Introduction
The epithelial-to-mesenchymal transition (EMT) is a
physiological process that occurs during embryogenesis and wound
healing in adults (1). During
cancer progression, EMT appears to promote dissemination of cells
from the tumor mass, facilitating tissue invasion and metastasis
(2). Epithelial ovarian cancer is
the most common cause of death from gynecologic tumors worldwide;
most cases are detected at advanced stages, which are characterized
by diverse metastatic lesions (3).
TLRs are usually expressed in immune cells, such as
macrophages and dendritic cells (4). However, in addition to their
expression in leukocytes, TLRs are found in multiple tumor types,
including in ovarian cancer (5).
Stimulation of specific TLRs is associated with carcinogenesis,
cancer progression, and site-specific metastasis (6,7).
Expression of TLR2, 3, 4, and 5 has been detected on the surface
epithelium of normal ovaries and benign and malignant ovarian
cancers (8). These data suggest
that the TLR-mediated signaling pathway plays a role in EMT and
metastasis of ovarian cancer; however, the underlying mechanisms of
ovarian cancer metastasis related to TLR stimulation remain
unclear.
The phosphatidylinositol 3-kinase (PI3K)/AKT pathway
is a critical signaling cascade in the progression and drug
resistance of human cancer cells (9,10).
TLR4-expressing metastatic colorectal cancer cells activate
PI3K/AKT signaling and metastatic capacity after stimulation with
lipopolysaccharide (LPS) (11). The
phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit α
(PIK3CA) gene encodes the catalytic subunit p110α of PI3K
class IA, which forms dimers with the regulatory subunit p85 of the
same enzyme (12).
Over-representation of the PIK3CA gene is one of the most
frequently reported abnormalities in ovarian carcinogenesis and is
thought to be an early event (13).
The p110β isoform is also significantly overexpressed both in
ovarian cancer patients and paclitaxel-resistant ovarian cancer
cell lines (14). However, whether
TLR-mediated PI3K-dependent signaling is correlated with invasive
capacity dependent of cancer stage and which downstream signaling
molecules are triggered in TLR-stimulated ovarian cancer cells
remain unclear.
In this study, we investigated the difference in
TLR-mediated PI3K signaling activity according to ovarian cancer
type using primary (Caov-3) and metastatic (SK-OV-3) ovarian cancer
cell lines (15). We also examined
which catalytic isoforms of class IA PI3Ks (p110α, p110β, and
p110δ) are involved in ovarian cancer metastasis.
Materials and methods
Cell lines
The human ovarian cancer cell lines Caov-3, OVCAR-3,
OV-90, and SK-OV-3 were purchased from the ATCC (Manassas, VA,
USA). Caov-3 and OVCAR-3 cells, representing primary cancer, were
harvested from human ovarian adenocarcinoma confined to the ovary.
OV-90 and SK-OV-3 cells, as representative metastatic cancer, were
established from ascites derived from ovarian cancer patients.
Caov-3 and OV-90 cells were maintained in DMEM medium (Corning
Incorporated, Corning, NY, USA) supplemented with 10% FBS (RMBIO,
Missoula, MT, USA), penicillin, streptomycin, and glutamine at 37°C
in 5% CO2. OVCAR-3 and SK-OV-3 cells were maintained in
RPMI-1640 medium (Corning Inc.) supplemented with 10% FBS (RMBIO),
penicillin, streptomycin, and glutamine at 37°C in 5%
CO2.
Chemicals
LPS (TLR4 ligand) and poly (I:C) (TLR3 ligand) were
obtained from Sigma-Aldrich (St. Louis, MO, USA). MALP-2 (TLR2/6
ligand) was purchased from Enzo Life Sciences (Farmingdale, NY,
USA). PP1 (Src inhibitor) and Bay 61–3606 (Syk inhibitor) were
purchased from Calbiochem (San Diego, CA, USA). A66 (p110α
inhibitor), TGX-221 (p110β inhibitor), CAL-101 (p110δ inhibitor),
LY294002 (pan-p110 inhibitor), Bay 80–6946 (p110α and p110β
inhibitor), and pictilisib (p110α and p110δ inhibitor) were
obtained from Selleckchem (Houston, TX, USA). Recombinant Gal-1 was
purchased from R&D Systems (Minneapolis, MN, USA).
RT-PCR
Total RNA was isolated using an RNeasy Mini kit
(Qiagen, Hilden, Germany). RNA was transcribed into cDNA using
oligo (dT) primers (Bioneer, Daejeon, Korea) and reverse
transcriptase. To investigate the expression of TLR genes in
ovarian cancer cells, PCR amplification was performed using
specific primer sets (Table I;
Bioneer) and Prime Taq Premix (GeNet Bio, Chungnam, Korea). PCR
products were analyzed via agarose gel electrophoresis and
visualized with ethidium bromide under UV light using the Multiple
Gel DOC system (Fujifilm, Tokyo, Japan). Data were analyzed using
ImageJ 1.38 software (National Institutes of Health, Bethesda, MD,
USA). Experiments were performed in triplicate.
 | Table I.Specific primer sequences used for
RT-PCR. |
Table I.
Specific primer sequences used for
RT-PCR.
|
| Primers, 5′→3′ |
|---|
|
|
|
|---|
| Target | Sense | Antisense |
|---|
| TLR1 |
CGTAAAACTGGAAGCTTGCAAGA |
CCTTGGGCCATTCCAAATAAGTCC |
| TLR2 |
GGCCAGCAAATTACCTGTGTG |
CCAGGTAGGTCTTGGTGTTCA |
| TLR3 |
ATTGGGTCTGGGAACATTTCTCTTC |
GTGAGATTTAAACATTCCTCTTCGC |
| TLR4 |
CTGCAATGGATCAAGGACCA |
TCCCACTCCAGGTAAGTGTT |
| TLR5 |
CATTGTATGCACTGTCACTC |
CCACCACCATGATGAGAGCA |
| TLR6 |
TAGGTCTCATGACGAAGGAT |
GGCCACTGCAAATAACTCCG |
| TLR7 |
AGTGTCTAAAGAACCTGG |
CTTGGCCTTACAGAAATG |
| TLR8 |
CAGAATAGCAGGCGTAACACATCA |
AATGTCACAGGTGCATTCAAAGGG |
| TLR9 |
TTATGGACTTCCTGCTGGAGGTGC |
CTGCGTTTTGTCGAAGACCA |
| TLR10 |
CAATCTAGAGAAGGAAGATGGTTC |
GCCCTTATAAACTTGTGAAGGTGT |
| β-actin |
ATCCACGAAACTACCTTCAA |
ATCCACACGGAGTACTTGC |
Western blotting
Cells were washed in PBS and lysed in NP-40 buffer
(Elpis Biotech, Daejeon, Korea) supplemented with a protease
inhibitor cocktail (Sigma-Aldrich). Protein phosphorylation states
were preserved through the addition of phosphatase inhibitors
(Cocktail II, Sigma-Aldrich) to the NP-40 buffer. Protein
concentrations were determined using a BCA assay kit (Pierce,
Rockford, IL, USA). Proteins (10 µg/sample) were resolved through
SDS-PAGE and then transferred to a nitrocellulose membrane
(Millipore Corp., Billerica, MA, USA). Membranes were blocked with
5% skim milk prior to western blot analysis. Chemiluminescence was
detected using an ECL kit (Advansta Corp., Menlo Park, CA, USA) and
the Multiple Gel DOC system (Fujifilm). The following primary
antibodies were used: E-cadherin, N-cadherin, Slug, Snail,
Vimentin, α-SMA, TCF8/Zeb1, β-actin, MMP2, MMP9, Myd88, p110α,
p110β, p110γ, p110δ, phosphor-p65, p65, Fyn, phospho-Lyn
(Tyr507), Lyn, phospho-Src (Tyr416), Src,
phospho-Syk (Tyr323), phospho-Syk
(Tyr525/526), and Syk (Cell Signaling Technology,
Beverly, MA, USA); α-SMA (Bioss, Woburn, MA, USA); and TLR2, TLR3,
TLR4, TLR5, TLR6, TLR7, galectin-1, phospho-Fyn (Thr12),
phospho-PTEN (Ser380/Thr382/383), and PTEN
(Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Small interfering RNA (siRNA)
transfection
Experimentally verified human TLR4- and
galectin-1-small interfering RNA (siRNA) duplex and negative
control-siRNA were obtained from Bioneer. Cells were seeded at a
concentration of 1×105 per well in a 96-well plate and
grown overnight. Cells were then transfected with 200 nM siRNA
using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer's instructions. Cells were used for
further experiments 48 h after transfection.
Quantification of human cytokines by
ELISA
A galectin-1 ELISA assay was performed as previously
described (16) using capture Ab
(200 ng/ml, 100 µl/well; AF1152; R&D Systems), detection Ab
(100 ng/ml, 100 µl/well; BAF1152; R&D Systems), and standard
recombinant galectin-1 (6–500 pg; R&D Systems). Active TGF-β1,
TNF-α, VEGF, IL-6, IL-8, and IL-10 were quantified using a single
cytokine ELISA assay kit (R&D Systems). Data are expressed as
the average of the biological replicates ± standard deviation
(SD).
Invasion assay
Invasion assay was performed using the CultreCoat
96-well Medium BME Cell Invasion assay kit (R&D Systems)
according to the manufacturer's protocol. Cells
(2.5×104) in serum-free RPMI-1640 or DMEM containing
0.1% FBS were seeded into the upper chamber, and the lower
compartment was filled with RPMI-1640 or DMEM containing 10% FBS as
a chemoattractant. After incubation for 24 h, non-invading cells on
the upper membrane surface were removed with a cotton swab. Invaded
cells were stained with calcein-AM and quantified using a
microplate reader.
Statistical analysis
Data are expressed as the mean ± standard deviation
(SD). Statistical analysis was conducted using one-way analysis of
variance. A p-value <0.05 was considered statistically
significant.
Results
Stimulation with TLR agonist induces
mesenchymal characteristics and invasion activity of SK-OV-3
cells
Using RT-PCR and western blots, we evaluated the
expression of TLRs in both primary (Caov-3 and OVCAR-3) and
metastatic (OV-90 and SK-OV-3) ovarian cancer cells. mRNA levels of
TLR2, 3, 4, 5, 6, 7, and 8 in SK-OV-3 cells were significantly
elevated compared to other ovarian cancer cells (Fig. 1A). The protein levels of TLR2, 3, 4,
5, and 6 in SK-OV-3 cells were also upregulated compared to Caov-3
cells (Fig. 1B). Next, we
investigated the effects of TLR2/6 agonist macrophage-activating
lipopeptide 2 (MALP-2), TLR4 agonist lipopolysaccharide (LPS), and
TLR3 agonist polyinosinic-polycytidylic acid (poly I:C) on changes
in EMT-related markers and invasion capacity in primary (Caov-3)
and metastatic (SK-OV-3) ovarian cancer cells. TLR activation with
various TLR agonists considerably increased mesenchymal
characteristics (N-cadherin, Slug, Vimentin, Snail, α-SMA, and
TCF8) in SK-OV-3 cells compared to Caov-3 cells (Fig. 1C). In addition, LPS stimulation of
SK-OV-3 cells increased the population of cells with spindle-shaped
morphology compared to LPS-treated Caov-3 cells (Fig. 1D). All TLR agonists significantly
increased the migratory capacity of SK-OV-3 cells, but not Caov-3
cells (Fig. 1E). These results
suggest that TLR-mediated signaling influences the migratory
capacity of metastatic ovarian cancer cell line SK-OV-3.
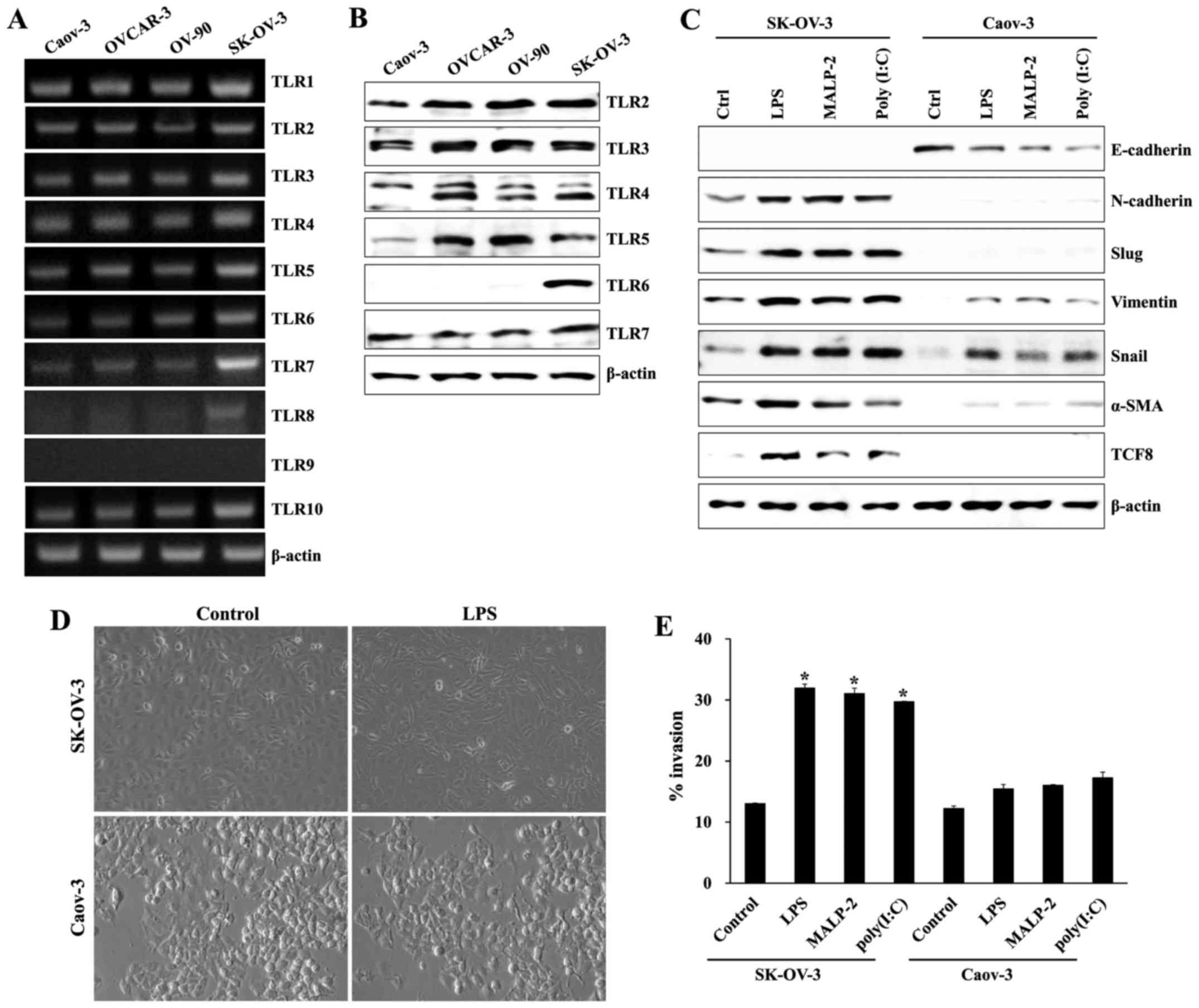 | Figure 1.TLR expression and the effects of TLR
agonist stimulation on ovarian cancer cells. (A) mRNA expression of
TLR1, 2, 3, 4, 5, 6, 7, 8, 9 and 10 in ovarian cancer cells was
measured using RT-PCR. (B) Protein expression levels of TLR2, 3, 4,
5, 6 and 7 in ovarian cancer cells were measured using western
blotting. β-actin was used as a loading control. (C) Cells
(1.5×105/well) were cultured with and without TLR
agonist [poly (I:C), LPS, or MALP2], and levels of EMT markers
(E-cadherin, Snail, α-SMA, Slug, TCF8, N-cadherin, and Vimentin)
were determined using western blot analysis. (D) Comparison of
morphologic changes in LPS-stimulated ovarian cancer cells.
Photographs were taken at ×100 magnification using a digital camera
under an inverted phase-contrast microscope (Olympus). (E) The
invasiveness of SK-OV-3 cells was enhanced by TLR agonists [poly
(I:C), LPS, and MALP2], as determined by a BME cell invasion assay
kit. Each value represented the mean ± standard deviation of three
measurements. *p<0.01 (SK-OV-3 vs. Caov-3). Results are
representative of three independent experiments. |
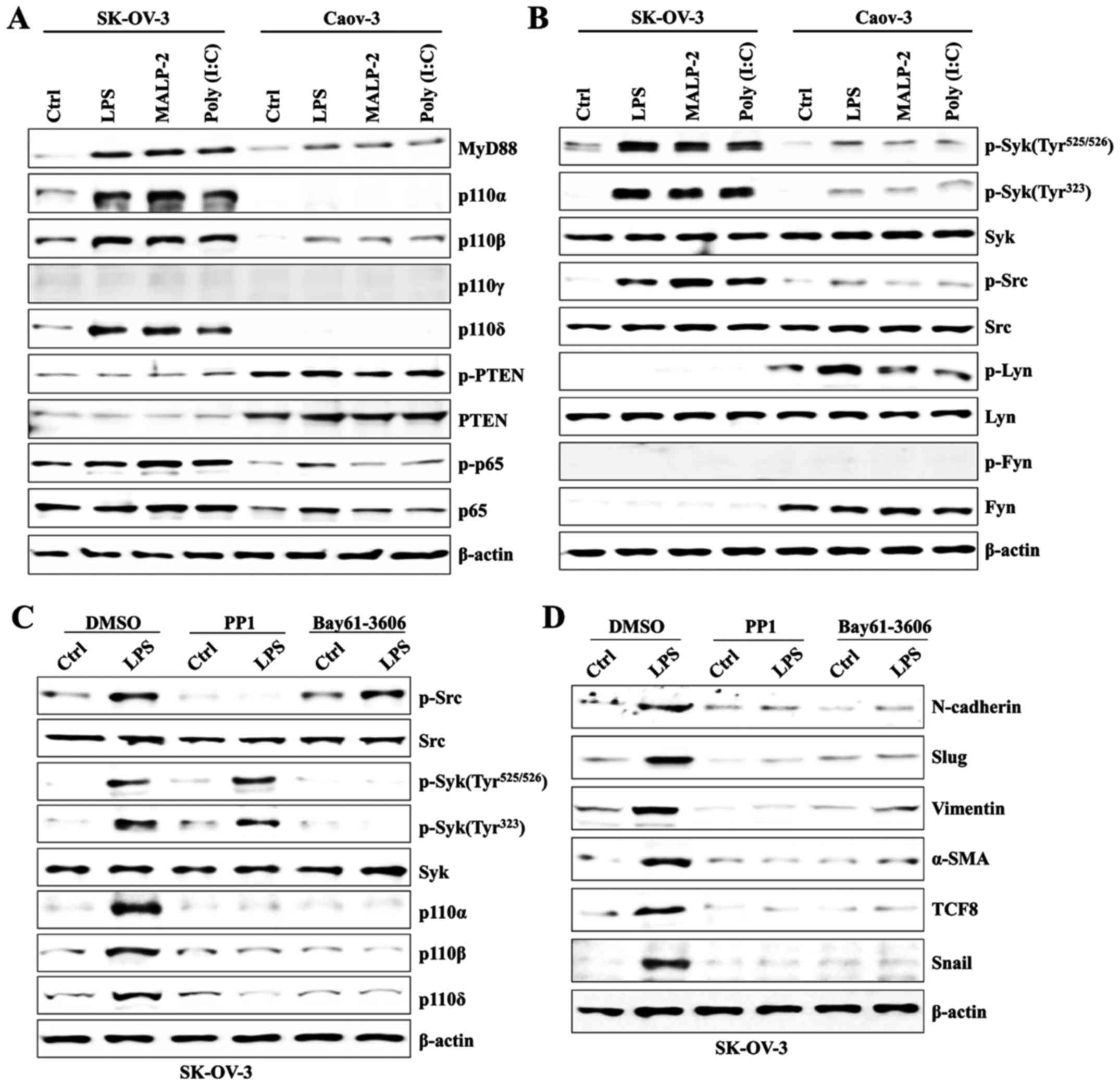
Syk/Src-dependent PI3K activation
induces mesenchymal markers in TLR-stimulated SK-OV-3 cells
Next, we investigated whether TLR stimulation with
specific ligands induces activation of PI3K. We also examined Syk-
and Src-family tyrosine kinase activity because PI3K signaling is
modulated via those tyrosine kinases (16,17).
The expression of class IA PI3Ks (p110α, p110β, and p110δ) in
SK-OV-3 cells was upregulated, as was the expression of MyD88 after
stimulation with several TLR ligands; however, MyD88 and the p110β
isoform were slightly increased in Caov-3 after engagement with
various TLR agonists (Fig. 2A).
Although the activity of Syk and Src tyrosine kinases was elevated
in LPS-activated SK-OV-3 cells (Fig.
2B), treatment with PP1 (an Src-specific inhibitor) and
Bay61-3606 (a Syk-specific inhibitor) efficiently blocked the
phosphorylation of tyrosine kinase and the activation of class IA
PI3Ks (Fig. 2C). In addition,
pharmacological inhibition of Syk and Src tyrosine kinases
significantly reduced the expression of EMT-related proteins
(Fig. 2D). Our data suggest that
Syk/Src-mediated PI3K signal transduction plays an important role
in TLR-induced EMT processes of SK-OV-3 cells.
TLR-mediated PI3K activation promotes
the secretion of EMT-related cytokines in SK-OV-3 cells
We found that SK-OV-3 cells stimulated by TLR
agonist activated class IA PI3Ks (p110α, p110β, and p110δ). Next,
we investigated the effects of specific PI3K inhibition on invasion
and secretion of EMT-related cytokines in LPS-stimulated SK-OV-3
cells. Pretreatment with a specific inhibitor (A66, p110α
inhibitor; TGX-221, p110β inhibitor; CAL-101, p110δ inhibitor;
LY294002, p110α/β/δ inhibitor; Pictilisib, p110α/δ inhibitor) for
each p110 isoform increased the expression of E-cadherin and
decreased the expression of mesenchymal markers in LPS-activated
SK-OV-3 cells (Fig. 3A). The
migratory activity of LPS-stimulated SK-OV-3 cells was
significantly suppressed after treatment with all p110 isoform
inhibitors (Fig. 3B). In addition,
TGX-221 (p110β inhibitor) and Bay-80-6946 (p110α/β inhibitor) had a
prominent influence on invasion attenuation (Fig. 3B). EMT, which is driven by a number
of growth factors and cytokines, results in loss of epithelial cell
polarities and connections (18).
To determine EMT-related cytokine secretion in ovarian cancer cells
after TLR stimulation, we compared the concentrations of
transforming growth factor β-1 (TGF-β1), vascular endothelial
growth factor (VEGF), interleukin-6 (IL-6), IL-8, IL-10, and tumor
necrosis factor-α (TNF-α) using a sandwich ELISA method with the
culture media in TLR agonist-triggered SK-OV-3 and Caov-3 cells.
TGF-β1 and TNF-α, critical cytokines for EMT, were markedly
increased in the culture media of SK-OV-3 cells after stimulation
with various TLR agonists, whereas those cytokines were barely
detectable in the Caov-3 culture media (Fig. 3C). Other cytokines (VEGF, IL-6,
IL-8, and IL-10) associated with invasive activity were also
~3-5-fold higher than those in non-TLR stimulated SK-OV-3 (Fig. 3C). Although all p110 isoform
inhibitors prevented the secretion of EMT-related cytokines,
treatment with TGX-221 or Bay-80-6946 prevented the secretion of
EMT-related cytokines most effectively, except for production of
IL-10, which was affected by CAL-101 or Pictilisib (Fig. 3D). Our data suggest that
TLR-mediated PI3K signaling influences the migratory capacity and
secretion of EMT-related cytokines in metastatic ovarian cancer
cells.
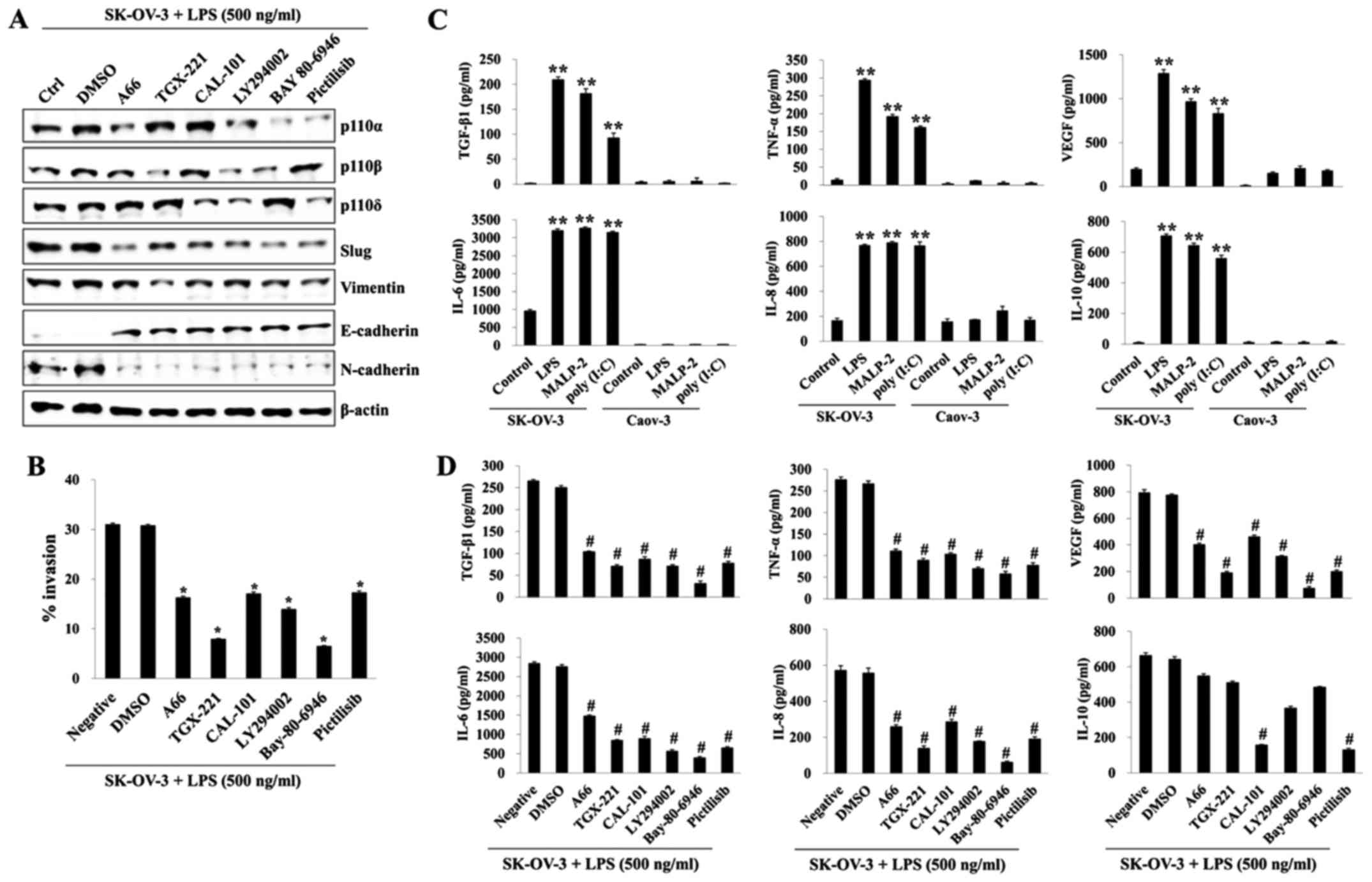 | Figure 3.TLR-induced PI3K activation promotes
invasion and EMT-related cytokine production in SK-OV-3 cells.
SK-OV-3 cells (1.5×105/well) were pretreated with p110α
inhibitor A66 (50 µM), p110β inhibitor TGX-221 (50 µM), p110δ
inhibitor CAL-101 (50 µM), Pan PI3K inhibitor LY294002 (50 µM),
p110α/β inhibitor Bay80-6946 (200 nM), or p110α/δ inhibitor
Pictilisib (200 nM) for 2 h. Then cells were treated with LPS (500
ng/ml) for 24 h. (A) Total cell lysates were immunoblotted with the
indicated antibodies. β-actin served as the loading control. (B)
The invasiveness of SK-OV-3 cells was inhibited by A66, TGX-221,
CAL-101, LY294002, Bay80-6946, or Pictilisib as detected by BME
cell invasion assay. Each value represents the mean ± SD of three
measurements. *p<0.01 (SK-OV-3+LPS vs. the group treated with
each inhibitor). (C) TLR agonists [poly (I:C), LPS, and MALP2]
induced the secretion of IL-8, IL-10, VEGF, IL-6, TNF-α, or active
TGF-β1. Cells were seeded onto 6-well plates
(1.5×105/well) and incubated overnight. Culture
supernatants were collected at 24 h after TLR agonist [poly (I:C),
LPS, and MALP2] stimulation, and the amounts of IL-8, IL-10, VEGF,
IL-6, TNF-α, or active TGF-β1 were determined by ELISA. Data are
representative of two independent experiments and show the mean ±
standard deviation of duplicate determinations. **p<0.001
(SK-OV-3 vs. Caov-3). (D) SK-OV-3 cells (1.5×105/well)
were pretreated with p110α inhibitor A66 (50 µM), p110β inhibitor
TGX-221 (50 µM), p110δ inhibitor CAL-101 (50 µM), Pan PI3K
inhibitor LY294002 (50 µM), p110α/β inhibitor Bay80-6946 (200 nM),
or p110α/δ inhibitor Pictilisib (200 nM) for 2 h, and then the
cells were stimulated with LPS for 24 h. The amounts of IL-8,
IL-10, VEGF, IL-6, TNF-α, or active TGF-β1 in culture supernatant
were determined by ELISA. Data are representative of two
independent experiments and show the mean ± standard deviation of
duplicate measurements. #p<0.005 (SK-OV-3+LPS vs. the
group treated with each inhibitor). Results are representative of
three independent experiments. |
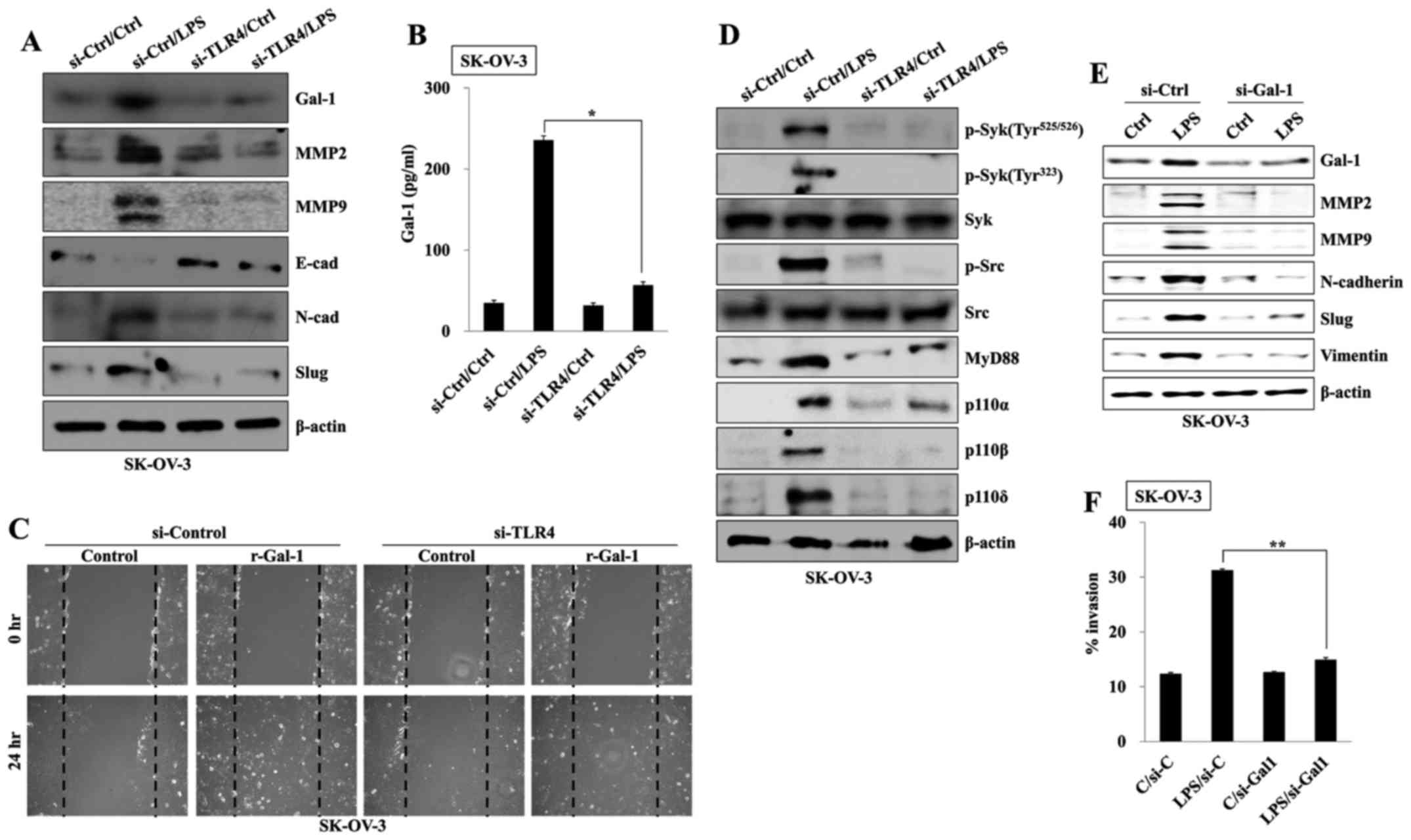
TLR4-mediated PI3K activation promotes
galectin-1 production in LPS-stimulated SK-OV-3
Galectin-1 is involved in cancer progression and is
associated with a poor prognosis in ovarian cancers (19). We determined the effects of TLR
agonists on the production of galectin-1 and investigated the
association between activation of PI3K and galectin-1 in ovarian
cancer cells. TLR activation using a specific ligand significantly
increased the expression of galectin-1, MMP2, and MMP9 in SK-OV-3
cells compared to Caov-3 cells (Fig. 4A
and B). Expression in LPS-stimulated SK-OV-3 was suppressed
after treatment with an inhibitor of Src (PP1) or Syk (Bay-61-3606)
tyrosine kinase (Fig. 4C).
Pharmacological inhibition of various p110 isoforms also prevented
the expression of galectin-1 (Fig.
4D). In particular, CAL-101 (p110δ inhibitor) and Pictilisib
(p110α/δ inhibitor) markedly blocked the production of galectin-1
in LPS-activated SK-OV-3 cells compared to the level of other p110
inhibitors (Fig. 4E). Our data
suggest that PI3K-dependent galectin-1 production is one of the
main pathways of ovarian cancer metastasis after TLR
stimulation.
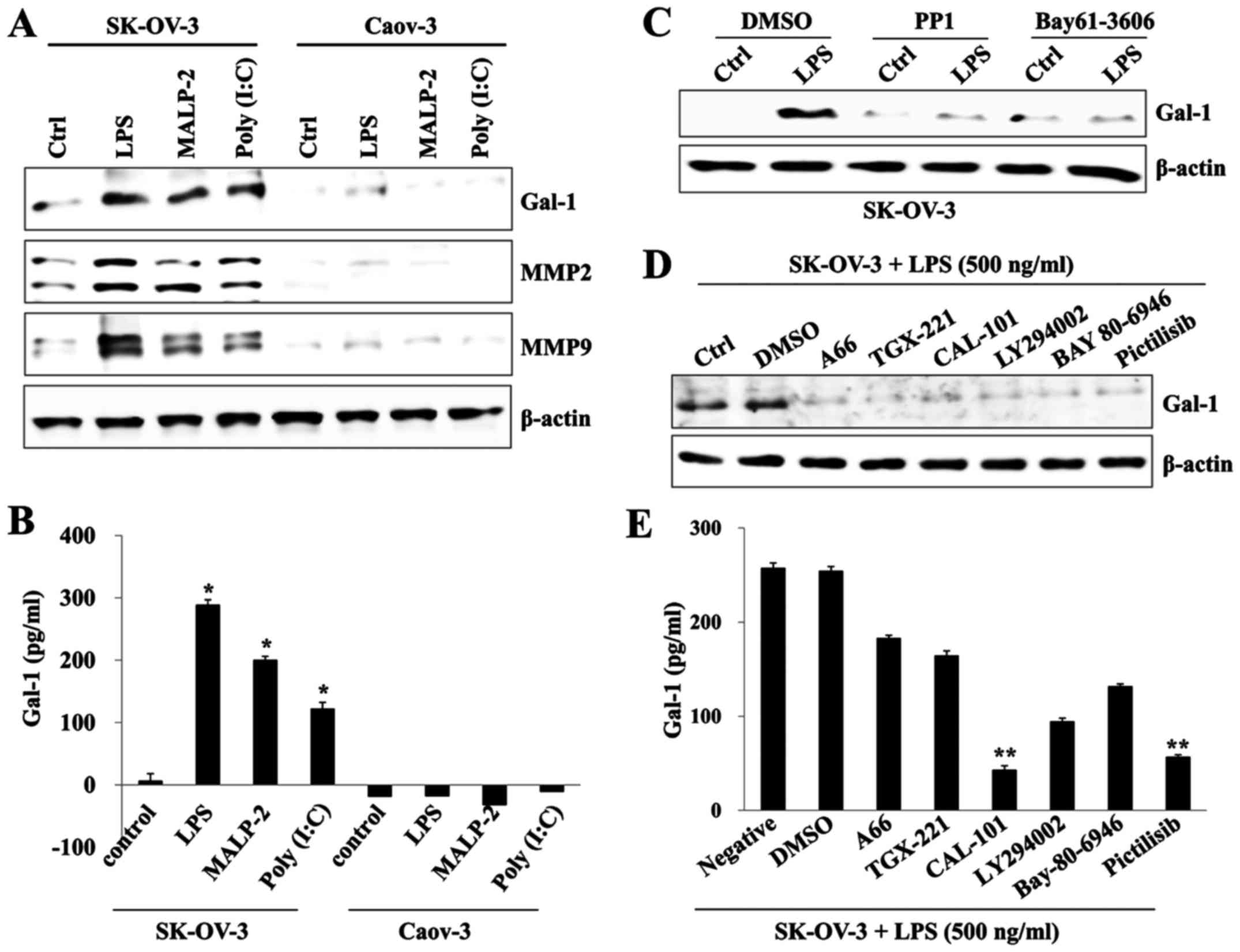 | Figure 4.TLR-stimulated PI3K signaling
regulates galectin-1 production in SK-OV-3 cells. (A) Cells
(1.5×105/well) were cultured with or without TLR3
agonist poly (I:C) (10 µg/ml), TLR4 agonist LPS (500 ng/ml), or
TLR2/6 agonist MALP2 (1 µg/ml) for 24 h. Total cell lysates were
immunoblotted with antibody against galectin-1 (Gal-1), MMP2, or
MMP9. (B) Culture supernatant was collected 24 h after TLR agonist
(poly (I:C), LPS, and MALP2) stimulation, and the amounts of
galectin-1 (Gal-1) were determined by ELISA. Data are
representative of two independent experiments and show the mean ±
standard deviation of duplicate measurements. *p<0.001 (SK-OV-3
vs. Caov-3). (C) Cells were pretreated with Src inhibitor PP1 (200
nM) or Syk inhibitor BAY-61-3606 (200 nM) and then treated with LPS
(500 ng/ml) for 24 h. Total cell lysates were immunoblotted with
antibody against galectin-1 (Gal-1). β-actin served as the loading
control. (D) Cells were pretreated with p110α inhibitor A66 (50
µM), p110β inhibitor TGX-221 (50 µM), p110δ inhibitor CAL-101 (50
µM), Pan PI3K inhibitor LY294002 (50 µM), p110α/β inhibitor
Bay80-6946 (200 nM), or p110α/δ inhibitor Pictilisib (200 nM) for 2
h and then treated with LPS (500 ng/ml) for 24 h. Total cell
lysates were immunoblotted with antibody against galectin-1
(Gal-1). β-actin was used as the loading control. (E) Culture
supernatant was collected 24 h after LPS stimulation, and the
amount of galectin-1 (Gal-1) was determined by ELISA. Data are
representative of two independent experiments and show the mean ±
standard deviation of duplicate measurements. **p<0.005
(SK-OV-3+LPS vs. the group treated with CAL-101 or Pictilisib). |
TLR4-mediated galectin-1 production
induces EMT of ovarian cancer cells
Next, we investigated whether a TLR4-induced
signaling pathway regulates galectin-1-mediated migration of
ovarian cancer cells. Stimulation with LPS of TLR4-knockdown
SK-OV-3 cells failed to increase the production of galectin-1 and
the expression of MMP2, MMP9, and mesenchymal markers (Figs. 5A and B). Treatment with recombinant
galectin-1 (r-Gal-1) enhanced the wound healing capacity of
TLR4-knockdown SK-OV-3 cells regardless of TLR4 expression or LPS
stimulation (Fig. 5C). In addition,
targeted inhibition of TLR4 blocked the phosphorylation of Src/Syk
kinase and the activation of PI3K in LPS-activated HCT116 cells
(Fig. 5D). Finally, we investigated
whether TLR4-mediated galectin-1 production affects migration and
the expression of mesenchymal markers. Downregulation of galectin-1
with small interfering RNA attenuated the activation of MMP2 and
MMP9, and the upregulation of mesenchymal markers in LPS-stimulated
SK-OV-3 cells (Fig. 5E).
Furthermore, the invasion capacity of galectin-1-knockdown SK-OV-3
cells was profoundly suppressed after stimulation with LPS
(Fig. 5F). These results suggest
that TLR4-mediated PI3K activation triggers the migratory and
invasive capacity of ovarian cancer cells through the production of
galectin-1.
Discussion
TLR activation in leukocytes at the tumor site can
stimulate immune cells that can actively fight tumors (20,21);
however, their activation can also have tumor-promoting effects
(22). Generally, high levels of
different TLRs in cancer cells are associated with disease
aggressiveness, drug resistance, and poor clinical outcomes
(23,24). Administration of paclitaxel with a
platinum regimen via an intravenous or intraperitoneal route is the
standard chemotherapy for ovarian cancer (25). Unfortunately, paclitaxel stimulates
the same signaling pathway via TLR4 to promote secretion of
pro-inflammatory cytokines (26).
LPS or paclitaxel binding to TLR4 in SK-OV-3 cells promotes IL-6,
IL-8, and VEGF production and resistance to drug-induced apoptosis
(27). In addition, our study
showed that the expression of TLR4 was more prominent in SK-OV-3
than in Caov-3 cells and stimulation with various TLR ligands
induced higher expression of mesenchymal markers and invasive
activity in SK-OV-3 than in Caov-3 cells. These results suggest
that remnant or resistant ovarian cancer cells could use this
anticancer drug to promote survival and growth after chemotherapy.
Based on these results, TLR levels and expression changes might be
critical diagnostic markers in metastatic ovarian cancer.
Stimulation of TLR4 with LPS results in an immediate
interaction between PI3K and MyD88, leading to the phosphorylation
of AKT (28). Aberrant activation
of this pathway has been widely reported in many human cancers,
including ovarian cancer (29).
Meanwhile, phosphatase and tensin homolog (PTEN), a negative
regulator of the AKT signaling pathway, delays the time to
progression in ovarian carcinomas and prolongs disease-free
survival (30). We observed that
Syk/Src-dependent class IA (p110α, p110β, and p110δ) PI3K
activation and expression of mesenchymal markers were significantly
increased in TLR-stimulated SK-OV-3 cells; however, the level of
PTEN in Caov-3 cells was maintained at a high level after
stimulation with TLR agonist. These results suggest that metastatic
or invasive ovarian cancer cell lines including SK-OV-3 cells are
sensitive to stimulation by TLR ligands, which could lead to the
development of aggressive phenotypes.
Although p110α expression is frequently observed in
ovarian cancer (13), the levels of
p110β, but not p110α, in ovarian cancer cells are critical factors
of drug resistance (14). Other
studies also have shown that manifestation of p110α has no clear
relationship with clinical outcome (31). SK-OV-3 cells secrete a higher level
of VEGF than Caov-3 cells (32).
TLR4-mediated class I PI3K activation in LPS-mediated SK-OV-3 cells
is positively correlated with the secretion of EMT-related
cytokines in SK-OV-3 cells. Interestingly, selective inhibitor of
p110α or p110β significantly reduced EMT-related cytokines and
targeted co-inhibition of p110α/β was the most effective method of
reducing EMT-inducing cytokines (TGF-β1, TNF-α, VEGF, IL-6 and
IL-8), except IL-10. IL-10 production was efficiently blocked by
CAL-101, a p110δ-specific inhibitor, compared to that of
LPS-activated SK-OV-3 cells treated with other inhibitors of the
p110 isoform. These results suggest that co-administration of
anticancer drugs with selective PI3K inhibitors during repeated
chemotherapy cycles according to serum cytokine levels in patients
could reduce the possibility of metastasis. Furthermore, we need to
investigate the signaling pathway to identify precise targets of
each condition and need to classify activated p110 isoform- and
EMT-associated cytokines in metastatic cancer cells for clinical
applications.
Galectin-1 is considered a prototypical galectin and
is expressed in many tumor types such as astrocytoma, melanoma, and
prostate, thyroid, colon, bladder, and ovarian cancers (33,34).
Based on these results, galectin-1 might be a promising candidate
for downstream targeting of the TLR/PI3K-mediated signaling pathway
in metastatic ovarian cancer. Pharmacological inhibition of PI3K
effectively blocked the secretion of galectin-1 in LPS-treated
SK-OV-3 cells. Furthermore, knockdown of TLR4 or galectin-1 with
siRNA not only suppressed the expression of mesenchymal markers
MMP2, and MMP9, but also inhibited invasion activity of
LPS-activated SK-OV-3 cells. These results suggest that
TLR/PI3K-induced galectin-1 production in ovarian cancer cells is
one of the key processes in further invasion and migration.
In conclusion, this study suggests that the TLR/PI3K
signaling axis plays a crucial role in facilitating ovarian cancer
cell metastasis, and the identification of new molecular targets
after TLR engagement is critical for predicting disease progression
and clinical outcome.
Acknowledgements
This study was supported by the Basic Science
Research Program of Ministry of Education (NRF-2015R1D1A1A
01056672) and Ministry of Science, ICT and Future Planning
(NRF-2015R1C1A2A01053732) through the National Research Foundation
(NRF) of Korea.
References
|
1
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sabe H: Cancer early dissemination:
Cancerous epithelial-mesenchymal transdifferentiation and
transforming growth factor β signalling. J Biochem. 149:633–639.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaughan S, Coward JI, Bast RC Jr, Berchuck
A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R,
Etemadmoghadam D, et al: Rethinking ovarian cancer: Recommendations
for improving outcomes. Nat Rev Cancer. 11:719–725. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Akira S, Takeda K and Kaisho T: Toll-like
receptors: Critical proteins linking innate and acquired immunity.
Nat Immunol. 2:675–680. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sato Y, Goto Y, Narita N and Hoon DS:
Cancer cells expressing toll-like receptors and the tumor
microenvironment. Cancer Microenviron. 2:(Suppl 1). 205–214. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chochi K, Ichikura T, Kinoshita M, Majima
T, Shinomiya N, Tsujimoto H, Kawabata T, Sugasawa H, Ono S, Seki S,
et al: Helicobacter pylori augments growth of gastric
cancers via the lipopolysaccharide-toll-like receptor 4 pathway
whereas its lipopolysaccharide attenuates antitumor activities of
human mononuclear cells. Clin Cancer Res. 14:2909–2917. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fukata M, Chen A, Vamadevan AS, Cohen J,
Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, et
al: Toll-like receptor-4 promotes the development of
colitis-associated colorectal tumors. Gastroenterology.
133:1869–1881. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou M, McFarland-Mancini MM, Funk HM,
Husseinzadeh N, Mounajjed T and Drew AF: Toll-like receptor
expression in normal ovary and ovarian tumors. Cancer Immunol
Immunother. 58:1375–1385. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu HG, Ai YW, Yu LL, Zhou XD, Liu J, Li
JH, Xu XM, Liu S, Chen J, Liu F, et al: Phosphoinositide
3-kinase/Akt pathway plays an important role in chemoresistance of
gastric cancer cells against etoposide and doxorubicin induced cell
death. Int J Cancer. 122:433–443. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsu RY, Chan CH, Spicer JD, Rousseau MC,
Giannias B, Rousseau S and Ferri LE: LPS-induced TLR4 signaling in
human colorectal cancer cells increases beta1 integrin-mediated
cell adhesion and liver metastasis. Cancer Res. 71:1989–1998. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shayesteh L, Lu Y, Kuo WL, Baldocchi R,
Godfrey T, Collins C, Pinkel D, Powell B, Mills GB and Gray JW:
PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet.
21:99–102. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sham JS, Tang TC, Fang Y, Sun L, Qin LX,
Wu QL, Xie D and Guan XY: Recurrent chromosome alterations in
primary ovarian carcinoma in Chinese women. Cancer Genet Cytogenet.
133:39–44. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jeong JY, Kim KS, Moon JS, Song JA, Choi
SH, Kim KI, Kim TH and An HJ: Targeted inhibition of phosphatidyl
inositol-3-kinase p110β, but not p110α, enhances apoptosis and
sensitivity to paclitaxel in chemoresistant ovarian cancers.
Apoptosis. 18:509–520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vergara D, Merlot B, Lucot JP, Collinet P,
Vinatier D, Fournier I and Salzet M: Epithelial-mesenchymal
transition in ovarian cancer. Cancer Lett. 291:59–66. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kurosaki T, Takata M, Yamanashi Y, Inazu
T, Taniguchi T, Yamamoto T and Yamamura H: Syk activation by the
Src-family tyrosine kinase in the B cell receptor signaling. J Exp
Med. 179:1725–1729. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Beitz LO, Fruman DA, Kurosaki T, Cantley
LC and Scharenberg AM: SYK is upstream of phosphoinositide 3-kinase
in B cell receptor signaling. J Biol Chem. 274:32662–32666. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim HJ, Jeon HK, Cho YJ, Park YA, Choi JJ,
Do IG, Song SY, Lee YY, Choi CH, Kim TJ, et al: High galectin-1
expression correlates with poor prognosis and is involved in
epithelial ovarian cancer proliferation and invasion. Eur J Cancer.
48:1914–1921. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC,
Martinez DG, Engle X, Gewirtz AT, Ahonen CL and Conejo-Garcia JR:
In situ stimulation of CD40 and Toll-like receptor 3 transforms
ovarian cancer-infiltrating dendritic cells from immunosuppressive
to immunostimulatory cells. Cancer Res. 69:7329–7337. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scarlett UK, Rutkowski MR, Rauwerdink AM,
Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC,
Gonzalez JL, Weaver J, et al: Ovarian cancer progression is
controlled by phenotypic changes in dendritic cells. J Exp Med.
209:495–506. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu WT, Jing YY, Yu GF, Han ZP, Yu DD, Fan
QM, Ye F, Li R, Gao L, Zhao QD, et al: Toll like receptor 4
facilitates invasion and migration as a cancer stem cell marker in
hepatocellular carcinoma. Cancer Lett. 358:136–143. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He W, Liu Q, Wang L, Chen W, Li N and Cao
X: TLR4 signaling promotes immune escape of human lung cancer cells
by inducing immunosuppressive cytokines and apoptosis resistance.
Mol Immunol. 44:2850–2859. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kelly MG, Alvero AB, Chen R, Silasi DA,
Abrahams VM, Chan S, Visintin I, Rutherford T and Mor G: TLR-4
signaling promotes tumor growth and paclitaxel chemoresistance in
ovarian cancer. Cancer Res. 66:3859–3868. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alberts DS, Liu PY, Hannigan EV, O'Toole
R, Williams SD, Young JA, Franklin EW, Clarke-Pearson DL, Malviya
VK, DuBeshter B, et al: Intraperitoneal cisplatin plus intravenous
cyclophosphamide versus intravenous cisplatin plus intravenous
cyclophosphamide for stage III ovarian cancer. N Engl J Med.
335:1950–1955. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Byrd-Leifer CA, Block EF, Takeda K, Akira
S and Ding A: The role of MyD88 and TLR4 in the LPS-mimetic
activity of Taxol. Eur J Immunol. 31:2448–2457. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Szajnik M, Szczepanski MJ, Czystowska M,
Elishaev E, Mandapathil M, Nowak-Markwitz E, Spaczynski M and
Whiteside TL: TLR4 signaling induced by lipopolysaccharide or
paclitaxel regulates tumor survival and chemoresistance in ovarian
cancer. Oncogene. 28:4353–4363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laird MH, Rhee SH, Perkins DJ, Medvedev
AE, Piao W, Fenton MJ and Vogel SN: TLR4/MyD88/PI3K interactions
regulate TLR4 signaling. J Leukoc Biol. 85:966–977. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kang S, Bader AG and Vogt PK:
Phosphatidylinositol 3-kinase mutations identified in human cancer
are oncogenic. Proc Natl Acad Sci USA. 102:802–807. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schöndorf T, Göhring UJ, Roth G, Middel I,
Becker M, Moser N, Valter MM and Hoopmann M: Time to progression is
dependent on the expression of the tumour suppressor PTEN in
ovarian cancer patients. Eur J Clin Invest. 33:256–260. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Kristensen GB, Helland A, Nesland
JM, Børresen-Dale AL and Holm R: Protein expression and prognostic
value of genes in the erb-b signaling pathway in advanced ovarian
carcinomas. Am J Clin Pathol. 124:392–401. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sher I, Adham SA, Petrik J and Coomber BL:
Autocrine VEGF-A/KDR loop protects epithelial ovarian carcinoma
cells from anoikis. Int J Cancer. 124:553–561. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Satelli A and Rao US: Galectin-1 is
silenced by promoter hypermethylation and its re-expression induces
apoptosis in human colorectal cancer cells. Cancer Lett. 301:38–46.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ito K and Ralph SJ: Inhibiting galectin-1
reduces murine lung metastasis with increased CD4(+) and CD8(+) T
cells and reduced cancer cell adherence. Clin Exp Metastasis.
29:561–572. 2012. View Article : Google Scholar : PubMed/NCBI
|