Introduction
Hepatocellular carcinoma (HCC) is the fifth most
commonly diagnosed cancer and second most frequent cause of
cancer-related deaths for males worldwide. In women, it is the
seventh most commonly diagnosed cancer and the sixth leading cause
of cancer-related deaths (1). HCC
is associated with a poor prognosis due to late diagnosis and a
lack of effective treatment options. Although great advances in
surgical techniques and medical care have been achieved over the
last several decades, the 5-year survival rate worldwide of HCC is
still less than 5%, mainly due to the high rate of recurrence and
metastasis (2). In fact, increasing
evidence suggests that HCC metastasis is a multistep process.
During progression to metastasis, cancer cells are thought to
acquire a mesenchymal phenotype, which allows them to leave the
site of the primary tumor, invade surrounding tissues, and migrate
to distant organs. After seeding, these cells switch back to an
epithelial phenotype and proliferate to form metastases (3,4). The
process by which cells switch from epithelial-mesenchymal (EMT)
phenotypes is known as EMT transition (5). EMT is a critical prognostic factor in
HCC and is involved in early recurrence or metastases after surgery
(6). Blocking EMT is currently
considered as a promising strategy to inhibit cancer metastasis and
improve patient survival.
Growing evidence suggests that inflammation promotes
EMT (7,8). Interleukin 6 (IL-6) a multifunctional
cytokine in the tumor microenvironment, has been regarded as the
main factor involved in EMT, contributing to tumor invasion and
metastasis (9–11). Previous studies revealed that IL-6
leads to the development of HCC (12,13),
as an independent predictor of HCC tumor recurrence, poor survival,
and tumor metastasis (14).
Furthermore, IL-6 appears to contribute as a potent factor to the
initiation of EMT via activation of Janus kinase (JAK) family
members (JAK1, JAK2, and TYK2), leading to the activation of
transcription factors of the signal transducer and activator of
transcription 3 (STAT3) signaling (15–17).
Emerging evidence suggests that STAT3 regulated the expression of
Twist by directly binding to the transcriptional starting site in
the Twist promoter (18). Moreover,
blocking STAT3 markedly suppressed the expression of Twist,
confirming the regulation of Twist by STAT3 (17). However, little is known about the
mechanisms of IL-6-induced EMT in HCC.
Increasing evidence has indicated that traditional
Chinese medicines contain anticancer ingredients. Norcantharidin
(NCTD; 7-oxabicyclo (2.2.1) heptane-2,3-dicarboxylic anhydride) is
a demethylated and low-cytotoxic analog of cantharidin, an active
ingredient of the Chinese blister beetle Mylabris, which has
been used in China to treat tumors, inflammation and many other
conditions for a long time (19,20).
Previous studies have reported that NCTD could modulate the
expression of Bcl-2 and Mcl-1 to effectively inhibit proliferation
and induce apoptosis in a variety of human tumor cells, as well as
producing fewer side effects and leukocytosis (21–27).
Several studies have suggested that NCTD inhibits cell migration
and invasion through the c-Jun N-terminal kinase (JNK) and
mitogen-activated protein (MAP) kinase signaling pathways in human
lung cancer cells and CRC cells (28–30).
However, the effects of NCTD on HCC metastasis have not been
elucidated thus far.
In the present study, we investigated the
anti-metastatic effects of NCTD using IL-6-treated HCC cells. We
found that NCTD inhibited IL-6-induced EMT and invasiveness through
the suppression of the JAK2 and STAT3 pathways to regulate TWIST
expression in HCC cell lines.
Materials and methods
Compound preparations and
reagents
NCTD (molecular formular:
C8H8O4, molecular weight:
168.1467, HPLC ≥98%) was purchased from the National Standard
Network (Beijing, China). NCTD was dissolved in dimethyl sulphoxide
(DMSO) to a concentration of 40 mg/ml as stock solution, stored at
room temperature and protected from the light. Serial
concentrations of NCTD were diluted in culture medium before use
(DMSO <1%, 0–120 µM). Cucurbitacin I (JSI-124, #C4493-1MG;
Sigma-Aldrich, St. Louis, MO, USA) a novel inhibitor of the
JAK2/STAT3 signaling pathway (molecular weight: 514.65, HPLC ≥95%)
(31), was dissolved in DMSO to a
concentration of 5 mg/ml as stock solution, stored at −20°C and
protected from the light. Then, cucurbitacin I was diluted to a
concentration of 0.5 µM in culture medium before use (DMSO <1%).
Human recombinant IL-6 (#I1395-50UG; Sigma-Aldrich) was dissolved
in phosphate-buffered saline (PBS) to a concentration of 100 µg/ml
as stock solution and stored at −20°C. The final concentration of
IL-6 was diluted to 100 ng/ml in culture medium before use.
Cell culture
HCC cell lines, HCCLM3 and SMMC-7721, were purchased
from the China Infrastructure of Cell Line Resources (Beijing,
China). Cells were cultured in Dulbecco's modified Eagles medium
(Gibco, Grand Island, NY, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Hyclone, Logan, Utah, USA) and
1% penicillin and streptomycin (Hyclone) in a 5% CO2
humidified incubator at 37°C.
Cell invasion assays
Transwell Matrigel invasion chambers (#3422; Corning
Costar Corporation, USA) with 8-µm membrane pores coated with 100
µl of 1:6 diluted Matrigel in serum-free DMEM (#356234; BD
Biosciences, CA, USA) were used for the cell invasion assay. Cells
were pretreated with 100 ng/ml of IL-6 for 48 h. Cells
(1×104) were plated in 100 µl of serum-free DMEM
containing NCTD (0, 30, 60 and 120 µM) and JSI-124 (0.5 µM) as the
positive control group into the upper chamber. The lower chamber
was filled with 600 µl of DMEM and 10% FBS medium. After incubation
at 37°C for 24 h in a 5% CO2 atmosphere, the non-invaded
cells in the inserts were removed with cotton swabs. The invaded
cells on the underside were treated with a fixative/staining
solution (0.1% crystal violet, 4% paraformaldehyde) for
visualization. The number of invading cells on the filters was
counted in 5 random fields per filter at an ×100 magnification in
triplicate wells for each group.
Immunofluorescence staining
Cells were treated with 100 ng/ml of IL-6 and grown
in 96-well plates. The cells were fixed for 15 min with 4%
paraformaldehyde, and permeabilized for 10 min in PBS containing
0.1% Triton X-100 at room temperature. The cells were rinsed in PBS
(pH 7.4) three times for 5 min. After blocking in Immunol staining
blocking buffer (Beyotime Biotechnology, Shanghai, China) for 1 h
at room temperature, the cells were probed with a primary antibody
at 4°C overnight. After rinsing in PBS, the cells were incubated
with secondary antibodies [anti-rabbit IgG-Alexa Fluor
555-conjugated (red) or anti-rabbit IgG-Alexa Fluor 488-conjugated
(green)] for 1 h at room temperature and the nuclei were stained
with DAPI (Beyotime Biotechnology) for 10 min. All of the images
were semi-quantitatively analyzed, and the 96-well plates were
mounted and viewed using ImageXpress Micro (high content analysis;
Molecular Devices, CA, USA). For analysis of E-cadherin, N-cadherin
and vimentin expression, fluorescence intensity was quantified by
assessing the intensity in the cells using Image-Pro Plus software,
version 6.0 (Media Cybernetics, Bethesda, MD, USA).
Western blot analysis
HCCLM3 cells were pretreated with 100 ng/ml of IL-6
for 48 h and then treated with NCTD (0, 30, 60, and 120 µM) for 24
h. The positive control group was subjected to JSI-124 treatment
(JAK2/STAT3 inhibitor, 0.5 µM) for 24 h. The cells were lysed in a
RIPA buffer with a protease inhibitor cocktail and a phosphatase
inhibitor cocktail (both from Beyotime Biotechnology), and then
clarified by centrifugation. Total cell lysates were resuspended in
SDS sample buffer and resolved by SDS-PAGE. Proteins were
transferred to polyvinylidene fluoride (PVDF) membranes (Millipore
Corp., Billerica, MA, USA), and then blocked with 5% non-fat milk
for 1 h at room temperature. The membranes were then incubated with
primary antibodies [E-cadherin (#3195), N-cadherin (#13116),
vimentin (#5741), JAK2 (#3230), pJAK2 (Tyr1007/1008, #3776), STAT3
(#12640) and pSTAT3 (Tyr705, #9145), (all from Cell Signaling
Technology, Inc., Danvers, MA, USA), and Twist (H-81, #sc-15393;
Santa Cruz Biotechnology, Dallas, Texas, USA)], overnight at 4°C
before incubation with the corresponding HRP-conjugated secondary
antibodies (1:10,000) diluted in TBST. Then, the membranes were
washed extensively with TBST and visualized using an enhanced
chemiluminescence detection system, followed by quantification
using the Image Quant LAS 4000 (GE Healthcare, Bucks, UK).
Quantitative RT-PCR
Total RNA was extracted from individual groups of
cells using TRIzol reagent and reversely transcribed to cDNA using
the First Strand cDNA Synthesis kit (Invitrogen). cDNA was
synthesized using 1 µg of total RNA in a 20-µl final volume by
reverse transcription utilizing SuperScript II Reverse
Transcriptase with oligo-dT(18)-primers (both from Invitrogen). The
relative levels of target gene mRNA transcripts were determined by
quantitative RT-PCR using the SYBR-Green Master Mix kit. RNA was
amplified using ABI Prism 7000 Sequence Detection System (Applied
Biosystems). The sequences of the primers are shown in Table I. The PCR amplification was
performed in triplicate at 95°C for 10 min and was subjected to 40
cycles of 95°C for 15 sec and 60°C for 30 sec. The relative levels
of each gene to GAPDH mRNA transcripts were calculated.
 | Table I.The sequences of primers. |
Table I.
The sequences of primers.
| Target gene | Primers
sequences |
|---|
| E-cadherin | F:
5′-CCCATCAGCTGCCCAGAAAATGAA-3′ |
|
| R:
5′-CTGTCACCTTCAGCCATCCTGTTT-3′ |
| N-cadherin | F:
5′-AAGAACGCCAGGCCAAACAAC-3′ |
|
| R:
5′-CTGGCTCAAGTCATAGTCCTGGTCT-3′ |
| Vimentin | F:
5′-GACAATGCGTCTCTGGCACGTCTT-3′ |
|
| R:
5′-TCCTCCGCCTCCTGCAGGTTCTT-3′ |
| JAK2 | F:
5′-TGGACAGAGAGAGAATTTTCTGAACT-3′ |
|
| R:
5′-TTCATTGCTTTCCTTTTTCACA-3′ |
| STAT3 | F:
5′-TTGCCAGTTGTGGTGATC-3′ |
|
| R:
5′-AGAACCCAGAAGGAGAAGC-3′ |
| TWIST | F:
5′-GTCCGCAGTCTTACGAGGAG-3′ |
|
| R:
5′-GCTTGAGGGTCTGAATCTTGCT-3′ |
| GAPDH | F:
5′-TGCACCACCAACTGCTTAGC-3′ |
|
| R:
5′-GGCATGGACTGTGGTCATGAG-3′ |
Statistical analysis
Data were expressed as the mean ± standard deviation
(SD) and statistical differences were determined by two-way
repeated ANOVA or by one-way ANOVA, followed by a Newman-Keuls test
using SPSS 13.0 statistical software. A p-value of <0.05 was
considered significant.
Results
IL-6 induces EMT in HCC cells
To investigate whether recombinant IL-6 could induce
EMT in HCC cells, we treated the human HCC cell lines, HCCLM3 and
SMMC-7721, which exhibit an epithelial phenotype, with various
concentrations (0, 50, and 100 ng/ml) of recombinant IL-6 for 48 h
or with 100 ng/ml of IL-6 for the indicated durations (0, 24, and
48 h). We assessed the expression of epithelial marker, E-cadherin,
and the mesenchymal markers, N-cadherin and vimentin. The results
demonstrated that E-cadherin expression was significantly
suppressed, although N-cadherin and vimentin expression was
increased with IL-6 treatment in HCCLM3 and SMMC-7721 cells as
compared to the control group (absence of IL-6) in a dose-dependent
manner (Fig. 1A). When the HCCLM3
and SMMC-7721 cells were treated with 100 ng/ml of IL-6 for 0, 24,
and 48 h, the data revealed that EMT had occurred in a
time-dependent manner (Fig. 1B). In
addition, we examined the expression of EMT markers in
hepatocellular cells by immunofluorescence staining. After the
HCCLM3 and SMMC-7721 cells were treated with IL-6 (100 ng/ml) for
48 h, similar effects of E-cadherin, N-cadherin and vimentin were
observed (Fig. 1C). We next
determined the mRNA expression levels of E-cadherin, N-cadherin and
vimentin in response to IL-6 (100 ng/ml) treatment for 0, 24 and 48
h by quantitative RT-PCR. The mRNA levels of E-cadherin were
significantly decreased in HCCLM3 and SMMC-7721 cells treated with
IL-6 as compared to the control cells without IL-6 treatment. In
contrast, the mRNA levels of N-cadherin and vimentin were markedly
increased in HCCLM3 and SMMC-7721 cells treated with IL-6 as
compared to the control cells without IL-6 treatment (Fig. 1D). In summary, these results
indicated that HCCLM3 and SMMC-7721 cells undergo EMT in response
to IL-6 exposure, via downregulation of epithelial marker
E-cadherin, and upregulation of mesenchymal markers N-cadherin and
vimentin.
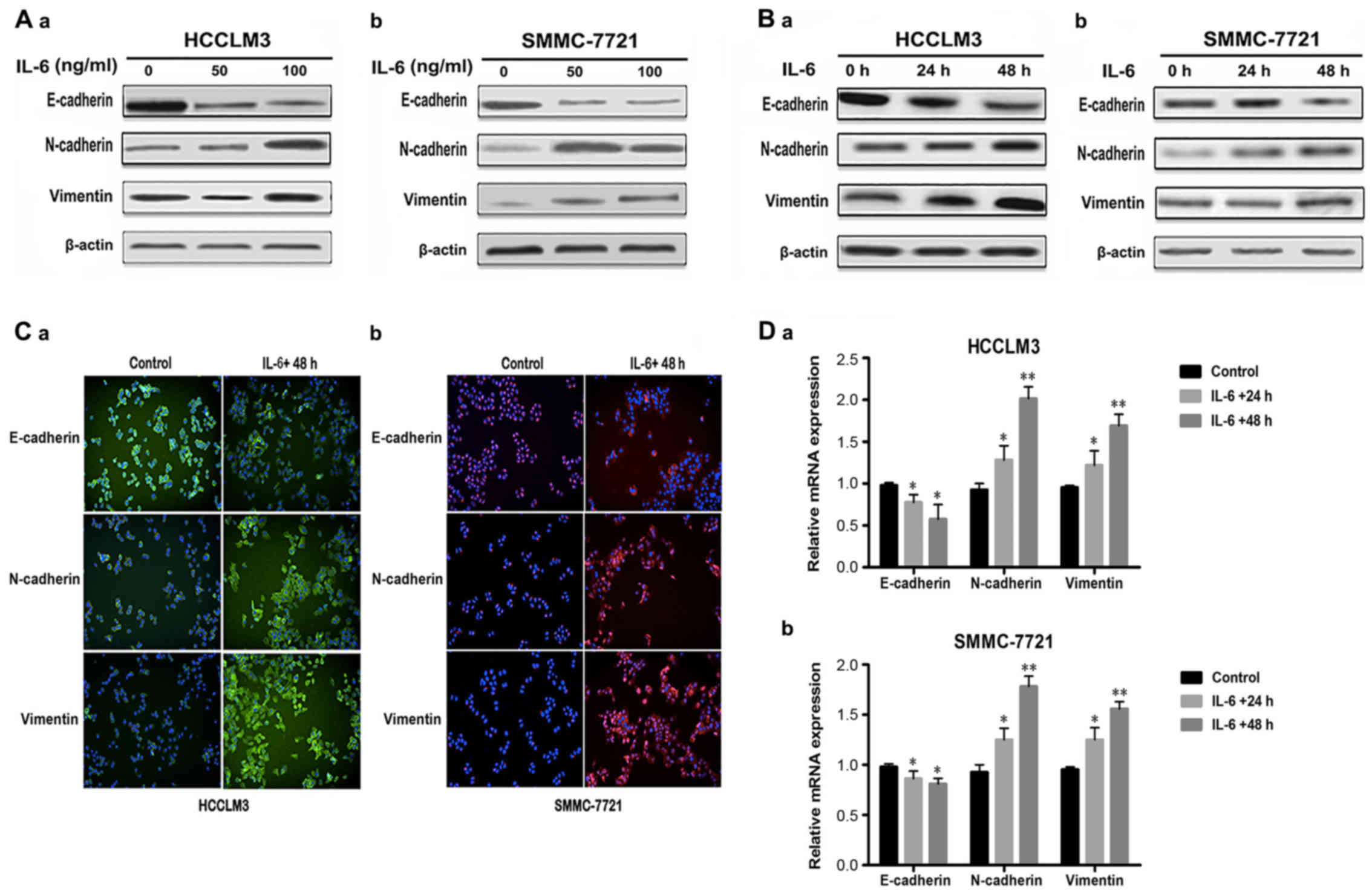 | Figure 1.IL-6 treatment induces EMT in HCC
cell lines. (A-a and b) Cells were treated with IL-6 (0, 50 and 100
ng/ml) for 48 h. The protein levels of EMT markers were assessed by
western blot analysis in HCCLM3 and SMMC-7721 cell lines. (B-a and
b) Cells were treated with IL-6 (100 ng/ml) for 0, 24 and 48 h. The
protein levels of EMT markers were assessed by western blot
analysis in HCCLM3 and SMMC-7721 cell lines. (C-a) HCCLM3 cells
were treated without or with IL-6 (100 ng/ml) for 48 h. Cells were
stained with primary antibodies against E-cadherin, N-cadherin and
vimentin, followed by Alexa Fluor 488-conjugated (green) secondary
antibodies. Nuclei were stained with DAPI (blue) (original
magnification, ×400). (C-b) SMMC-7721 cells were treated without or
with IL-6 (100 ng/ml) for 48 h. Cells were stained with primary
antibodies against E-cadherin, N-cadherin and vimentin, followed by
Alexa Fluor 555-conjugated (red) secondary antibodies. Nuclei were
stained with DAPI (blue) (original magnification, ×400). (D-a and
b) Cells were exposed to 100 ng/ml of IL-6 for 24 and 48 h. The
mRNA expression levels of E-cadherin, N-cadherin and vimentin were
determined by quantitative RT-PCR (mean values ± SD, n=3, are
provided. *P<0.05, **P<0.01). IL-6, interleukin 6; EMT,
epithelial-mesenchymal transition; HCC, hepatocellular
carcinoma. |
IL-6-induced EMT is mediated by the
activation of the JAK2/STAT3/TWIST pathway in HCC cells
JAK2/STAT3 signaling regulates different cellular
functions, including the EMT process, in multiple cancer types.
Herein, western blot results revealed that STAT3 was phosphorylated
and activated to upregulate EMT transcription factor, TWIST, after
IL-6 (100 ng/ml) treatment for the indicated durations (0, 24 and
48 h) in HCC cell lines HCCLM3 and SMMC-7721 (Fig. 2A-a and b). We hypothesized that
JAK2/STAT3 signaling is required for IL-6-mediated EMT in HCC. To
test this hypothesis, we used a JAK2/STAT3 specific inhibitor
cucurbitacin I (JSI-124) to suppress the activation of STAT3.
JSI-124 decreased STAT3 phosphorylation and TWIST expression
induced by IL-6, respectively, in HCCLM3 and SMMC-7721 (Fig. 2A-c and d). We further examined the
role of JSI-124 in IL-6-mediated EMT in HCCLM3 and SMMC-7721 cells.
As observed, JSI-124 significantly reversed IL-6-mediated
downregulation of E-cadherin, upregulation of N-cadherin and
vimentin, and decreased the mRNA levels of Twist (Fig. 2B).
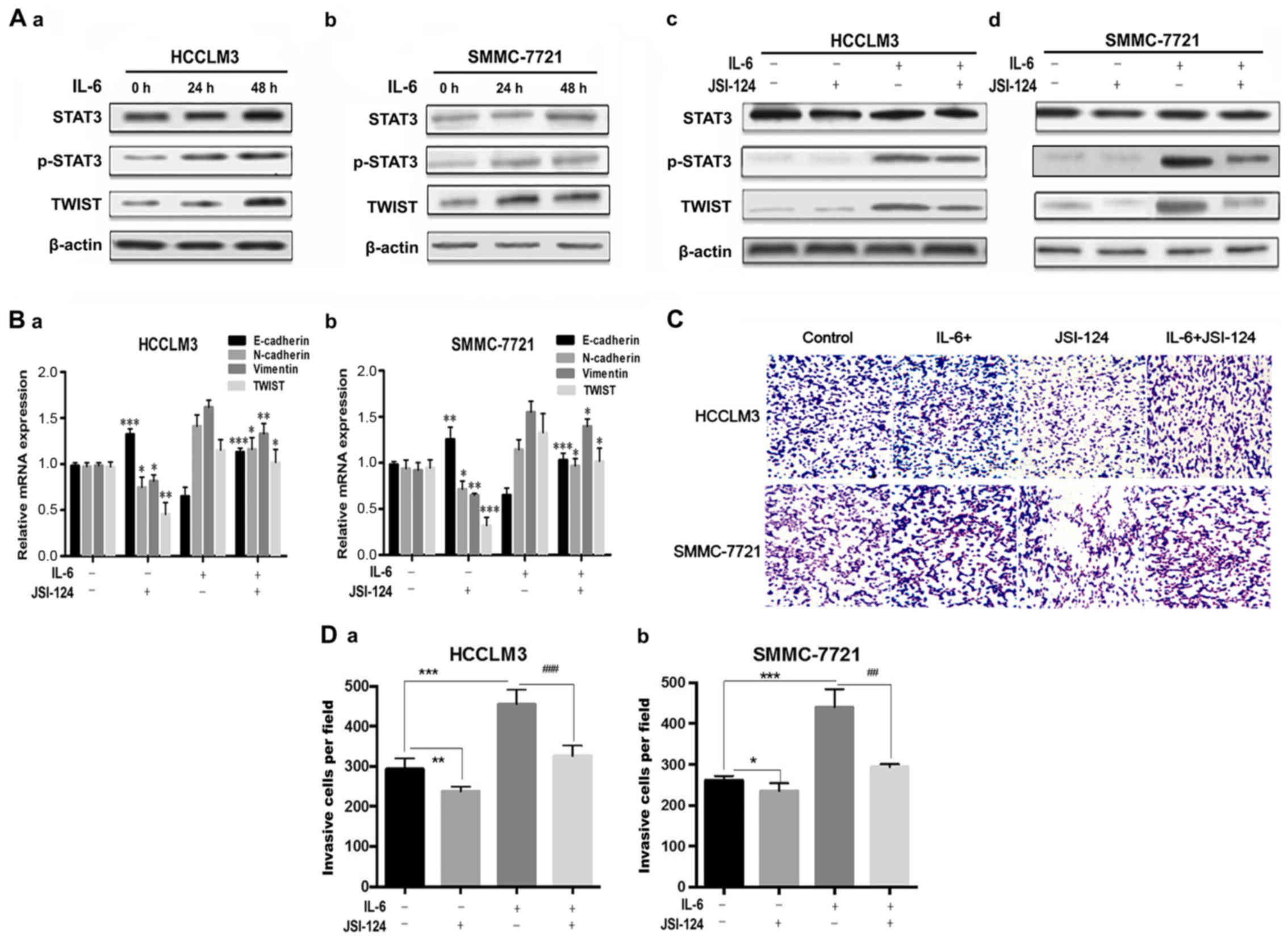 | Figure 2.JAK2/STAT3/TWIST signaling is
required for IL-6-induced EMT in HCC cell lines. (A-a and b)
Western blot analysis of p-STAT3-Y705, STAT3 and TWIST in HCCLM3
and SMMC-7721 cells after IL-6 treatment for the indicated periods
(0, 24 and 48 h). (A-c and d) JSI-124 inhibited IL-6-induced STAT3
phosphorylation and TWIST expression in HCCLM3 and SMMC-7721 cell
lines. (B-a and b) Cells were pretreated with 100 ng/ml of IL-6 for
48 h and then exposed to 0.5 µM of JAK2/STAT3 inhibitor (JSI-124)
for 24 h. The mRNA levels of E-cadherin, N-cadherin, vimentin and
Twist were determined by RT-PCR in HCCLM3 and SMMC-7721 cell lines.
JSI-124 significantly reversed IL-6-mediated downregulation of
E-cadherin, upregulation of N-cadherin and vimentin, and decreased
mRNA levels of Twist. (C and D-a and b) JSI-124 inhibited
IL-6-induced cell invasion in HCCLM3 and SMMC-7721 cells. Cells
were treated with 100 ng/ml of IL-6 and/or 0.5 µM of JSI-124 for 24
h, and allowed to pass through 8-µm membrane pores coated with 100
µl of Matrigel in Transwells. Invasive cells through the pores were
stained with 1% crystal violet (magnification, ×200). The mean
values ± SD, n=3 are provided. *P<0.05, **P<0.01,
***P<0.001 vs. the control group, #P<0.05,
##P<0.01, ###P<0.001 vs. the IL-6+ and
JSI-124-). JAK, Janus tyrosine kinase; STAT3, signal transducer and
activator of transcription 3; IL-6, interleukin 6; EMT,
epithelial-mesenchymal transition; HCC, hepatocellular
carcinoma. |
We used Transwell assays to further investigate the
invasive capacities of HCCLM3 and SMMC-7721 cells. IL-6 treatment
with 100 ng/ml for 24 h enhanced cell invasiveness, whereas
incubation with JSI-124 markedly suppressed the IL-6-induced
increase of invaded cells. The results revealed that IL-6 enhanced
the invasion abilities of HCCLM3 and SMMC-7721 cell lines, and that
the JAK2/STAT3 inhibitor (JSI-124) suppressed IL-6-induced cell
invasion (Fig. 2C and D). These
findings demonstrated that upregulation of TWIST by STAT3 was
required for IL-6-induced EMT and invasion in the HCCLM3 and
SMMC-7721 cell lines.
Norcantharidin suppresses IL-6-induced
EMT in HCC cells
To investigate the effect of NCTD treatment on
IL-6-induced EMT in the HCCLM3 and SMMC-7721 cell lines, the cells
were treated with 100 ng/ml of IL-6 for 48 h. Western blot analysis
revealed that the expression of epithelial marker, E-cadherin, was
significantly increased, while mesenchymal markers, N-cadherin and
vimentin, were downregulated following incubation with various NCTD
concentrations (0, 30, 60 and 120 µM) for 24 h when compared to the
IL-6-treated group (0 µM of NCTD) (Fig.
3A and B). Furthermore, the relative mRNA levels of EMT markers
from the NCTD groups after treatment with 100 ng/ml of IL-6 for 48
h, were analyzed by quantitative RT-PCR. E-cadherin mRNA was
suppressed after IL-6 treatment. However, this suppression was
reversed by treatment with different NCTD concentrations (60 and
120 µM). In contrast, the relative levels of N-cadherin and
vimentin mRNA transcripts were increased after IL-6 treatment.
However, this induction was prevented in the NCTD-treated (120 µM)
group (Fig. 3C and D).
Collectively, the data demonstrated that NCTD treatment reversed
the EMT process induced by IL-6 in the HCCLM3 and SMMC-7721 cell
lines.
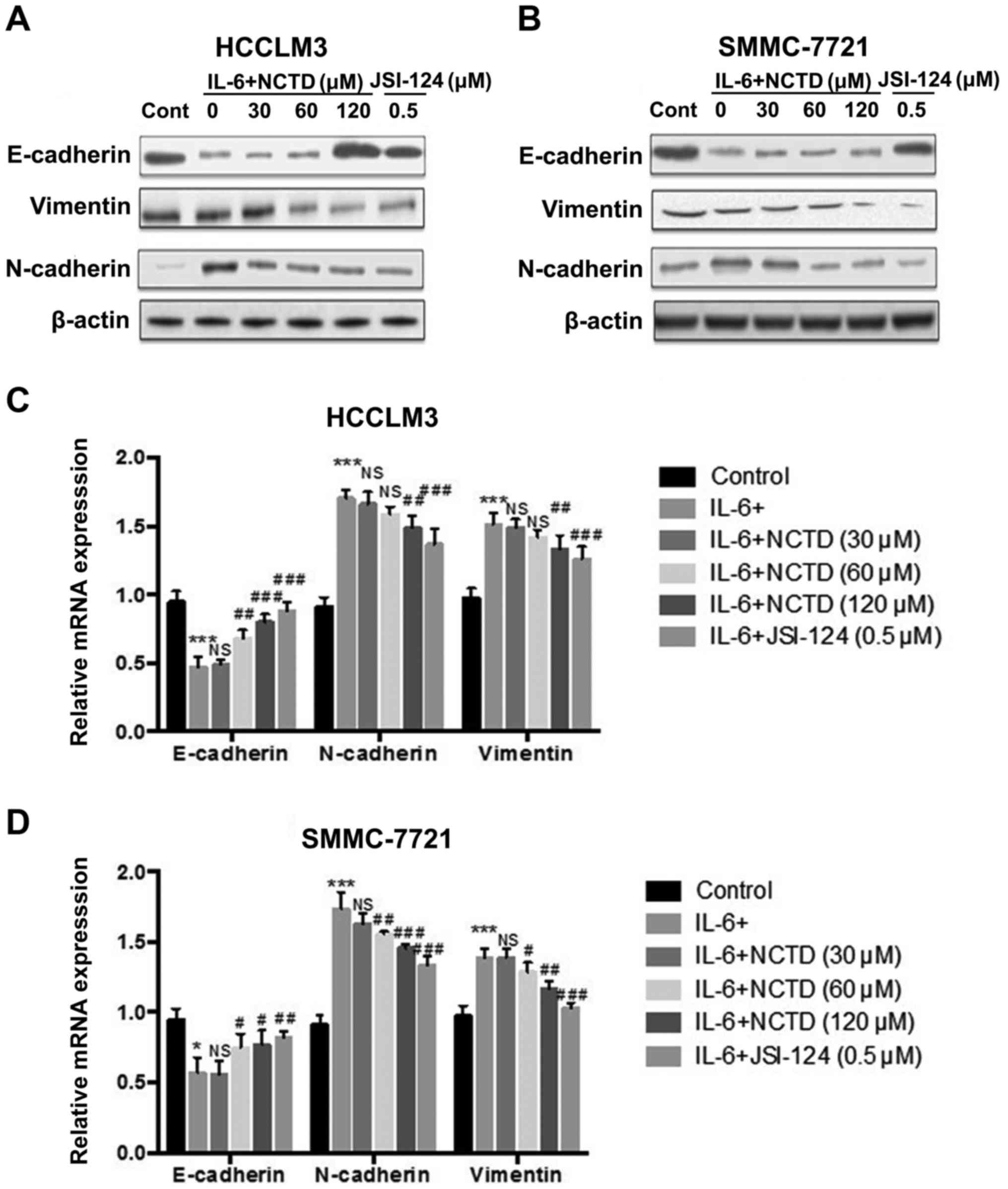 | Figure 3.NCTD effectively blocks the IL-6
induced EMT in HCC cell lines. (A and B) After pretreatment with
IL-6 (100 ng/ml) for 48 h, western blot analysis of E-cadherin,
N-cadherin and vimentin protein expression was performed in HCCLM3
and SMMC-7721 cells after exposure to various concentrations of
NCTD (30, 60 and 120 µM) or JSI-124 (0.5 µM) for 24 h. (C and D)
Cells were pretreated with IL-6 (100 ng/ml) for 48 h and the levels
of E-cadherin, N-cadherin and vimentin mRNA were determined by
RT-PCR after exposure to various concentrations of NCTD (30, 60 and
120 µM) or JSI-124 (0.5 µM) for 24 h in HCCLM3 and SMMC-7721 cell
lines. E-cadherin was upregulated, whereas N-cadherin and vimentin
were downregulated following incubation with NCTD for 24 h as
compared to the IL-6-treated group (0 µM of NCTD). The mean values
± SD, n=3 are provided. *P<0.05, **P<0.01, ***P<0.001 vs.
the control group, NS P>0.05, #P<0.05,
##P<0.01, ###P<0.001 vs. the IL-6
group). NCTD, norcantharidin; IL-6, interleukin 6; EMT,
epithelial-mesenchymal transition; HCC, hepatocellular
carcinoma. |
NCTD inhibits IL-6-induced cell
invasion in HCC cells
The effects of NCTD on IL-6-induced cell invasion in
the HCCLM3 and SMMC-7721 cell lines was further investigated by
Transwell assays. The results revealed that IL-6 significantly
enhanced the invasion ability of the two cell lines (P<0.001),
whereas concomitant incubation with NCTD or JSI-124 suppressed the
IL-6-induced invasion ability of the cells (Fig. 4). Thus, these findings demonstrated
that NCTD suppressed IL-6-induced cellular invasiveness in a
dose-dependent manner.
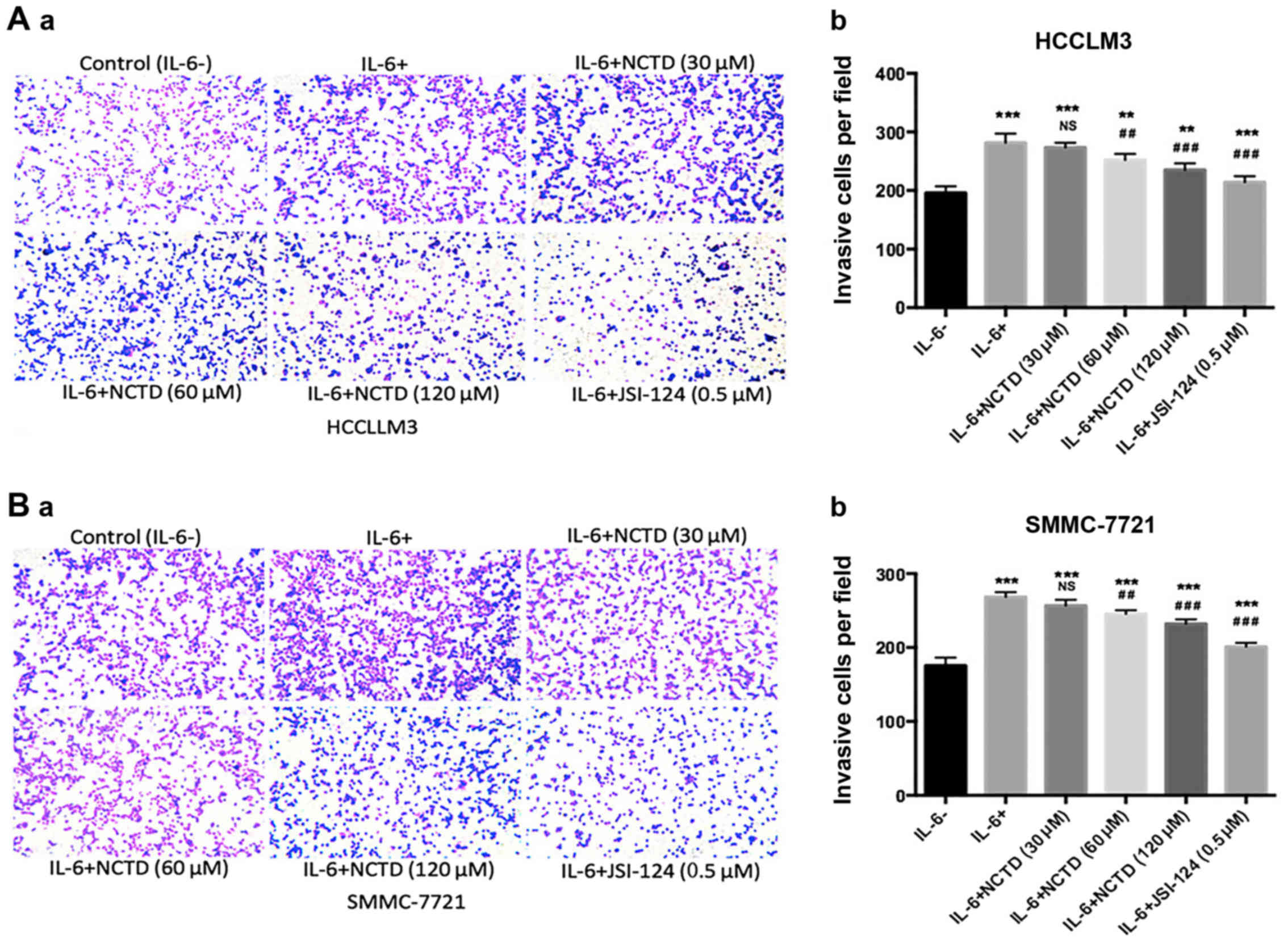 | Figure 4.NCTD significantly suppresses the
IL-6-induced invasiveness in HCC cell lines. (A-a and b and B-a and
b) Cell invasive abilities were assessed by Transwell assay with
different treatments in HCCLM3 and SMMC-7721 cells (magnification,
×200). IL-6 significantly enhanced the invasive abilities after
pretreatment (100 ng/ml of IL-6) for 48 h. However, NCTD and
JSI-124 decreased cell invasion after exposure to various
concentrations of NCTD (30, 60 and 120 µM) and JSI-124 (0.5 µM),
for 24 h. The mean values ± SD, n=3 are provided. *P<0.05,
**P<0.01, ***P<0.001 vs. the control group, NS P>0.05,
#P<0.05, ##P<0.01,
###P<0.001 vs. the IL-6 group). NCTD, norcantharidin;
IL-6, interleukin 6; HCC, hepatocellular carcinoma. |
NCTD inhibits IL-6-induced EMT through
the JAK/STAT3/TWIST signaling pathway in HCC cells
To explore the underlying mechanisms of the
inhibitory activities of NCTD on IL-6-induced EMT, we determined
its effect on the activation of the JAK/STAT3/TWIST pathway. The
expression of JAK2, phosphorylated JAK2 (p-JAK2), STAT3,
phosphorylated STAT3 (p-STAT3) and TWIST after IL-6 treatment in
the different concentration NCTD-treated groups was analyzed by
western blot analysis. Notably, the expression of p-JAK2 and
p-STAT3 increased after exposure of HCCLM3 and SMMC-7721 cells to
IL-6. Furthermore, the expression of p-JAK2 and p-STAT3 decreased
after treatment with NCTD in a dose-dependent manner (Fig. 5A-a and b). Notably, the JAK2/STAT3
inhibitor JSI-124 significantly suppressed the p-JAK2 and p-STAT3
levels. Treatment with NCTD and JSI-124 also markedly decreased
JAK2 and STAT3 phosphorylation induced by IL-6 (Fig. 5B).
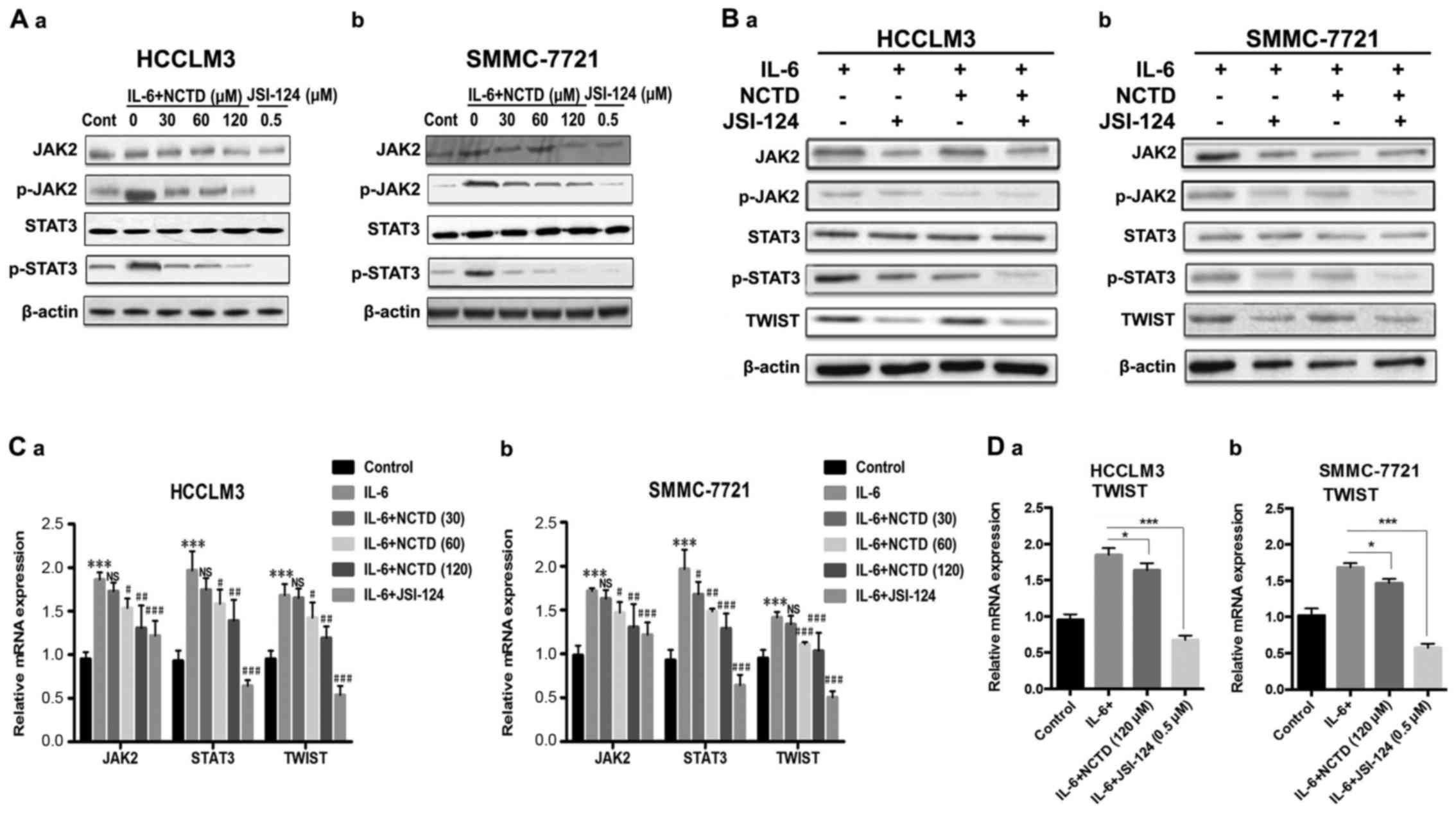 | Figure 5.NCTD inhibits JAK2/STAT3/TWIST
signaling in HCC cell lines. (A-a and b) After pretreatment with
IL-6 (100 ng/ml) for 48 h, western blot analysis of the protein
expression of JAK2, phosphorylated JAK2 (p-JAK2), STAT3, and
phosphorylated STAT3 (p-STAT3) was performed in HCCLM3 and
SMMC-7721 cells after exposure to various concentrations of NCTD
(30, 60 and 120 µM) or JSI-124 (0.5 µM) for 24 h. (B-a and b) Cells
were pretreated with IL-6 (100 ng/ml) for 48 h, and the protein
levels of JAK2, p-JAK2, STAT3, p-STAT3 and TWIST were determined by
western blot analysis after exposure to NCTD (120 µM) or JSI-124
(0.5 µM) for 24 h in HCCLM3 and SMMC-7721 cell lines. (C-a and b)
Cells were pretreated with IL-6 (100 ng/ml) for 48 h, and the
levels of JAK2, STAT3, TWIST mRNA were determined by RT-PCR after
exposure to various concentrations of NCTD or JSI-124 (0.5 µM) for
24 h in HCCLM3 and SMMC-7721 cell lines. (D-a and b) Cells were
pretreated with IL-6 (100 ng/ml) for 48 h, the mRNA levels of TWIST
were determined by RT-PCR after exposure to NCTD (120 µM) or
JSI-124 (0.5 µM) for 24 h in HCCLM3 and SMMC-7721 cell lines. (Mean
values ± SD, n=3, are provided. *P<0.05, **P<0.01,
***P<0.001 vs. the control group, NS P>0.05,
#P<0.05, ##P<0.01,
###P<0.001 vs. the IL-6 group). NCTD, norcantharidin;
JAK, Janus tyrosine kinase; STAT3, signal transducer and activator
of transcription 3; IL-6, interleukin 6; HCC, hepatocellular
carcinoma. |
Finally quantitative RT-PCR analysis revealed that
the levels of JAK2 and STAT3 increased after IL-6 treatment,
whereas after treatment with different concentrations of NCTD, the
relative levels of JAK2, STAT3 and TWIST decreased in a
dose-dependent manner in HCCLM3 and SMMC-7721 cells. Notably, the
decrease in the expression of JAK2, STAT3 and TWIST with the NCTD
(30 µM) treatment was not in a statistically significant manner in
the HCCLM3 cell lines (Fig. 5C).
Moreover, NCTD (120 µM) treatment resulted in a significant
decrease of IL-6-mediated TWIST expression. Likewise the expression
of TWIST in the two cell lines was compared to the suppression of
JAK2/STAT3, suggesting that NCTD directly counteracted the
IL-6-mediated induction of TWIST (Fig.
5D). In summary, these results demonstrated that NCTD inhibited
IL-6-induced EMT via the downregulation of the TWIST transcription
factor through JAK2/STAT3 signaling.
Discussion
EMT has been recognized as one of the universal
mechanisms by which cancer cells acquire migratory and invasive
capacity (5,32–34).
Invasion is a key step to progression toward a malignant phenotype,
and occurs when tumor cells translocate from the relatively
constrained initial neoplastic mass into neighboring host tissues.
During the process of EMT, epithelial cells acquire a fibroblastoid
appearance due to downregulation of epithelial markers and
upregulation of mesenchymal markers, thus generating a migratory
phenotype (35,36). EMT which has been actively
investigated in HCC (37–39), can be induced by numerous cytokines
and growth factors, including IL-6 (40). Previous studies revealed that IL-6
induces a complex reciprocally regulated cytokine network in tumor
cells which leads to the development of malignant and invasive
tumors (40–42). IL-6 was shown to promote EMT changes
in mesenchymal tumors from HCC patients as well as mesenchymal HCC
cell lines (6). HCCLM3 and
SMMC-7721 cell lines have highly invasive properties and were used
in metastasis studies (43,44). In this study, we established the
IL-6-induced EMT model in HCC cell lines (HCCLM3 and SMMC-7721),
and demonstrated that IL-6 stimulated EMT in a time- and
dose-dependent manner accompanied by downregulation of E-cadherin
and upregulation of N-cadherin and vimentin in HCCLM3 and SMMC-7722
cells. In addition, administration of IL-6 significantly enhanced
the invasion potential of HCC cell lines as a result of EMT.
Previous studies have suggested that IL-6 can induce EMT changes by
activating its downstream protein STAT3 within the tumor
microenvironment, and plays an important role in the pathogenesis
of human cervical carcinoma and breast cancer (41,45).
Whether STAT3 plays a role in EMT induction in human HCC still
remains to be determined. JSI-124 is a selective inhibitor of
JAK2/STAT3 and has been demonstrated to exert anti-proliferative
and antitumor effects both in vitro and in vivo
(46). In the present study, we
found that the JAK2/STAT3 inhibitor (JSI-124) reversed the
IL-6-induced EMT process and STAT3 phosphorylation in HCC cell
lines, suggesting that JAK2/STAT3 may be involved in IL-6-induced
EMT change and may be one of the mechanisms responsible for this
change. Twist has been reported as a major transcriptional
suppressor downregulating E-cadherin expression and thus resulting
in initial EMT and ultimate tumor metastasis (47,48).
Emerging evidence suggests that STAT3 activation upregulates Twist
expression (17,41). Our data revealed that Twist, as an
EMT activator, was significantly increased during EMT induction by
IL-6. However, blocking STAT3 activation markedly suppressed the
expression of Twist, confirming the regulation of Twist by
STAT3.
In addition to EMT change, both HCCLM3 and SMMC-7721
cells after treatment with IL-6 gained invasive capacities. Using
the cell line model, we investigated the effects of NCTD on
IL-6-induced EMT and cell invasion. The results revealed that NCTD
hindered the upregulation of N-cadherin and vimentin, and the
downregulation of E-cadherin induced by IL-6 in HCCLM3 and
SMMC-7721 cells, as well as the increased cell invasiveness induced
by IL-6. IL-6-mediated activation of JAK2/STAT3 implicated in EMT
and metastasis, and inhibition of JAK2 or blockade of activated
STAT3 significantly suppressed the EMT process, cell migration and
invasion in colon and pancreatic cancer (49,50).
During the investigation process into the effects of NCTD on the
levels of p-JAK2 and p-STAT3 in liver cancer cells after the
treatment of IL-6, we found that NCTD significantly inhibited the
activation of JAK2/STAT3 induced by IL-6, although there was a
slight variation in the levels of JAK2 in the presence of NCTD or
JSI-124. The variation could be caused by different cell activity
states. Moreover, we observed that the EMT-related transcription
factor TWIST was downregulated after NCTD treatment, both at the
mRNA and protein levels. All of these results clearly demonstrated
that NCTD could block IL-6-induced EMT via the JAK2/STAT3/TWIST
signaling pathway, which is an important mechanism underlying HCC
development and metastasis.
To conclude, IL-6 promotes the EMT process during
HCC development. NCTD can effectively target cell EMT by
suppressing JAK2/STAT3/TWIST signaling under IL-6 stimulation.
These findings warrant further assessment of NCTD, as an anticancer
drug, in clinically relevant cancer models to explore its potential
role in the treatment of HCC patients.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (Beijing, China; grant no.
81403346), the Natural Science Foundation of Shandong Province
(Shandong, China; grant no. ZR2014HL095), and the China
Postdoctoral Science Foundation (Beijing, China; grant no.
2014M550132).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang H and Chen L: Tumor microenviroment
and hepatocellular carcinoma metastasis. J Gastroenterol Hepatol.
28:(Suppl 1). 43–48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iwatsuki M, Mimori K, Yokobori T, Ishi H,
Beppu T, Nakamori S, Baba H and Mori M: Epithelial-mesenchymal
transition in cancer development and its clinical significance.
Cancer Sci. 101:293–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamada S, Okumura N, Wei L, Fuchs BC,
Fujii T, Sugimoto H, Nomoto S, Takeda S, Tanabe KK and Kodera Y:
Epithelial to mesenchymal transition is associated with shorter
disease-free survival in hepatocellular carcinoma. Ann Surg Oncol.
21:3882–3890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hussain SP and Harris CC: Inflammation and
cancer: An ancient link with novel potentials. Int J Cancer.
121:2373–2380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
López-Novoa JM and Nieto MA: Inflammation
and EMT: An alliance towards organ fibrosis and cancer progression.
EMBO Mol Med. 1:303–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rose-John S, Waetzig GH, Scheller J,
Grötzinger J and Seegert D: The IL-6/sIL-6R complex as a novel
target for therapeutic approaches. Expert Opin Ther Targets.
11:613–624. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grivennikov SI and Karin M: Inflammatory
cytokines in cancer: Tumour necrosis factor and interleukin 6 take
the stage. Ann Rheum Dis. 70:(Suppl 1). i104–i108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rokavec M, Wu W and Luo JL: IL6-mediated
suppression of miR-200c directs constitutive activation of
inflammatory signaling circuit driving transformation and
tumorigenesis. Mol Cell. 45:777–789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Porta C, De Amici M, Quaglini S, Paglino
C, Tagliani F, Boncimino A, Moratti R and Corazza GR: Circulating
interleukin-6 as a tumor marker for hepatocellular carcinoma. Ann
Oncol. 19:353–358. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shackel NA, McCaughan GW and Warner FJ:
Hepatocellular carcinoma development requires hepatic stem cells
with altered transforming growth factor and interleukin-6
signaling. Hepatology. 47:2134–2136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wong VW, Yu J, Cheng AS, Wong GL, Chan HY,
Chu ES, Ng EK, Chan FK, Sung JJ and Chan HL: High serum
interleukin-6 level predicts future hepatocellular carcinoma
development in patients with chronic hepatitis B. Int J Cancer.
124:2766–2770. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Colomiere M, Ward AC, Riley C, Trenerry
MK, Cameron-Smith D, Findlay J, Ackland L and Ahmed N: Cross talk
of signals between EGFR and IL-6R through JAK2/STAT3 mediate
epithelial-mesenchymal transition in ovarian carcinomas. Br J
Cancer. 100:134–144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen W, Gao Q, Han S, Pan F and Fan W: The
CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal
transition by cooperatively activating STAT3-Twist signaling.
Tumour Biol. 36:973–981. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola
D, Mansour M, Xu LM, Costanzo C, Cheng JQ and Wang LH: Twist is
transcriptionally induced by activation of STAT3 and mediates STAT3
oncogenic function. J Biol Chem. 283:14665–14673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ho YP, To KK, Au-Yeung SC, Wang X, Lin G
and Han X: Potential new antitumor agents from an innovative
combination of demethylcantharidin, a modified traditional Chinese
medicine, with a platinum moiety. J Med Chem. 44:2065–2068. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang GS: Medical uses of mylabris in
ancient China and recent studies. J Ethnopharmacol. 26:147–162.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen YN, Chen JC, Yin SC, Wang GS, Tsauer
W, Hsu SF and Hsu SL: Effector mechanisms of norcantharidin-induced
mitotic arrest and apoptosis in human hepatoma cells. Int J Cancer.
100:158–165. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fan YZ, Zhao ZM, Fu JY, Chen CQ and Sun W:
Norcantharidin inhibits growth of human gallbladder carcinoma
xenografted tumors in nude mice by inducing apoptosis and blocking
the cell cycle in vivo. Hepatobiliary Pancreat Dis Int. 9:414–422.
2010.PubMed/NCBI
|
|
23
|
Fan YZ, Fu JY, Zhao ZM and Chen CQ:
Inhibitory effect of norcantharidin on the growth of human
gallbladder carcinoma GBC-SD cells in vitro. Hepatobiliary Pancreat
Dis Int. 6:72–80. 2007.PubMed/NCBI
|
|
24
|
Kok SH, Cheng SJ, Hong CY, Lee JJ, Lin SK,
Kuo YS, Chiang CP and Kuo MY: Norcantharidin-induced apoptosis in
oral cancer cells is associated with an increase of proapoptotic to
antiapoptotic protein ratio. Cancer Lett. 217:43–52. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu CC, Ko FY, Yu CS, Lin CC, Huang YP,
Yang JS, Lin JP and Chung JG: Norcantharidin triggers cell death
and DNA damage through S-phase arrest and ROS-modulated apoptotic
pathways in TSGH 8301 human urinary bladder carcinoma cells. Int J
Oncol. 41:1050–1060. 2012.PubMed/NCBI
|
|
26
|
Yang PY, Chen MF, Kao YH, Hu DN, Chang FR
and Wu YC: Norcantharidin induces apoptosis of breast cancer cells:
Involvement of activities of mitogen activated protein kinases and
signal transducers and activators of transcription. Toxicol In
Vitro. 25:699–707. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang Y, Liu Q, Liu K, Yagasaki K and
Zhang G: Suppression of growth of highly-metastatic human breast
cancer cells by norcantharidin and its mechanisms of action.
Cytotechnology. 59:2092009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen YJ, Kuo CD, Tsai YM, Yu CC, Wang GS
and Liao HF: Norcantharidin induces anoikis through Jun-N-terminal
kinase activation in CT26 colorectal cancer cells. Anticancer
Drugs. 19:55–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luan J, Duan H, Liu Q, Yagasaki K and
Zhang G: Inhibitory effects of norcantharidin against human lung
cancer cell growth and migration. Cytotechnology. 62:349–355. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Q, Duan H, Luan J, Yagasaki K and
Zhang G: Effects of theanine on growth of human lung cancer and
leukemia cells as well as migration and invasion of human lung
cancer cells. Cytotechnology. 59:211–217. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Blaskovich MA, Sun J, Cantor A, Turkson J,
Jove R and Sebti SM: Discovery of JSI-124 (cucurbitacin I), a
selective Janus kinase/signal transducer and activator of
transcription 3 signaling pathway inhibitor with potent antitumor
activity against human and murine cancer cells in mice. Cancer Res.
63:1270–1279. 2003.PubMed/NCBI
|
|
32
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nieto MA: The ins and outs of the
epithelial to mesenchymal transition in health and disease. Annu
Rev Cell Dev Biol. 27:347–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nieto MA and Cano A: The
epithelial-mesenchymal transition under control: Global programs to
regulate epithelial plasticity. Semin Cancer Biol. 22:361–368.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee TK, Poon RT, Yuen AP, Ling MT, Kwok
WK, Wang XH, Wong YC, Guan XY, Man K, Chau KL, et al: Twist
overexpression correlates with hepatocellular carcinoma metastasis
through induction of epithelial-mesenchymal transition. Clin Cancer
Res. 12:5369–5376. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee TK, Man K, Poon RT, Lo CM, Yuen AP, Ng
IO, Ng KT, Leonard W and Fan ST: Signal transducers and activators
of transcription 5b activation enhances hepatocellular carcinoma
aggressiveness through induction of epithelial-mesenchymal
transition. Cancer Res. 66:9948–9956. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giannelli G, Bergamini C, Fransvea E,
Sgarra C and Antonaci S: Laminin-5 with transforming growth
factor-beta1 induces epithelial to mesenchymal transition in
hepatocellular carcinoma. Gastroenterology. 129:1375–1383. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lederle W, Depner S, Schnur S, Obermueller
E, Catone N, Just A, Fusenig NE and Mueller MM: IL-6 promotes
malignant growth of skin SCCs by regulating a network of autocrine
and paracrine cytokines. Int J Cancer. 128:2803–2814. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y, Tian B, Yang J, Zhao L, Wu X, Ye SL,
Liu YK and Tang ZY: Stepwise metastatic human hepatocellular
carcinoma cell model system with multiple metastatic potentials
established through consecutive in vivo selection and studies on
metastatic characteristics. J Cancer Res Clin Oncol. 130:460–468.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yao J, Liang L, Huang S, Ding J, Tan N,
Zhao Y, Yan M, Ge C, Zhang Z, Chen T, et al: MicroRNA-30d promotes
tumor invasion and metastasis by targeting Galphai2 in
hepatocellular carcinoma. Hepatology. 51:846–856. 2010.PubMed/NCBI
|
|
45
|
Miao JW, Liu LJ and Huang J:
Interleukin-6-induced epithelial-mesenchymal transition through
signal transducer and activator of transcription 3 in human
cervical carcinoma. Int J Oncol. 45:165–176. 2014.PubMed/NCBI
|
|
46
|
Su Y, Li G, Zhang X, Gu J, Zhang C, Tian Z
and Zhang J: JSI-124 inhibits glioblastoma multiforme cell
proliferation through G(2)/M cell cycle arrest and apoptosis
augment. Cancer Biol Ther. 7:1243–1249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu AN, Zhu ZH, Chang SJ and Hang XS:
Twist expression associated with the epithelial-mesenchymal
transition in gastric cancer. Mol Cell Biochem. 367:195–203. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sasaki K, Natsugoe S, Ishigami S,
Matsumoto M, Okumura H, Setoyama T, Uchikado Y, Kita Y, Tamotsu K,
Sakamoto A, et al: Significance of Twist expression and its
association with E-cadherin in esophageal squamous cell carcinoma.
J Exp Clin Cancer Res. 28:1582009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bromberg J and Wang TC: Inflammation and
cancer: IL-6 and STAT3 complete the link. Cancer Cell. 15:79–80.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huang C, Yang G, Jiang T, Zhu G, Li H and
Qiu Z: The effects and mechanisms of blockage of STAT3 signaling
pathway on IL-6 inducing EMT in human pancreatic cancer cells in
vitro. Neoplasma. 58:396–405. 2011. View Article : Google Scholar : PubMed/NCBI
|














