Introduction
Natural killer (NK) cells are part of the innate
immune system and are one of the primary cell types involved in
anticancer immunosurveillance. At the onset of neoplastic growth,
tumor cells upregulate surface antigens, some of which are ligands
for NK cell activating receptors, including NKG2D. Once NK cells
are activated, cytotoxic granules are exocytosed into the
extracellular space between the NK cell and the target cell and
apoptosis of target cells is induced (1). As the cancer progresses, however,
advanced tumor cells have found ways to downregulate surface NKG2D
ligands. It has been shown that the extracellular domains of MHC
class-I related molecule A (MICA), a human NKG2D ligand, can be
shed by tumor cells by proteolytic cleavages, thus, preventing
activation of cytotoxic immune cells (2). The immune system may also aid in
selecting against tumor cells that maintain NKG2D ligand expression
(3). Due to these properties, MICA
serves as an excellent target for anticancer gene therapy.
Interleukin 12 (IL-12), also known as NK cell
stimulatory factor 2, is an activator and initiator of NK cell
proliferation. The activity of IL-12 is more efficient than either
IL-2 or the IFNs, requiring picomolar instead of nanomolar
concentrations (4). In fact, NK
cells genetically engineered to express IL-12 in a membrane
anchored form by mouse sonic hedgehog C-terminal domain showed
reduced (>10-fold) dependency on IL-2 and a significantly
prolonged survival time for tumor- bearing mice receiving an
intravenous injection of these cells (5). IL-12 has been shown consistently to
have potent antitumor activity primarily through the activity of
IFN-γ, the activation of CD8+ T cells, and the
inhibition of angiogenesis (6,7).
A previous study in our laboratory showed that a
bifunctional fusion protein containing the mouse NKG2D ligand mouse
UL-16 binding protein-like transcript 1 (MULT1) and mouse IL-12 can
effectively activate mouse NK cells both in vitro and in
vivo leading to a reduction in tumor size. It is believed that
the enhanced antitumor effect is due to the simultaneous activation
of NK cells and other NKG2D expressing killer cells through the
MULT1/NKG2D pathway and the IL-12/IL-12R pathway. In addition,
MULT1 may also function as a carrier to deliver IL-12 directly to
the killer cells. When the MULT1 domain binds to an NKG2D receptor
on the surface of an NK cell, the IL-12 may also be held in close
proximity to IL-12 receptors on the same cell, which will maintain
a high level of IL-12 in the tumor microenvironment while providing
low systemic IL-12 levels and reduced toxicity (8).
For this work to be translated for clinical use, the
protein needs to be adapted for use in humans. The purpose of the
present study is to develop a bifunctional fusion protein
containing a human NKG2D ligand, MICA and IL-12. Since mouse IL-12
is fully effective in engaging the human IL-12 receptor, a
recombinant mouse IL-12 gene is used in this model study (9). This human version of the fusion
protein was first examined by transfecting the fusion gene into the
human lung carcinoma cell line A549. Stable clones of tumor cells
expressing the fusion protein and control IL-12 or MICA proteins
were evaluated in a series of in vitro assays to determine
their ability to activate the human natural killer cell line NK92
and isolated PBMCs.
Materials and methods
Cells
The human lung carcinoma cell line A549 (ATCC no.
CRM-CCL-185) was cultured in F-12K medium containing 10% fetal
bovine serum (FBS) and 100 µg/ml gentamicin at 37°C with 5%
CO2. Human natural killer cell line (CRL-2407)
NoGFP-CD16.NK92 (PTA-6967) and the freshly isolated human PBMCs
were cultured in α-MEM with 2 mM L-glutamine, 1.5 g/l sodium
bicarbonate, 0.2 mM inositol, 0.1 mM β2-mercaptoethanol, 0.02 mM
folic acid, 200 U/ml recombinant IL-2, 12.5% horse serum, 12.5% FBS
and 100 µg/ml gentamicin at 37°C with 5% CO2. The blood
samples were procured following a GHS IRB approved protocol (CC/ORI
07-02) and the patients consented that their blood materials would
be used for research purposes.
Construction of fusion gene and
control gene vectors
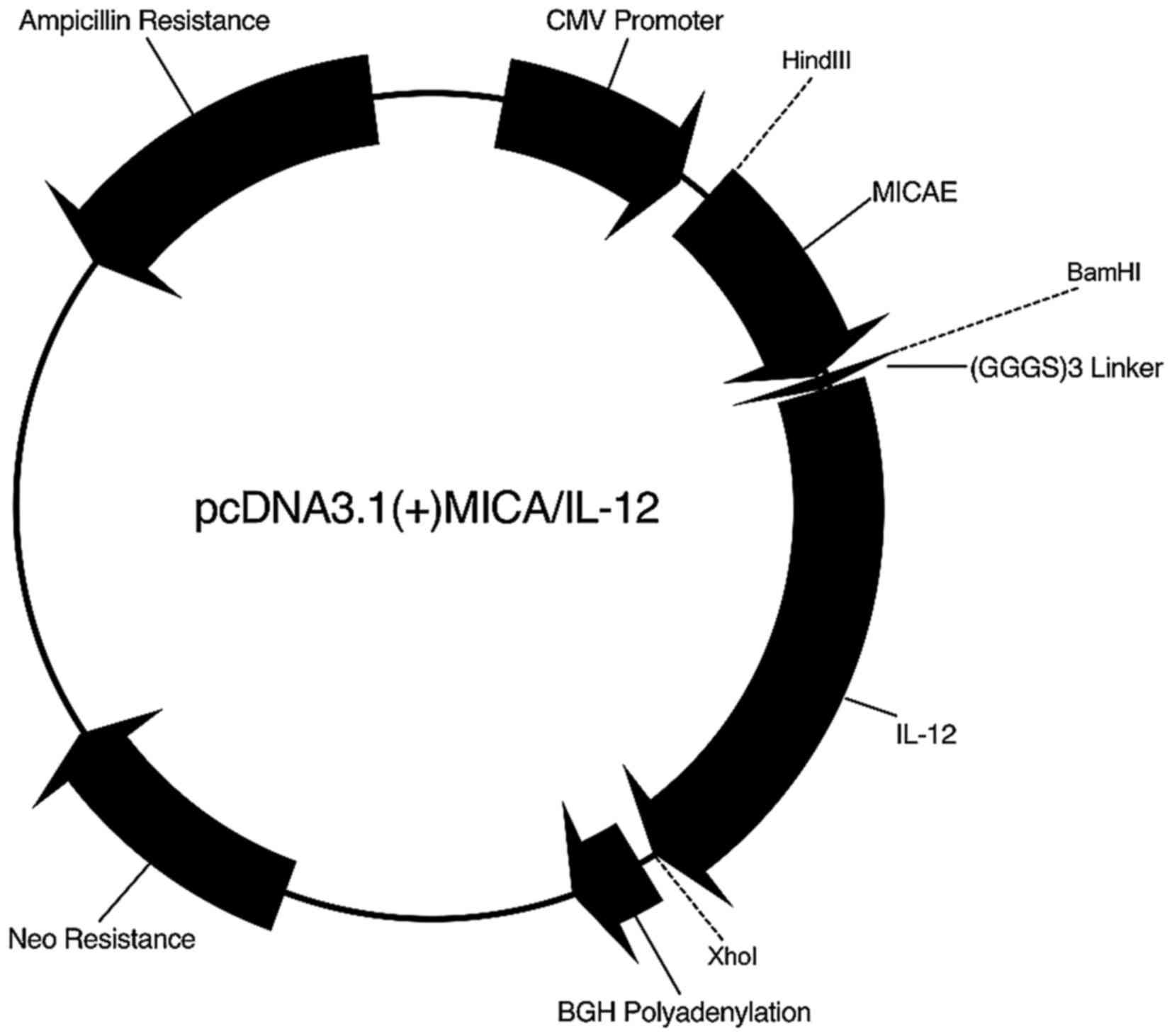
pcDNA3.1(+)MICA/IL-12
The MICA extracellular domain sequence was amplified
from pCMV-SPORT6-MICA (Open Biosystems, Lafayette, CO, USA) using
the 5 primer CCCAA GCTTGAGAGGGTGGCGACGTCGGGG, the 3′ primer CG
GGATCCCTGCCAATGACTCTGAAGCACC and Phusion High-Fidelity DNA
polymerase. The 5 primer contains a restriction cut site for
HindIII and the 3 primer contains the restriction cut site
for BamHI. The PCR fragment was excised and gel purified
using a gel purification kit. Double enzyme digestion using
HindIII and BamHI was performed on the purified
fragment. The plasmid pcDNA3.1(+)MULT1E/mIL-12, created earlier in
our laboratory (8), was also
digested with HindIII and BamHI to remove the MULT1E
and allowing for the insertion of MICA directly upstream of a
(GGGS)3 linker and IL-12 in frame. The linearized
plasmid was excised and gel purified using a gel purification kit.
The enzyme digested MICA PCR fragment was then ligated to the
vector creating the new plasmid pcDNA3.1(+)MICA/IL-12 (Fig. 1). The sequence was confirmed using
DNA sequencing.
pcDNA3.1(+)IL-12 vector
To make a control IL-12 vector, the IL-12 sequence
was amplified from pORF-mIL-12 using the 5 primer
CCAAGCTTCCATGGGTCAATCACGCTACCTCC with a HindIII cut site and
the 3 primer CCTCGAGCTAG GATCGGACCCTGCAGGG with a XhoI
restriction cut site using Phusion High-Fidelity DNA polymerase.
The fragment was excised and gel purified using a gel purification
kit. Double enzyme digestion was performed on the purified fragment
using HindIII and XhoI. pcDNA3.1(+)Neo was digested
with HindIII and XhoI allowing for the insertion of
IL-12. The fragment was excised and gel purified using a gel
purification kit. The IL-12 sequence was then ligated to
pcDNA3.1(+)Neo creating the new plasmid pcDNA3.1(+)IL-12. The
sequence was confirmed using DNA sequencing.
pcDNA3.1(+) MICA vector
To make a control MICA vector encoding a secretable
form of the extracellular domain of MICA, the extracellular MICA
sequence was amplified from pcDNA3.1(+)MICA/IL-12. The 5 primer was
the same used for the construction of pcDNA3.1(+)MICA/IL-12 and the
3 primer (CCTCGAGCTACTGCCAATGACTCT) included a stop codon and the
restriction site for XhoI. PCR was performed to amplify MICA
using Phusion High-Fidelity DNA polymerase. The fragment was
excised and gel purified using a gel purification kit. Double
enzyme digestion was performed on the purified fragment using
HindIII and XhoI. pcDNA3.1(+)Neo was digested with
HindIII and XhoI allowing for the insertion of MICA.
The fragment was excised and gel purified using a gel purification
kit. The MICA sequence was then ligated to the pcDNA3.1(+)Neo
creating the new plasmid pcDNA3.1(+)MICA. The sequence was
confirmed using DNA sequencing.
Cell transfection
A549 cells were transfected with either
pcDNA3.1(+)MICA/IL-12, pcDNA3.1(+)IL-12 or pcDNA3.1(+)MICA using
Lipofectamine as directed by the manufacturer. To obtain stable
clones, the transfected A549 cells were cultured in medium
containing 250 µg/ml of G418. Drug-resistant clones were collected
and subcultured in the presence of the appropriate drug.
Proliferation assay
To determine if the selected clones grow at the same
rate as the untransfected parental A549 cells, 0.05×106
cells/well were plated and cultured on 96-well plates in
triplicates and counted every day for 5 days. At each time point,
CellTiter 96 Aqueous non-radioactive cell proliferation assay
(Promega, Madison, WI, USA) was used following the manufacturers
instructions to determine the relative cell count. Clones were
compared using a two-way ANOVA with the Tukeys post-test.
RT-PCR
Total RNA was extracted from each clone using an
RNeasy Plus Mini kit following the manufacturers directions. RT-PCR
was run with 1 µg of the total RNA using the Phusion RT-PCR kit
following the manufacturers directions. To amplify a 177-bp segment
from MICA the 5 primer CCTTG GCCATGAACGTCAGG and the 3 primer
CCTCTGAGG CCTCGCTGCG were used. To amplify an 805 bp sequence of
IL-12 the 5 primer GGGTGATGGGCTATCTGAGC and the 3 primer
AACTTGAGGGAGAAGTAGGAATGG were used. To amplify a 1.2 kb portion of
the MICA/IL-12 fusion gene containing 446 bp of MICA, the
(GGGS)3 linker, and 708 bp of IL-12 the 5 primer
CCTTGGCCATGAACGTCAGG and the 3 primer GGGAGTCCAGTCCACCTCTA were
used. All reactions included the GAPDH housekeeping gene using the
5 primer ATGACATCAAGAAGGTGGTG and the 3 primer
CATACCAGGAAATGAGCTTG.
Detection of secreted protein
Cells of different clones were plated at
2×106 cells in 6-well plates with normal growth media on
day one. On day two, the media was removed, the cells were rinsed
with phosphate-buffered saline (PBS) and serum-free F-12K was
added. On day three, the serum-free RPMI was removed and
centrifuged at 1000 × g for 15 min. The supernatant was then tested
using an appropriate immunofluorescence or ELISA protocol.
To detect the presence of MICA/IL-12, 96-well plates
were coated with 2 µg/ml rhNKG2D/Fc chimera in carbonate coating
buffer and incubated overnight at 4°C. The plates were rinsed with
PBS and blocked with 10% FBS for 1.5 h. After washing three times
with PBS, 100 µl of the cell supernatant was added to the plate in
triplicates. The samples were incubated at room temperature for 3
h. After removing the samples and washing the plates three times
with PBS, 1 µg/ml PE rat anti-mouse IL-12 (p40/p70) in blocking
solution was added to the plates. The plates were then incubated
for 1.5 h at room temperature. After washing five times with PBS,
the plates were read with a BioTek Synergy H1 hybrid reader. Clones
were compared using a one-way ANOVA with the Tukeys post-test.
Corrected RFU is calculated by subtracting background RFU from each
value.
To detect the presence of secreted IL-12 from the
cells, a mIL-12 p70 Ready-SET-Go ELISA kit was used following the
manufacturers directions. Plates were read with a BioTek EON
microplate reader. Clones were compared using a one-way ANOVA with
the Tukeys post-test.
NK92 and A549 co-culture
NK92 cells were plated with A549 cells or clones
expressing the fusion protein, IL-12, or MICA on a 96-well plate at
a ratio of 10:1 in 250 µl NK media and incubated for 48 h. After
the incubation, 100 µl of media was removed and analyzed for the
presence of IFN-γ using human IFN-γ ELISA reagent kit (Thermo
Fisher Scientific, Waltham, MA, USA) following the manufacturers
directions. The rest of the media were removed from the cells, the
NK suspension cells were carefully rinsed off, and 100 µl fresh NK
media was added. The remaining tumor cell number was determined
using CellTiter 96 AQueous non-radioactive cell proliferation assay
(Promega) following the manufacturers instructions. Clones were
compared using a one-way ANOVA with the Tukeys post-test.
Primed NK92 cytotoxicity
The A549 clones were plated on a 6-well plate in 5
ml NK media at a density of 0.5×106 cells/well and
incubated for 48 h. After the incubation, the supernatant was
collected from these cells and used to plate 4×105 NK92
cells on a 6-well plate. After a 48 h priming period, the NK92
cells were removed and plated on a 96-well plate at a density of
5×104 cells/well with 0.5×104 parental A549
cells and incubated for 24 h. After the incubation, the media were
removed from the cells and the wells were carefully washed four
times to remove the non-adherant NK92 cells. A total of 100 µl
fresh NK media was added. Cell proliferation was determined using
CellTiter 96 AQueous non-radioactive cell proliferation assay
(Promega) following the manufacturers instructions. Clones were
compared using a one-way ANOVA with the Tukeys post-test. Corrected
OD is calculated by subtracting background OD from each value.
Preparation of human PBMCs
PBMCs were isolated from leukapheresis product using
the Ficoll-Paque method. Briefly, the blood product was mixed 1:4
with PBS and loaded on top of 30 ml lymphocyte separation media and
was centrifuged for 15 min. The PBMC layer was then removed and
rinsed with PBS. ACK lysing buffer was then used to lyse any
remaining RBCs. After a final PBS rinse, cells were counted and
used.
Supernatant transfer to PBMC
The A549 clones were plated on a 6-well plate in 1.5
ml PBMC media at a density of 0.6×106 cells/well and
incubated for 48 h. After the incubation, the supernatant was
collected from these cells and used to plate 2×105
freshly isolated PBMCs on a 96-well plate and the cells were
incubated for 10 days. After the incubation, 100 µl of media was
removed and analyzed for the presence of IFN-γ using human IFN-γ
ELISA reagent kit (Thermo Fisher Scientific) following the
manufacturers directions. The rest of the media was removed from
the cells and 100 µl fresh PBMC media were added. Cell
proliferation was determined using CellTiter 96 AQueous
non-radioactive cell proliferation assay (Promega) following the
manufacturers instructions. Clones were compared using a one-way
ANOVA with the Tukeys post-test.
Statistical analysis
GraphPad software was used to plot graphs and run
statistics. The significance was represented as P<0.05,
P<0.01 or P<0.001.
Results
Construction of expression
vectors
To construct the plasmid containing the MICA/IL-12
fusion gene, the MICA extracellular domain cDNA sequence was
amplified from pCMV-SPORT6-MICA and used to replace MULT1E in the
plasmid pcDNA3.1(+)MULT1E/mIL-12 that had been constructed earlier
in our laboratory (1). MICA was
inserted directly upstream of a (GGGS)3 linker, allowing
flexibility, and IL-12 in frame creating the new plasmid
pcDNA3.1(+)MICA/IL-12 (Fig. 1). The
IL-12 control plasmid, pcDNA3.1(+)IL-12, was constructed by
amplifying the IL-12 sequence from pORF-mIL-12 and ligating it into
the pcDNA3.1(+) vector. Since the fusion protein is secretable and
only contains the extracellular domain of MICA, PCR was used to
amplify MICA from pcDNA3.1(+)MICA/IL-12 and add a stop codon at the
end of the extracellular domain. Since MICA is a type I
transmembrane protein and the signal sequence is conserved, the
resulting protein should be transported outside the cell (10).
Transfection and stable clone
selection
A549 cells were transfected with plasmids
pcDNA3.1(+)MICA/IL-12 (Fig. 1),
pcDNA3.1(+)MICA, or pcDNA3.1(+)IL-12, respectively, using
Lipofectamine. Multiple clones from each transfection that were
geneticin resistant were selected. RT-PCR and ELISA analysis (see
below) were used to screen these clones and clones M-IL/4 and
M-IL/19 were selected for fusion protein MICA/IL-12, clone IL/14
for IL-12 and clone M/19 for MICA. An in vitro cell
proliferation assay showed that all clones grow at a similar rate
(data not shown).
Stable clones express the fusion
protein
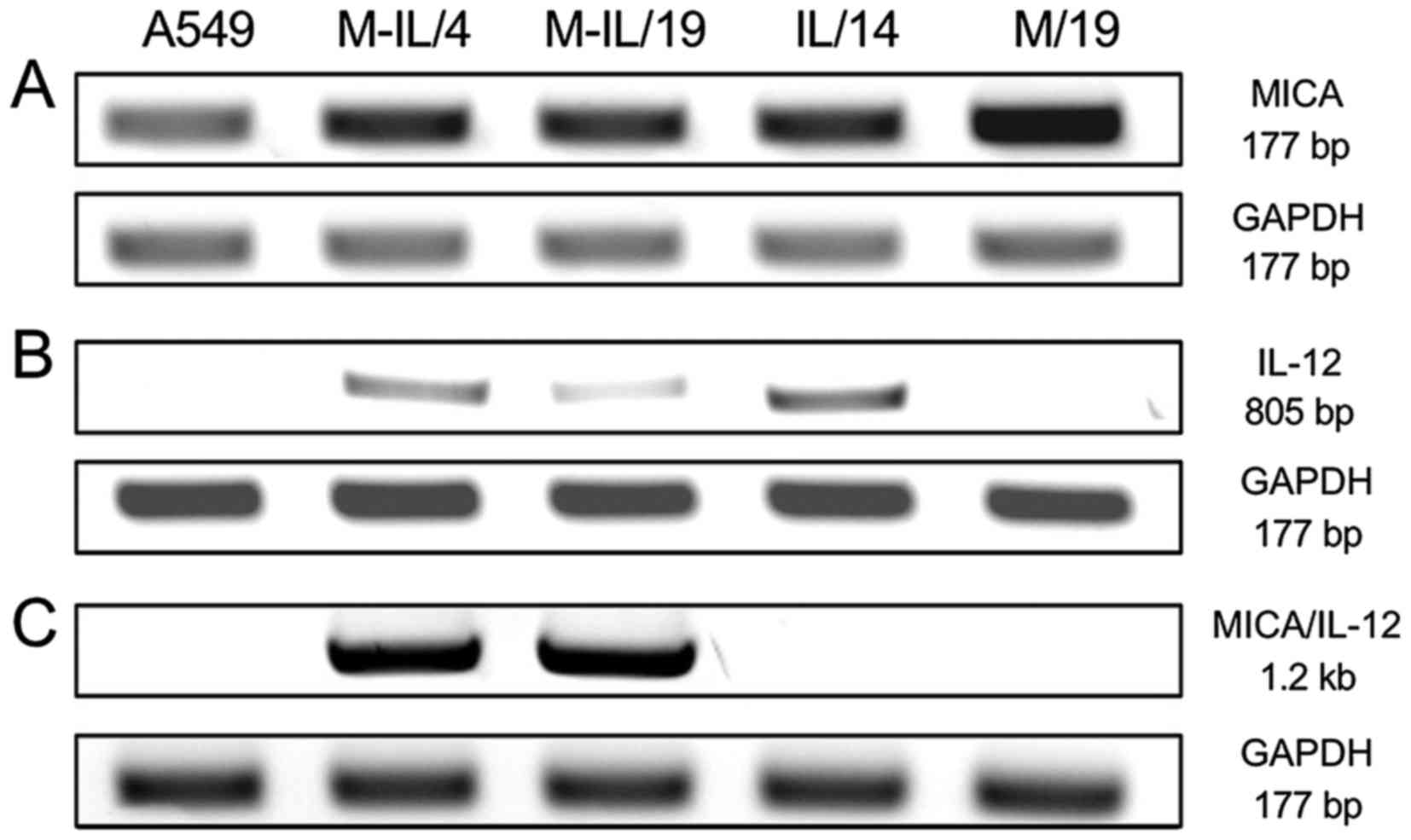
The fusion protein expression was first examined by
reverse transcription-PCR (RT-PCR) on total RNA samples collected
from the clones using three pairs of primers. The first pair was
specific to a 177 bp portion of the extracellular domain of MICA.
The second pair amplified an 805 bp sequence of IL-12. The final
primer pair amplified a 1.2 kb portion of the MICA/IL-12 fusion
gene containing 446 bp of MICA, the (GGGS)3 linker, and
708 bp of IL-12. All cells, including the A549 control cells and
IL/14, contain mRNA transcripts for MICA (Fig. 2A). The fusion gene clones, along
with the IL/14 clone contain mRNA transcripts for IL-12 (Fig. 2B). Only clones M-IL/4 and M-IL/19
contain mRNA transcripts for the MICA/IL-12 fusion gene (Fig. 2C).
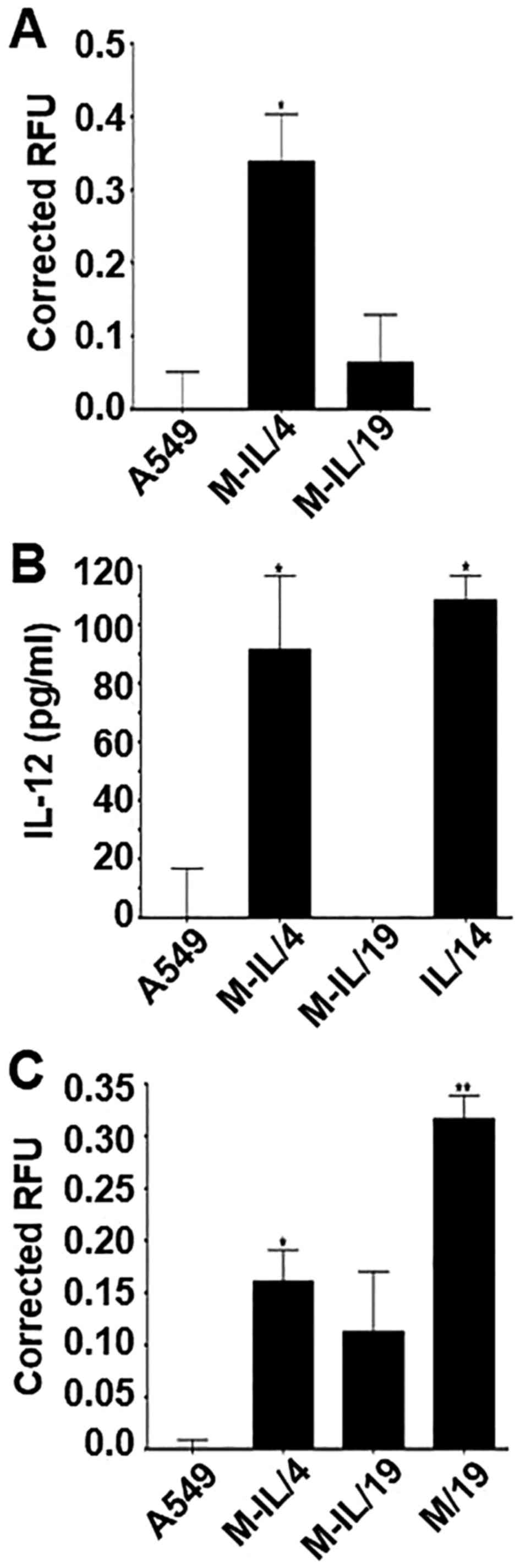
A sandwich ELISA was used to detect the secretion of
the transgene products. The rhNKG2D/Fc chimera was used to coat the
plate and anti-mouse IL-12 conjugated with PE was used as the
detecting antibody. Clone M-IL/4 produced significant levels of the
MICA/IL-12 fusion protein and successfully released it into the
supernatant. Clone M-IL/19 did not produce detectable amounts of
the fusion protein (Fig. 3A). ELISA
was also performed to detect IL-12 or MICA separately. Clones
M-IL/4 and IL/14 both produced similar amounts of IL-12 (Fig. 3B). M-IL/19 produced no detectable
levels of IL-12. The secreted form of MICA was detected at
significant levels in clones M-IL/4 and M/19 (Fig. 3C).
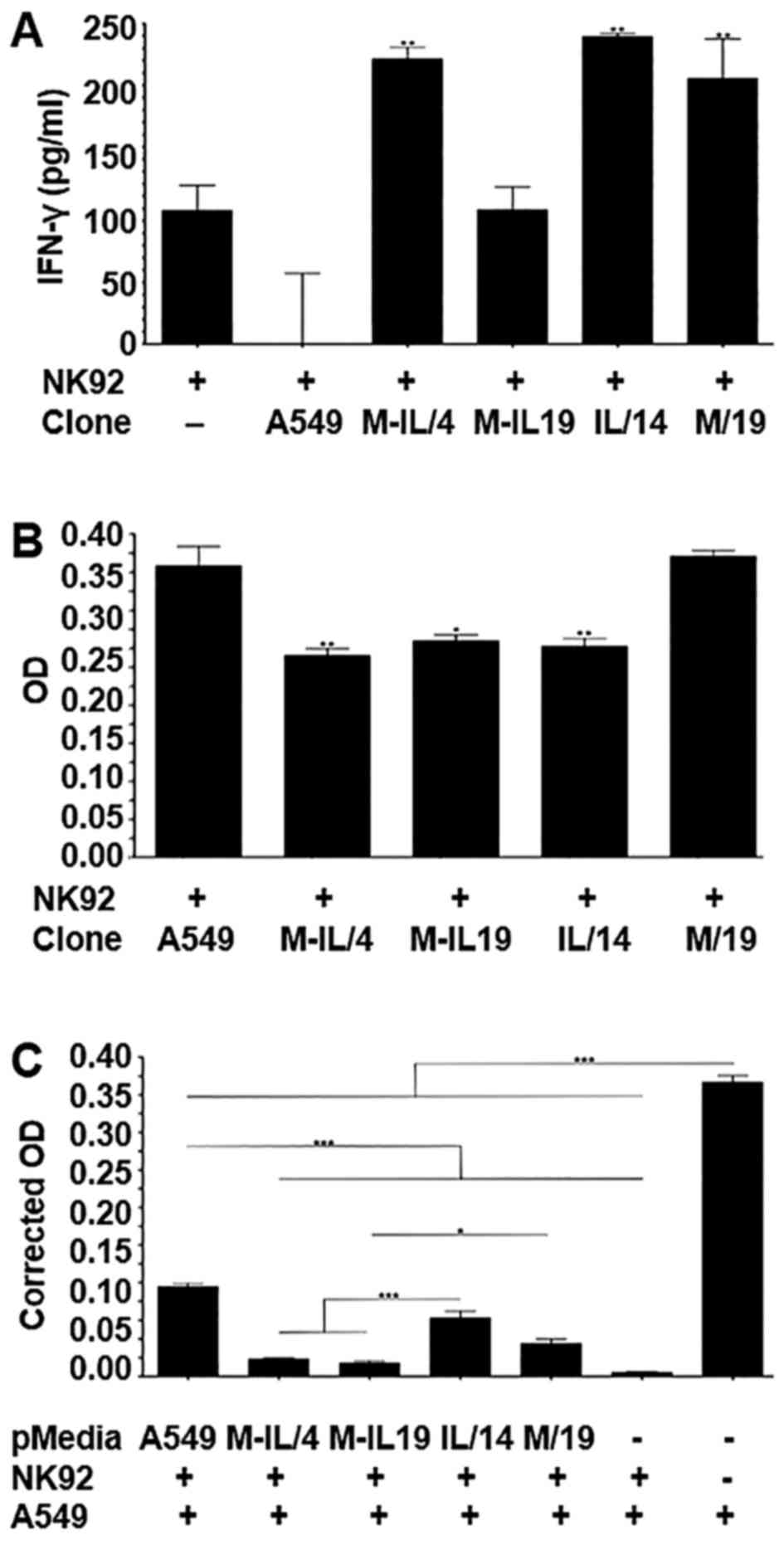
MICA/IL-12 activates NK92 cells
Human NK92 cells were co-cultured with the A549
cells and clones M-IL/4, M-IL/19, I/14, or M/19 for 48 h. To
determine if the proteins produced by these clones could activate
the NK92 cells, two assays were performed. First, 100 µl of the
media was removed and screened using an IFN-γ ELISA. The NK92 cells
co-cultured with clones M-IL/4, IL/14 and M/19 showed significantly
higher IFN-γ production than NK92 cells co-cultured with A549
parental cells. M-IL/19 was not able to significantly increase
IFN-γ production (Fig. 4A).
The second assay measured the primed NK92 cell
cytotoxicity towards the tumor cell clones. After removing the
supernatants from the co-cultures, the wells were gently rinsed
five times with PBS to remove the remaining suspension NK92 cells
and leave behind the adherent tumor cells. The remaining cells were
then measured using an MTS cell proliferation assay. There were
significantly fewer M-IL/4, M-IL/19 and IL/14 cells when compared
to the A549 parental cells. There was no significant difference in
the number of M/19 cells compared to the A549 parental cells
(Fig. 4B).
We assessed whether the pre-activation or priming of
NK92 cells with supernatant collected from the clones expressing
the fusion protein, IL-12, or MICA can enhance the killing of
parental A549 cells. A549 parental cells and the clones M-IL/4,
M-IL/19, IL/14 or M/19 were cultured in NK media for 48 h before
collecting the supernatant. NK92 cells were then plated in the
collected supernatants and allowed to incubate for 48 h. These
primed NK92 cells were then plated with parental A549 cells at a
ratio of 10:1 and incubated for 24 h. At the end of the incubation,
the culture media were removed and the wells were gently rinsed
five times to remove the suspension NK92 cells and leave behind the
adherent A549 cells. The remaining cells were measured using an MTS
cell proliferation assay. Although all NK92 cells, whether primed
or not, were able to significantly reduce the tumor cell number
compared to the control, M-IL/4 and M-IL/19 primed NK92 cells were
able to kill significantly more tumor cells than either A549, IL-4,
or M/19 primed NK92 cells. Notably, unprimed NK92 cells were the
most effective killers of A549 cells (Fig. 4C).
MICA/IL-12 activates human PBMCs
Human peripheral blood mononuclear cells (PBMCs)
were isolated from leukapheresis product using the Ficoll-Paque
method. These cells were then cultured with supernatants collected
from A549 cells or clones M-IL/4, M-IL/19, M/19, or IL/14 for 10
days. To determine if the supernatants could promote PBMC
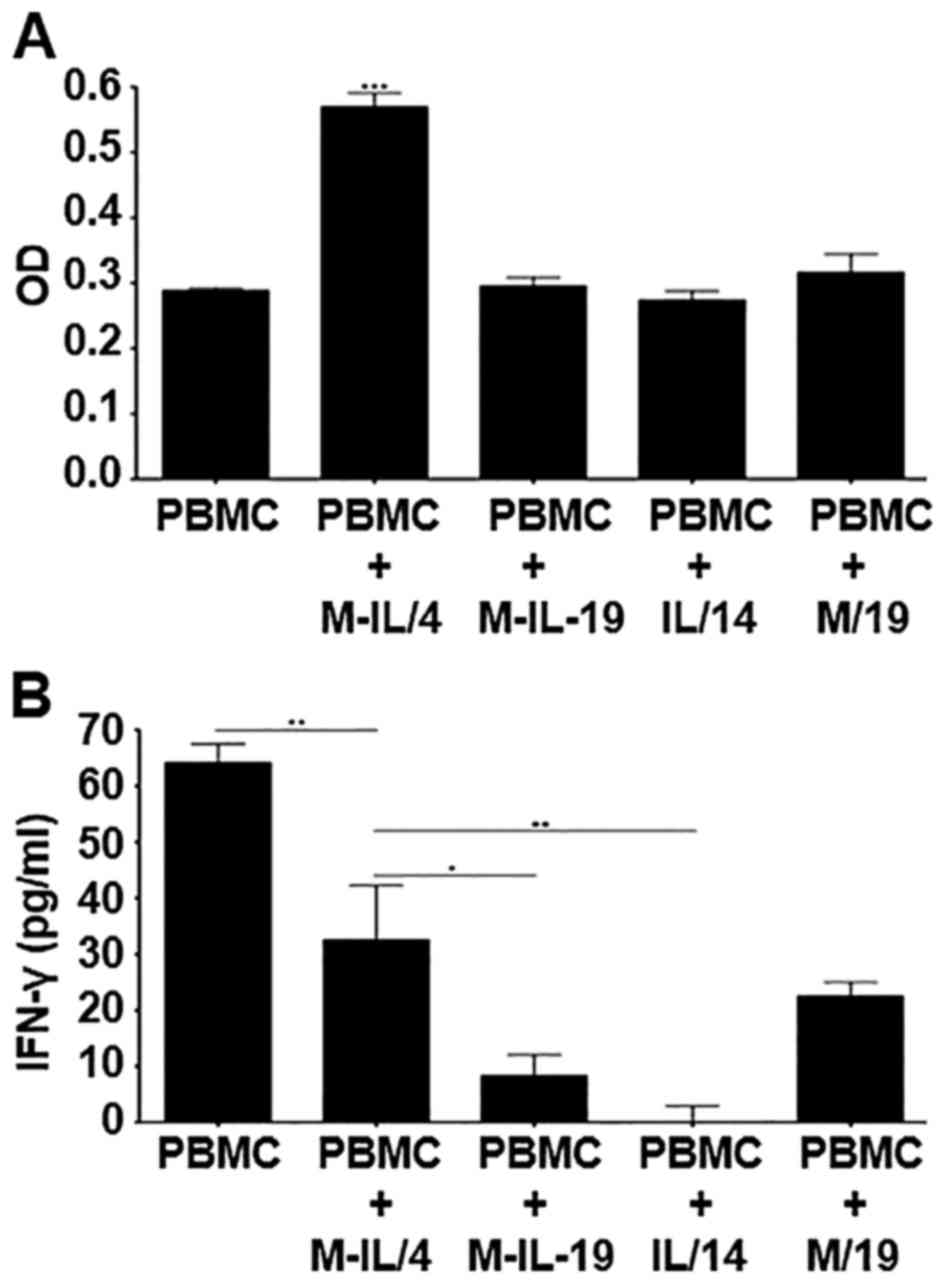
proliferation, an MTS cell proliferation assay was performed. Only
supernatant collected from clone M-IL/4 that expresses the fusion
protein at a relatively high level was able to significantly
increase PBMC cell number (Fig.
5A). An IFN-γ ELISA was also used to detect killer cell
activation. Clone M-IL/4 was also able to induce a significantly
higher production of IFN-γ when compared to clones M-IL/19 and
IL/14. Clone M/19 also induced production of a large amount of
IFN-γ (Fig. 5B). Interestingly,
PBMCs without co-culture expressed the highest level of IFN-γ.
Discussion
Since NK cells play an important role in tumor
immunosurveilance, many strategies have been employed to harness
their ability to prevent neoplastic growth. In the present study we
developed a bifunctional fusion gene encoding MICA/IL-12, and
hypothesized that the bifunctional protein, when expressed by tumor
cells, would effectively activate NK cells and other NKG2D
expressing killer cells in the tumor environment and increase
cytotoxicity.
RT-PCR indicates that all clones contained mRNA for
MICA (Fig. 2A). This is not
surprising since MICA is commonly expressed in lung carcinomas
(11). IL-12 mRNA is detected in
the two fusion gene clones (M-IL/4 and M-IL/19) and the IL-12 clone
(IL/14) (Fig. 2B). Finally, a
portion of the MICA/IL-12 fusion protein including the linker
segment was detected only in the fusion gene clones M-IL/4 and
M-IL/19. The data indicate that the clones have in fact taken up
the transgenes and transcription of the genes of interest can be
detected.
It is important to demonstrate that the fusion
protein is produced and being released from the cell into the tumor
microenvironment where it can interact with killer cells. To test
this, supernatants were collected from all the clones together with
A549 cells and screened using ELISA. As Fig. 3 shows, the engineered proteins are
produced by the respective clones and released from the cells into
the supernatants. Clone M-IL/4 produces a relatively high level of
fusion protein MICA/IL-12, but it is not detectable in the
supernatant collected from M-IL/19 (Fig. 3A). Because there was a clear band
detected after RT-PCR indicating that M-IL/19 does contain the
fusion gene, it is possible that the protein amount produced by
this clone is beneath the detection threshold of the methods used.
Both clones M-IL/4 and IL/14 produce significant levels of IL-12.
It should also be noted that the amount of IL-12 detected in the
clone M-IL/4 supernatant is similar to the amount of IL-12 detected
in the clone IL/14 supernatant. This indicates that clone IL/14 is
a good control for this experiment (Fig. 3B). As expected, significant levels
of MICA were detected in the supernatants from clones M-IL/4 and
M/19. Since there is not a significant difference between the MICA
levels of clone M-IL/4 and M/19, clone M/19 is a good control for
this experiment (Fig. 3C).
To determine if fusion protein MICA/IL-12 is
functional and has enhanced ability to activate killer cells, the
human natural killer cell line NK92, was co-cultured with A549
cells and clones M-IL/4, M-IL/19, IL/14 and M/19 for 48 h at a
ratio of 10:1. The amount of IFN-γ produced by the NK92 cells was
evaluated with ELISA to determine the level of NK cell activation.
The NK92 cells co-cultured with clones M-IL/4, IL/14 and M/19
showed significantly higher IFN-γ production than NK92 cells
co-cultured with A549 parental cells. Clone IL/14 was also able to
increase IFN-γ production significantly compared to NK92 cells that
were not cultured with any A549 cells. M-IL/19 was not able to
significantly increase IFN-γ production compared to NK92 cells
alone (Fig. 4A). M-IL/4 stimulated
production at similar levels of IFN-γ as clone IL/14, which is
expected because they both have similar IL-12 expression levels as
shown in the IL-12 ELISA (Fig. 3B).
It is interesting that NK92 cells co-cultured with A549 parental
cells produced much less IFN-γ when compared with NK92 cells that
were not co-cultured with any cells. This is possibly due to either
the production of or the shedding of inhibitory ligands from A549
tumor cells into the supernatant, a phenomenon commonly seen in
tumors, which can reduce the activation of the NK92 cells by
engaging the inhibitory receptors, such as NKG2A/B, KIR2DL4 and
ILT-2 (12–16). The results not only confirm the
notion that the activation of NK cells and other killer cells
depends on the balance of inhibitory receptors and activating
receptors, but also demonstrate that the fusion protein MICA/IL-12
can change the balance towards activation.
To determine if the fusion protein could enhance
NK92 cytotoxicity toward the tumor cells, the number of A549 cells
remaining after the co-culture was measured. There were
significantly fewer cells of clones M-IL/4, M-IL/19 and IL/14 when
compared to the A549 parental cells or clone M/19 cells (Fig. 4B). Although the cytotoxicities among
clones M-IL/4, M-IL/19 and IL/14 are not statistically different,
clone M-IL/4 shows a slightly higher activity. This indicates that
clone M-IL/19 does produce some MICA/IL-12 even though it was
undetectable using ELISA (Fig. 3C).
This suggests that when compared to a larger amount of IL-12, even
a small amount of the fusion protein can produce similar cytotoxic
results. This is also interesting because clone M-IL/19 was not
able to increase IFN-γ production of the NK92 cells whereas M-IL/4
and IL/14 did (Fig. 4A). This
suggests that the addition of the MICA portion of the protein
increases the cytotoxic activity of the NK92 cells.
One of the current hurdles for gene therapy is the
low efficiency of gene delivery (17). With this in mind, it is important to
determine if MICA/IL-12 can also activate NK92 cells to kill A549
cells that do not express the protein. NK92 cells that were primed
for 48 h with the supernatants from A549 cells and clones of
M-IL/4, M-IL/19, IL/14 and M/19 were co-cultured with parental A549
cells at a ratio of 10:1 and incubated for 24 h. After the removal
of the suspension NK92 cells, the remaining tumor cells were
measured using an MTS cell proliferation assay. All NK92 cells,
whether primed or not, were able to significantly reduce the A549
cell number compared to the control without NK92 cells. This is not
a surprise as these cells are highly cytolytic. Both clones M-IL/4
and M-IL/19 primed NK92 cells were able to kill significantly more
A549 cells than either A549, IL-4, or M/19 primed NK92 cells. This
suggests that priming with MICA/IL-12 is more effective than
priming with either IL-12 or MICA alone, even when the amount of
MICA/IL-12 is small as seen in clone M-IL/19. Notably, the unprimed
NK92 cells killed significantly more A549 cells than all other NK92
cells except the NK92 cells primed with MICA/IL-12 (Fig. 4C). This suggests that MICA/IL-12
expression, even in small amounts, was able to overcome the
inhibitory signals from A549 cells during NK92 priming.
Although the NK92 cell line is a good model for
human NK cells, it does not give a complete picture of the in
vivo effects of fusion protein MICA/IL-12. To better understand
how MICA/IL-12 will behave in a patient, further experiments were
conducted using human PBMCs isolated from leukapheresis products.
Supernatants collected from clones M-IL/4, M-IL/19, IL/14, or M/19
were used to treat PBMCs for 10 days and then a relative cell
number was determined. Only supernatant from clone M-IL/4, that
expresses the highest level of the fusion protein, is able to
significantly increase PBMC cell number (Fig. 5A), indicating that MICA/IL-12 is
able to either increase proliferation and/or increase
sustainability of these cells.
An IFN-γ ELISA was also used to detect NK and other
killer cell activation in PBMC culture. As Fig. 5B shows, supernatant from clone
M-IL/4 stimulates PBMCs to produce the most IFN-γ, although
supernatant from clone M/19 also stimulates PBMCs to produce
relatively high levels of IFN-γ. M-IL/4 induced a significantly
higher production of IFN-γ when compared to M-IL/19 and IL/14
(Fig. 5C).
An interesting observation from the present study is
that the IFN-γ production by unprimed PBMCs is significantly higher
than those primed with a clone. This, again, is possibly due to
either the production of or the shedding of inhibitory ligands into
the supernatant by the tumor cells (12–15).
This effect is more pronounced with the PBMCs compared to NK92
cells (Fig. 4A) as the IFN-γ
production by unprimed PBMCs is higher than that of PBMCs primed
with supernatants from any of the tumor cell clones (Fig. 5B), while IFN-γ production by NK92
cells primed with clones M-IL/4, IL/14 or M/19 is higher than that
of the unprimed NK92 cells. This is probably due to the larger
repertoire of PBMC inhibitory receptors. Although the fusion
protein MICA/IL-12 did not completely recover the inhibitory
effects as it does in NK92 culture, it is much more effective than
IL-12 or MICA alone.
Another interesting observation seen when comparing
the NK92 and the PBMC results is the effectiveness of the IL/14 and
M/19 clones. In the experiments conducted using the NK92 cells, the
clone that produced only IL-12 (IL/14) performed better than the
clone that produced only MICA (M/19). The IL-12 was able to
initiate production of more IFN-γ by NK92 cells and resulted in
more cell death during the co-culture compared to M/19 (Fig. 4A and B). However, in the PBMC
experiments, clone M/19 initiated the production of more IFN-γ by
NK92 cells than was initiated by clone IL/14 (Fig. 5B). This suggests that the IL-12 may
be a more effective activator of NK cells, but the MICA can
activate other killer cell types within the heterogeneous PBMC
population. The fusion protein, however, consistently outperforms
both individual components when tested separately.
In conclusion, a fusion protein containing the
extracellular domain of the NKG2D ligand MICA and IL-12 was
successfully created and can be stably expressed by A549 tumor
cells. The fusion protein can increase the production of IFN-γ by
NK92 cells and also increase the cytolytic activity of these cells
toward A549 cells expressing MICA/IL-12. The bystander effect was
also demonstrated when MICA/IL-12 primed NK92 cells were able to
kill parental A549 cells significantly more than NK92 cells primed
with either IL-12 or MICA alone. This result was also an indication
that the activation by MICA/IL-12 was able to overcome inhibitory
signals by the A549 cells. Preliminary studies also indicate that
MICA/IL-12 can increase proliferation and/or sustainability of
isolated human PBMCs. The data suggest that MICA/IL-12 is effective
at augmenting the IFN-γ production of cells cultured with soluble
inhibitory factors produced by A549 cells. Although the study is
preliminary, the data, along with the data from the previous mouse
studies suggest that the MICA/IL-12 bifunctional fusion protein is
an effective activator of killer cells for cancer treatment.
References
|
1
|
Berke G: The binding and lysis of target
cells by cytotoxic lymphocytes: Molecular and cellular aspects.
Annu Rev Immunol. 12:735–773. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Salih HR, Rammensee H-G and Steinle A:
Cutting edge: Down-regulation of MICA on human tumors by
proteolytic shedding. J Immunol. 169:4098–4102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dunn GP, Old LJ and Schreiber RD: The
three Es of cancer immunoediting. Annu Rev Immunol. 22:329–360.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Robertson MJ, Soiffer RJ, Wolf SF, Manley
TJ, Donahue C, Young D, Herrmann SH and Ritz J: Response of human
natural killer (NK) cells to NK cell stimulatory factor (NKSF):
Cytolytic activity and proliferation of NK cells are differentially
regulated by NKSF. J Exp Med. 175:779–788. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu L, Zhao Z, Wei Y, Marcotte W Jr,
Wagner TE and Yu X: An IL-12/Shh-C domain fusion protein-based
IL-12 autocrine loop for sustained natural killer cell activation.
Int J Oncol. 41:661–669. 2012.PubMed/NCBI
|
|
6
|
Brunda MJ, Luistro L, Warrier RR, Wright
RB, Hubbard BR, Murphy M, Wolf SF and Gately MK: Antitumor and
antimetastatic activity of interleukin 12 against murine tumors. J
Exp Med. 178:1223–1230. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Voest EE, Kenyon BM, OReilly MS, Truitt G,
DAmato RJ and Folkman J: Inhibition of angiogenesis in vivo by
interleukin 12. J Natl Cancer Inst. 87:581–586. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tietje A, Li J, Yu X and Wei Y:
MULT1E/mIL-12: A novel bifunctional protein for natural killer cell
activation. Gene Ther. 21:468–475. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schoenhaut DS, Chua AO, Wolitzky AG, Quinn
PM, Dwyer CM, McComas W, Familletti PC, Gately MK and Gubler U:
Cloning and expression of murine IL-12. J Immunol. 148:3433–3440.
1992.PubMed/NCBI
|
|
10
|
Halenius A, Gerke C and Hengel H:
Classical and non-classical MHC I molecule manipulation by human
cytomegalovirus: So many targets-but how many arrows in the quiver?
Cell Mol Immunol. 12:139–153. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Groh V, Rhinehart R, Secrist H, Bauer S,
Grabstein KH and Spies T: Broad tumor-associated expression and
recognition by tumor-derived gamma delta T cells of MICA and MICB.
Proc Natl Acad Sci USA. 96:6879–6884. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baltz KM, Krusch M, Baessler T, Schmiedel
BJ, Bringmann A, Brossart P and Salih HR: Neutralization of
tumor-derived soluble glucocorticoid-induced TNFR-related protein
ligand increases NK cell anti-tumor reactivity. Blood.
112:3735–3743. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia
Y, Kappes JC, Barnes S, Kimberly RP, Grizzle WE, et al: Murine
mammary carcinoma exosomes promote tumor growth by suppression of
NK cell function. J Immunol. 176:1375–1385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Buggins AG, Milojkovic D, Arno MJ, Lea NC,
Mufti GJ, Thomas NS and Hirst WJ: Microenvironment produced by
acute myeloid leukemia cells prevents T cell activation and
proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways.
J Immunol. 167:6021–6030. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Urosevic M and Dummer R: HLA-G and IL-10
expression in human cancer-different stories with the same message.
Semin Cancer Biol. 13:337–342. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maki G, Klingemann HG, Martinson JA and
Tam YK: Factors regulating the cytotoxic activity of the human
natural killer cell line, NK-92. J Hematother Stem Cell Res.
10:369–383. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin L, Zeng X, Liu M, Deng Y and He N:
Current progress in gene delivery technology based on chemical
methods and nano-carriers. Theranostics. 4:240–255. 2014.
View Article : Google Scholar : PubMed/NCBI
|