-GW-IH
retroviral expression vector using the Gateway LR reaction (Thermo
Fisher Scientific, Waltham, MA, USA).
Cell culture
MCF 10A cells were purchased from the American Type
Culture Collection (ATCC, Manassas, VA, USA). The isogenic MCF 10A
cell lines with EGFR mutations (L858R/+ and T790M/+) and their
parental cells, used to create the mutant isogenic cells, were
obtained from Horizon Discovery (Cambridge, UK). The GP2-293
packaging cell line was provided by Takara Bio.
MCF 10A cells were cultured in Dulbeccos modified
Eagles medium (DMEM)/Ham's F-12 (Wako, Tokyo, Japan) supplemented
with 5% heat-inactivated horse serum (Gibco, Thermo Fisher
Scientific), 10 µg/ml insulin (human, recombinant), 5 µM forskolin
(both from Wako), 0.5 µg/ml hydrocortisone (Sigma-Aldrich, St.
Louis, MO, USA), 20 ng/ml EGF (human, recombinant), 100 U/ml
penicillin, and 100 µg/ml streptomycin (all from Wako) at 37°C in a
humidified incubator with 5% CO2. GP2-293 packaging
cells were cultured in DMEM (Wako) containing 10% heat-inactivated
fetal bovine serum (Gibco) at 37°C in a humidified incubator with
5% CO2. Cell number and viability were measured using
trypan blue dye exclusion with a Vi-Cell counter (Beckman-Coulter,
Brea, CA, USA). To estimate confluency and doubling time (DT), the
cells were seeded in 96-well plates (cat. CLS3595; Corning Inc.,
Corning, NY, USA) at 5×103 cells/well and measured using
an IncuCyte ZOOM live cell imaging system (Essen BioScience, Ann
Arbor, MI, USA) and IncuCyte ZOOM software.
Retroviral packaging and
infection
GP2-293 packaging cells were seeded into 6-well
plates at a density of 4×105 cells/well. The next day,
the cells were transfected with 0.2 µg plasmid containing the
ecotropic envelope (env) gene and 2 µg retroviral plasmid
mixed with 7.2 µg polyethylenimine (cat. 24765-2; Polysciences,
Warrington, PA, USA). The culture supernatant was replaced with
fresh medium at 7.5 h after transfection and again the following
day. At 60 h after transfection, the virus-containing culture
supernatant was harvested and centrifuged at 1,000 × g for 5 min to
remove cell debris. This was used as the virus solution.
MCF 10A cells (ATCC) expressing the ecotropic
receptor gene were seeded into 6-well culture plates at a
concentration of 7×105 cells and were infected with 1 ml
appropriately diluted virus solution containing 8 µg/ml
hexadimethrine bromide (Sigma) the following day. At 24 h after
infection, the cells were expanded onto a 10-cm dish and cultured
in medium containing 20 µg/ml hygromycin B (Wako).
Measurement of total and
phosphorylated EGFR
Total and phosphorylated EGFR levels were detected
by immunofluorescence staining. Cells were seeded at
4×103 cells/well onto 384-well bottom clear black plates
(cat. 781096; Greiner Bio-One, Frickenhausen, Germany). After 24 h
culture, the cells were fixed with 4% paraformaldehyde phosphate
buffer solution (Wako). After blocking with 5% bovine serum albumin
in Tris-buffered saline solution for 30 min, the cells were
incubated with primary antibodies against EGFR (mouse monoclonal
clone AT2H8) and phospho-(Y1173)-EGFR (rabbit monoclonal clone
E124) (both from Abcam, Cambridge, MA, USA) in 1:100 dilution at
37°C for 4 h. The cells were then washed 3 times with Tris-buffered
saline containing 0.05% Tween-20 solution followed by incubation
with anti-mouse or anti-rabbit IgG antibodies conjugated to
DyLight488 or DyLigh594 (NovusBio, Littleton, CO, USA),
respectively, in 1:1,000 dilution and stained with Hoechst 33342
solution (Dojindo Co., Kumamoto, Japan) in 1:1,000 dilution to
visualize the nuclei at 37°C for 1 h. The cells were imaged with a
×10 objective and a fluorescein filter using the Operetta High
Content Imaging system and analyzed with the Columbus Image Data
Storage and Analysis system (both from PerkinElmer, Waltham, MA,
USA) to identify the nucleus, EGFR and phospho-EGFR.
To test the inhibition of EGFR phosphorylation
mediated by EGFR TKIs, the cells were seeded at 2×104
cells/well onto 96-well bottom clear black plates (cat. 6005182;
PerkinElmer), cultured for 24 h, and then treated with gefitinib,
afatinib, or osimertinib for 1 h. The cells were then fixed and
stained as indicated above. The vehicle solvent (DMSO) was used as
a control at a concentration of 0.1%.
Growth inhibition assay
The growth inhibitory activity of EGFR TKIs against
cells was assayed by measuring the amount of ATP in the cells using
CellTiter-Glo (Promega, Madison, WI, USA). The cells were incubated
in 384-well plates at a density of 1×103 cells/well with
a medium volume of 40 µl for 4 h. The cells were then treated with
0.1 µl EGFR TKI solutions at final concentration ranges of 25 µM to
1.3 nM (10-point dose) using an ADS-348-8 Multistage-dispense
station (Biotec, Tokyo, Japan). The vehicle solvent (DMSO) was used
as a control at a maximum concentration of 0.25%. After 72 h, 10 µl
CellTiter-Glo reagent solution was added to the medium and the
plate was mixed with a plate mixture and incubated for 10 min at
30°C. The luminescence was measured using an EnSpire plate reader
(PerkinElmer). The IC50 values were analyzed using
Morphit software (The Edge Software Consultancy, Guildford,
UK).
Results
Cells overexpressing EGFR mutant
genes
To develop a cell-based assay system for evaluating
EGFR TKIs, MCF 10A cells (ATCC), which are non-tumorigenic
immortalized breast epithelial cells and EGF
proliferation-dependent, were used in this study. Firstly, MCF 10A
cells expressing ecotropic receptor were created and used as a
control to establish MCF 10A cells overexpressing wild-type EGFR
(WT) and EGFR mutants (L858R and T790M) using a retroviral system.
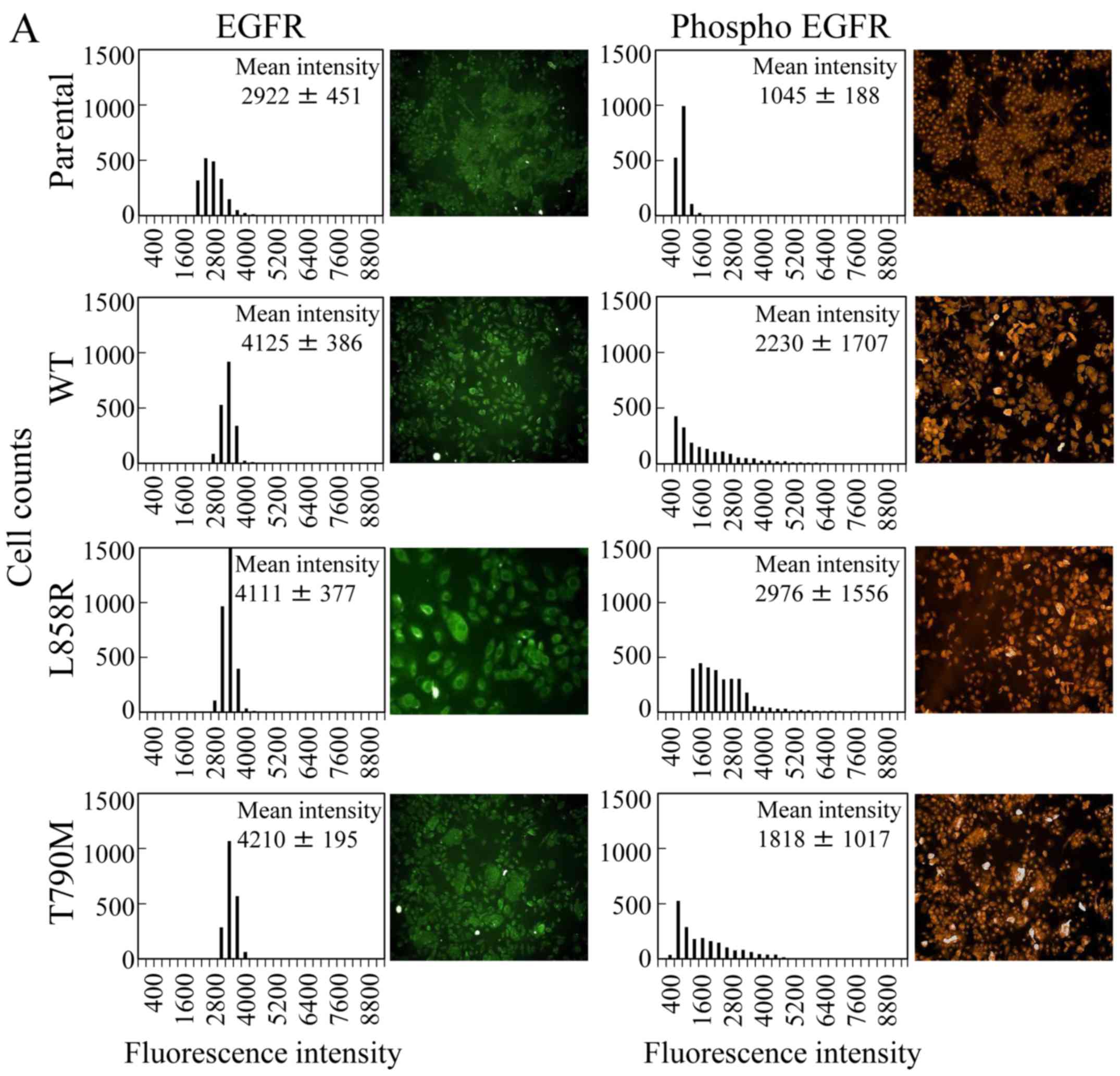
The levels of total and phosphorylated EGFR (pY1173) were detected
with immunofluorescence. As predicted, the mean intensities of
green fluorescence corresponding to EGFR in the overexpressing
cells were ~1.5 times higher than that of the MCF 10A cell line
expressing the ecotropic receptor (Fig.
1A). The scattering in the EGFR expression histograms was
narrow (SD, 195–451), suggesting that the amount of EGFR in the
cells was constant. The levels of phosphorylated EGFR (red) in WT
and T790M cells were ~2 times that of the control line, whereas the
constitutively active EGFR mutant L858R showed a 3-fold increase
compared with the control line (Fig.
1A). However, the scattering in the histograms of the
phosphorylated EGFR levels in the gene-overexpressing cells was
wide (SD, 1017–1707), suggesting that phosphorylated EGFR
accumulated in the cells over time.
Next, we examined whether the depletion of EGF
affected the proliferation of the various cells. The cells were
cultured in medium with or without EGF for 72 h. The confluency and
DT of the cells were measured using the IncuCyte ZOOM live cell
imaging system. DTs of WT, L858R, and T790M (16–19 h) cells were
similar to the control line (20 h) in medium with EGF (Fig. 1B). In the absence of EGF, the
control, WT and T790M lines did not grow, indicating that their
growth is dependent on EGF (Fig.
1B). On the other hand, the constitutively active EGFR mutant
L858R was able to proliferate in the absence of EGF; DTs of L858R
with and without EGF were 16 and 25 h, respectively.
To confirm the relationship between EGFR
phosphorylation and EGF stimulation, the cells were cultured
without EGF for 3 h and the phosphorylated receptor levels were
measured (Fig. 1C). A high level of
phosphorylated EGFR was found in the L858R overexpressing line that
exhibits growth under these conditions of EGF depletion as was
observed in culture with EGF, whereas the levels of phosphorylated
EGFR of the other cells were low compared with the respective
EGF-containing cultures (Fig.
1C).
Growth inhibitory effects of EGFR TKIs
depend on the underlying EFGR mutation
To investigate the sensitivity of EGFR TKIs to L858R
and T790M, a growth inhibitory assessment was conducted for the
various cells treated with these agents. Each generation of EGFR
TKIs under clinical use and in development was examined in these
experiments (Table I). The cells
were treated with the drugs at 4 h after seeding and incubated for
72 h. Then, the numbers of viable cells were calculated by
measuring the amounts of ATP in the cells using CellTiter-Glo
reagents. The IC50 values of 21 EGFR TKIs against each
cell type are shown in Table II.
WT demonstrated greater resistance against all EGFR TKIs than the
control cells. The IC50 values of the drugs against WT
were 28 nM-4.5 µM. Afatinib, which is a representative
second-generation drug, exhibited the strongest inhibitory effect
in 21 EGFR TKIs. No major difference was observed in the inhibitory
effects between each generation of EGFR TKIs.
| Table II.IC50 values of EGFR TKIs
against control- and various EGFR-expressing cell lines. |
Table II.
IC50 values of EGFR TKIs
against control- and various EGFR-expressing cell lines.
| Compound | Generation | Control | WT | L858R | T790M |
|---|
| Erlotinib | First | 0.221 | 0.379 | 0.031 (0.08) | 4.315 (11.39) |
| Gefitinib | First | 0.180 | 0.371 | 0.034 (0.09) | 5.781 (15.58) |
| AEE-788 | First | 0.157 | 0.342 | 0.032 (0.09) | 2.488 (7.27) |
| AG-1478 | First | 0.088 | 0.163 | 0.018 (0.11) | 8.697 (53.36) |
| Icotinib | First | 0.524 | 0.601 | 0.043 (0.07) | 14.639 (24.36) |
| WHI-P154 | First | 0.267 | 0.447 | 0.080 (0.18) | 8.110 (18.14) |
| PD 153035 | First | 0.095 | 0.190 | 0.021 (0.11) | 8.382 (44.12) |
| Afatinib | Second | 0.009 | 0.028 | 0.002 (0.07) | 0.365 (13.04) |
| Lapatinib | Second | 1.612 | 4.502 | 2.590 (0.58) | 7.564 (1.68) |
| AC 480 | Second | 1.159 | 1.637 | 1.452 (0.89) | 10.274 (6.28) |
| Dacomitinib | Second | 0.011 | 0.037 | 0.002 (0.05) | 1.060 (28.65) |
| Pelitinib | Second | 0.014 | 0.059 | 0.005 (0.08) | 0.189 (3.20) |
| Varlitinib | Second | 1.550 | 2.483 | 1.217 (0.49) | 8.331 (3.36) |
| AZD 8931 | Second | 0.013 | 0.040 | 0.004 (0.10) | 4.183 (104.58) |
| AST-1306 | Second | 0.029 | 0.093 | 0.025 (0.27) | 0.471 (5.06) |
| WZ3146 | Third | 0.114 | 0.309 | 0.028 (0.09) | 0.156 (0.50) |
| WZ4002 | Third | 0.557 | 1.525 | 0.090 (0.06) | 0.643 (0.42) |
| WZ8040 | Third | 0.096 | 0.285 | 0.016 (0.06) | 0.102 (0.36) |
| Osimertinib | Third | 0.197 | 0.628 | 0.065 (0.10) | 0.419 (0.67) |
| Rociletinib | Third | 0.754 | 1.667 | 0.246 (0.15) | 0.778 (0.47) |
| CUDC-101 | Multitarget | 0.051 | 0.084 | 0.025 (0.30) | 0.056 (0.67) |
Next, the sensitivity of EGFR TKIs to L858R and
T790M was evaluated. The relative ratios (mutant/WT) of the
IC50 values of L858R and T790M were calculated (Table II). The IC50 values of
all drugs against the constitutively active EGFR mutant L858R were
significantly lower than those of the control and WT lines. In
addition, these high sensitivities (ratios, 0.06–0.18) were
especially evident in the first and third-generations of EGFR TKIs.
On the other hand, T790M exhibited remarkable resistance against
the first-generation agents. Although the relative ratios of the
second-generation varied (1.68–104.58), a weaker inhibitory effect
was observed against T790M than against the WT receptor. As
expected, the inhibitory effect of the third-generation agents
against T790M was the strongest compared to the first and
second-generations.
Inhibition of EGFR phosphorylation
with EGFR TKIs depends on the underlying EGFR mutation
Inhibition of EGFR phosphorylation in WT, L858R and
T790M by treatment with gefitinib, afatinib and osimertinib was
analyzed by immunofluorescence. The cells were cultured on 96-well
plates for 24 h, treated with EGFR TKIs for 1 h, fixed, and stained
with the antibody against phosphorylated EGFR (pY1173). The
inhibitory rates of EGFR phosphorylation in L858R markedly
increased at low concentrations (3–30 nM) of gefitinib, afatinib
and osimertinib compared with WT (Fig.
2). On the other hand, the levels of phosphorylated EGFR in
T790M did not decrease at a high concentration (3 µM) of gefitinib
or afatinib (Fig. 2), whereas a
third-generation inhibitor, osimertinib, decreased the levels of
phosphorylated EGFR in T790M to levels similar to WT (Fig. 2D). These results were identical to
the growth inhibitory effects of these agents.
Comparison between the overexpressing and isogenic
cells with EGFR mutations. To compare our mutant-expressing cells
with the introduced mutant cells created by using isogenic cell
line technology, we investigated the sensitivity of EGFR TKIs
including each generation (Table
III) toward the L858R and T790M mutant isogenic cells. First,
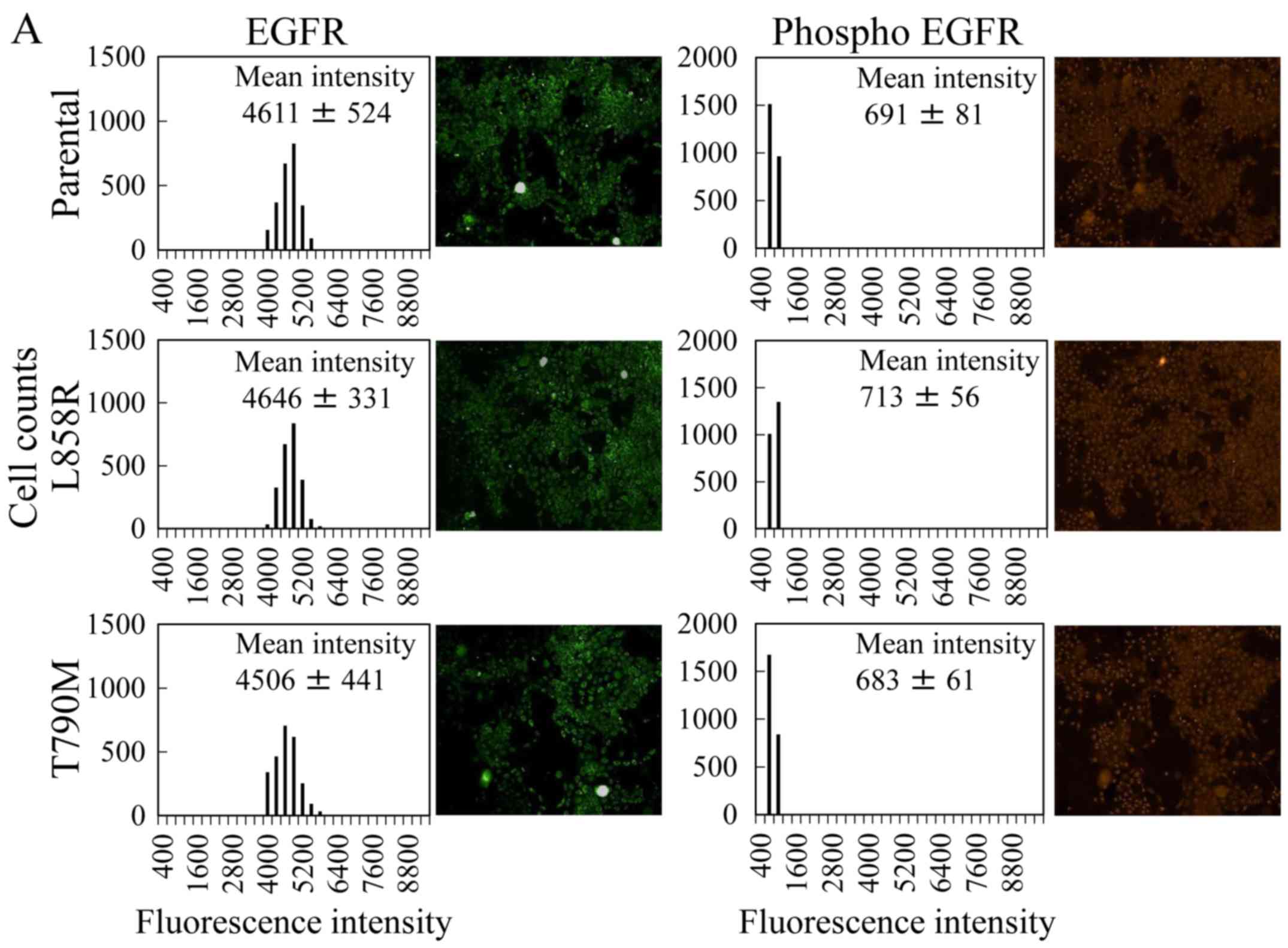
we evaluated the levels of total and phosphorylated EGFR (pY1173)
by immunofluorescence. The levels of total EGFR in the MCF 10A cell
lines between two cell lines derived from ATCC and Horizon
Discovery were different. The mean intensities of EGFR in the
parental cells from Horizon Discovery (Fig. 3A) were ~1.5 times higher than those
of the control cells from ATCC (Fig.
1A). The mean intensities and histograms of the fluorescence in
the isogenic cells were similar to those of the parental cells
(Fig. 3A). Although the levels of
EGFR in the isogenic and mutant-expressing cells were equivalent,
the levels of phosphorylated EFGR in the isogenic cells were lower
than those in the mutant-expressing cells. Notably, the level of
phosphorylated EGFR in the constitutively active EGFR mutant L858R
did not increase, unlike the observation in the L858R-expressing
cells.
| Table III.IC50 values of EGFR TKIs
against the isogenic cells. |
Table III.
IC50 values of EGFR TKIs
against the isogenic cells.
| Compound | Generation | Parental | L858R | T790M |
|---|
| Erlotinib | First | 0.282 | 0.051 (0.18) | 1.750 (6.21) |
| Gefitinib | First | 0.218 | 0.045 (0.21) | 0.846 (3.88) |
| PD 153035 | First | 0.147 | 0.025 (0.17) | 0.520 (3.54) |
| WHI-P154 | First | 0.362 | 0.112 (0.31) | 1.427 (3.94) |
| Afatinib | Second | 0.016 | 0.002 (0.13) | 0.017 (1.06) |
| Lapatinib | Second | 2.085 | 2.330 (1.12) | 4.823 (2.31) |
| AC 480 | Second | 1.545 | 1.839 (1.19) | 4.708 (3.05) |
| Dacomitinib | Second | 0.023 | 0.003 (0.13) | 0.031 (1.35) |
| Osimertinib | Third | 0.323 | 0.084 (0.26) | 0.301 (0.93) |
| Rociletinib | Third | 1.167 | 0.303 (0.26) | 0.504 (0.43) |
| CUDC-101 | Multitarget | 0.066 | 0.038 (0.58) | 0.078 (1.18) |
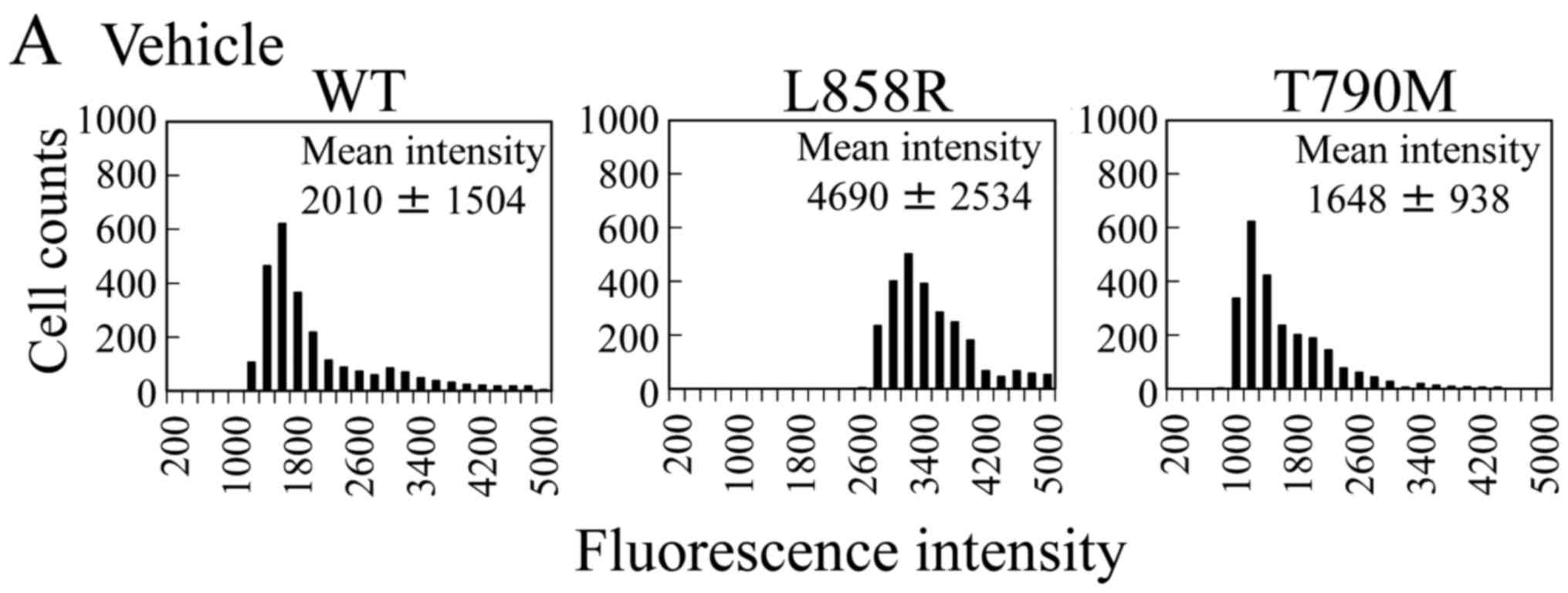
Next, we examined whether the growth of isogenic
cells was dependent on EGF. We found that the parental (MCF 10A
from Horizon Discovery) and the T790M and L858R mutant isogenic
lines showed EGF dependency. In contrast to the L858R-expressing
cells, the L858R mutant isogenic cells did not grow without EGF
unlike the L858R-expressing cells (Fig.
3B), because the phosphorylated EGFR level in the L858R mutant
isogenic cells was similar to that of the parental cells and was
not elevated (Fig. 3A).
The growth inhibitory effects of 11 EGFR TKIs
against parental and L858R and T790M mutant isogenic cells were
tested. The IC50 values are shown in Table III. Although the EGFR TKI
sensitivities against the isogenic cells were similar to those of
the mutant EGFR-expressing cells, the relative ratios (mutant/WT)
of the IC50 values differed. The IC50 values
of gefitinib against parental, L858R and T790M isogenic cells were
218, 45 and 846 nM, respectively (Table III and Fig. 3C). The relative ratios compared to
parental cells were 0.21 for L858R and 3.88 for T790M (Table III). These results showed that the
drug responsiveness against isogenic mutants was lower than that
for mutant-expressing cells; 0.09 for L858R and 15.58 for T790M
(Table II and Fig. 3C). In addition, the IC50
values of afatinib against the isogenic cells were 16 (parental), 2
(L858R), and 17 nM (T790M), demonstrating that the parental and
T790M mutant isogenic cells exhibited similar values (Table III and Fig. 3C). On the contrary, the
IC50 value (365 nM) of afatinib against T790M-expressing
cells was higher than for cells expressing WT (28 nM) (Table II and Fig. 3C). These results indicated that our
mutant gene-expressing cells may possess higher drug sensitivity
than isogenic cells.
Discussion
We established MCF 10A cells overexpressing EGFR
mutants to construct a novel cell-based assay for the evaluation of
EGFR TKIs, which could not be correctly evaluated (i.e., in a
clinically relevant manner) using available T790M-mutated tumor
cell lines or xenograft models based on these cell lines (10–15)
because of differences in genetic backgrounds (e.g., genome
sequence and gene expression) among WT and T790M-mutated tumor cell
lines. Unlike in these cell lines, the comparative analysis between
WT and mutant EGFR-expressing cells is straightforward. As
expected, L858R-expressing cells showed an increase of EGFR TKI
sensitivity compared with cells expressing WT, whereas
T790M-expressing cells showed resistance against EGFR TKIs
(Table II). In addition, the third
generation of EGFR TKIs inhibited the cell growth of
T790M-expressing cells that were more resistant to the first and
second generation than were WT. For example, the third-generation
agent osimertinib (currently marketed) exhibited substantially
higher inhibitory activity against the cell growth of
T790M-expressing cells than erlotinib and gefitinib. These results
were identical with the clinical findings for these drugs (1,7),
suggesting the possibility of evaluation reflecting the state of
cancer tissue in an organism using this mutant-expressing cell
line. Furthermore, our results suggest the additional possibility
of the utility of this mutant EGFR-expressing line for the
evaluation of mutant-selective inhibitors and drug screening.
The isogenic cell line technology used to develop
the lines studied here was initially developed by Di Nicolantonio
et al, wherein a panel of isogenic human cell lines was
created by employing homologous recombination (27). The isogenic cells (T790M and L858R
mutants) exhibited sensitivity toward EGFR TKIs as expected;
however, our mutant gene-expressing cells exhibited higher drug
sensitivity than these isogenic cells (Fig. 3C). In addition, L858R mutant
isogenic cells did not show growth in medium without EGF (Fig. 3B). We consider that the difference
in drug sensitivity and cell growth is due to the levels of mutant
EGFR and of EGFR phosphorylation, as the EGFR mutant
gene-expressing cells showed higher levels of EGFR expression and
phosphorylation than the control line (Fig. 1), whereas mutant isogenic cell EGFR
expression was equivalent to the parental line and phosphorylated
EGFR did not accumulate (Fig.
3).
In conclusion, the results of this study demonstrate
that our mutant gene-expressing cell model is superior to isogenic
cells for the evaluation of anti-EGFR drug efficacy against EGFR
mutation.
Acknowledgements
This study was partially supported by grants for
translational research programs from Fukushima Prefecture.
References
|
1
|
Russo A, Franchina T, Ricciardi GR, Picone
A, Ferraro G, Zanghì M, Toscano G, Giordano A and Adamo V: A decade
of EGFR inhibition in EGFR-mutated non small cell lung cancer
(NSCLC): Old successes and future perspectives. Oncotarget.
6:26814–26825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gazdar AF: Activating and resistance
mutations of EGFR in non-small-cell lung cancer: Role in clinical
response to EGFR tyrosine kinase inhibitors. Oncogene. 28:(Suppl
1). S24–S31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moran C: Importance of molecular features
of non-small cell lung cancer for choice of treatment. Am J Pathol.
178:1940–1948. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Massarelli E, Johnson FM, Erickson HS,
Wistuba II and Papadimitrakopoulou V: Uncommon epidermal growth
factor receptor mutations in non-small cell lung cancer and their
mechanisms of EGFR tyrosine kinase inhibitors sensitivity and
resistance. Lung Cancer. 80:235–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Keedy VL, Temin S, Somerfield MR, Beasley
MB, Johnson DH, McShane LM, Milton DT, Strawn JR, Wakelee HA and
Giaccone G: American Society of Clinical Oncology provisional
clinical opinion: Epidermal growth factor receptor (EGFR) Mutation
testing for patients with advanced non-small-cell lung cancer
considering first-line EGFR tyrosine kinase inhibitor therapy. J
Clin Oncol. 29:2121–2127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stinchcombe TE: Novel agents in
development for advanced non-small cell lung cancer. Ther Adv Med
Oncol. 6:240–253. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwak EL, Sordella R, Bell DW,
Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR,
Fidias P, Lynch TJ, et al: Irreversible inhibitors of the EGF
receptor may circumvent acquired resistance to gefitinib. Proc Natl
Acad Sci USA. 102:7665–7670. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engelman JA, Zejnullahu K, Gale CM,
Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov
GN, Bradner JE, et al: PF00299804, an irreversible pan-ERBB
inhibitor, is effective in lung cancer models with EGFR and ERBB2
mutations that are resistant to gefitinib. Cancer Res.
67:11924–11932. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li D, Ambrogio L, Shimamura T, Kubo S,
Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A,
Himmelsbach F, et al: BIBW2992, an irreversible EGFR/HER2 inhibitor
highly effective in preclinical lung cancer models. Oncogene.
27:4702–4711. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miller VA, Hirsh V, Cadranel J, Chen YM,
Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al: Afatinib
versus placebo for patients with advanced, metastatic
non-small-cell lung cancer after failure of erlotinib, gefitinib,
or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase
2b/3 randomised trial. Lancet Oncol. 13:528–538. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sequist LV, Besse B, Lynch TJ, Miller VA,
Wong KK, Gitlitz B, Eaton K, Zacharchuk C, Freyman A, Powell C, et
al: Neratinib, an irreversible pan-ErbB receptor tyrosine kinase
inhibitor: Results of a phase II trial in patients with advanced
non-small-cell lung cancer. J Clin Oncol. 28:3076–3083. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reckamp KL, Giaccone G, Camidge DR,
Gadgeel SM, Khuri FR, Engelman JA, Koczywas M, Rajan A, Campbell
AK, Gernhardt D, et al: A phase 2 trial of dacomitinib
(PF-00299804), an oral, irreversible pan-HER (human epidermal
growth factor receptor) inhibitor, in patients with advanced
non-small cell lung cancer after failure of prior chemotherapy and
erlotinib. Cancer. 120:1145–1154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Walter AO, Sjin RT, Haringsma HJ, Ohashi
K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, et al:
Discovery of a mutant-selective covalent inhibitor of EGFR that
overcomes T790M-mediated resistance in NSCLC. Cancer Discov.
3:1404–1415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sequist LV, Soria JC, Goldman JW, Wakelee
HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard
GR, Dziadziuszko R, et al: Rociletinib in EGFR-mutated
non-small-cell lung cancer. N Engl J Med. 372:1700–1709. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jänne PA, Yang JC, Kim DW, Planchard D,
Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al: AZD9291
in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J
Med. 372:1689–1699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gendreau SB, Ventura R, Keast P, Laird AD,
Yakes FM, Zhang W, Bentzien F, Cancilla B, Lutman J, Chu F, et al:
Inhibition of the T790M gatekeeper mutant of the epidermal growth
factor receptor by EXEL-7647. Clin Cancer Res. 13:3713–3723. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Somwar R, Shum D, Djaballah H and Varmus
H: Identification and preliminary characterization of novel small
molecules that inhibit growth of human lung adenocarcinoma cells. J
Biomol Screen. 14:1176–1184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang HY, Hsu MK, Wang KH, Tseng CP, Chen
FC and Hsu JT: Non-small-cell lung cancer cells combat epidermal
growth factor receptor tyrosine kinase inhibition through immediate
adhesion-related responses. Onco Targets Ther. 9:2961–2973. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Greulich H, Chen TH, Feng W, Jänne PA,
Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR,
et al: Oncogenic transformation by inhibitor-sensitive and
-resistant EGFR mutants. PLoS Med. 2:e3132005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carey KD, Garton AJ, Romero MS, Kahler J,
Thomson S, Ross S, Park F, Haley JD, Gibson N and Sliwkowski MX:
Kinetic analysis of epidermal growth factor receptor somatic mutant
proteins shows increased sensitivity to the epidermal growth factor
receptor tyrosine kinase inhibitor, erlotinib. Cancer Res.
66:8163–8171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin WH, Song JS, Lien TW, Chang CY, Wu SH,
Huang YW, Chang TY, Fang MY, Yen KJ, Chen CH, et al: A
high-throughput cell-based screening for L858R/T790M mutant
epidermal growth factor receptor inhibitors. Anticancer Res.
32:147–151. 2012.PubMed/NCBI
|
|
26
|
Goshima N, Kawamura Y, Fukumoto A, Miura
A, Honma R, Satoh R, Wakamatsu A, Yamamoto J, Kimura K, Nishikawa
T, et al: Human protein factory for converting the transcriptome
into an in vitro-expressed proteome. Nat Methods. 5:1011–1017.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Di Nicolantonio F, Arena S, Gallicchio M
and Bardelli A: Isogenic mutant human cells: A new tool for
personalized cancer medicine. Cell Cycle. 9:20–21. 2010. View Article : Google Scholar : PubMed/NCBI
|