Introduction
Oral cancer is a rare cancer that represents 1% of
solid cancers. Squamous cell carcinoma (SCC) represents 90% of oral
cancer. The incidence rate depends on populations because oral
squamous cell carcinoma (OSCC) is associated with tobacco use and
alcohol consumption (1,2). There is no specific biomarker and the
standard treatment for OSCC is dissection (3). Although several cancer genes are known
as drivers in head and neck squamous cell carcinoma (HNSCC), there
is no specific therapeutic molecule (4–6).
Recent sequencing studies examined HNSCC, including OSCC occurring
at a heterogeneous set of anatomical sites (1,7–9).
NOTCH1 mutations are frequently detected in OSCC in addition
to TP53, CDKN2A, PIK3CA, and HRAS mutations. These
mutations, including NOTCH1 mutations, are known in solid
cancers (1,7,8,10).
Studies on Caucasian populations indicate that NOTCH1
mutations might be associated with worse prognosis, but studies on
Asian populations indicate an oncogenic role for NOTCH1
(1).
NOTCH family includes four members (termed
NOTCH1-4), which are type 1 transmembrane receptors. NOTCH1 affects
proliferation, differentiation, and apoptosis of diverse cell types
in various organisms (11). NOTCH
proteins are composed of an extracellular domain (NECD) and an
intracellular domain (NICD). The NECD contains epidermal growth
factor repeats (EGFr) 1–36 and EGFr 12 is the ligand-binding
domain. When EGFr 12 binds to its ligands, jagged and delta family
proteins, the NOTCH receptor is cleaved. After cleavage of the
NOTCH receptor, the NICD is released from the cell membrane and it
migrates to the nucleus. The NICD interacts with RBP-J, which is a
DNA-binding protein. Finally, these complexes activate HES or HEY
family of transcription genes, thereby inducing downstream factors
(11). NOTCH signaling presents an
oncogenic or tumor suppressive effect, owing to crosstalk with the
EGF receptor (EGFR) pathway and the subsequent activation of the
PI3K-AKT pathway, KRAS, CCND1, and matrix metalloproteinase 9
(MMP9) (11).
NOTCH1 is a well-known oncogene in blood
cancer; 50% of patients with T-cells acute lymphoblastic leukemia
(T-ALL) present NOTCH1 mutations (12). However, the mutation spectrum of
NOTCH1 in OSCC is different from that of T-ALL. Therefore,
the function of NOTCH1 in solid cancer is unclear. Recent
studies suggested that NOTCH1 may play an oncogenic or tumor
suppressive role in solid cancer because the NOTCH1 pathway
regulates various oncogenes and tumor suppressor genes such as
c-Myc, PI3K, RAS family, EGFR, PTEN, and TP53.
For example, both overexpression and downregulation of NOTCH and
ligands have been implicated in several human cancers, including
OSCC, in clinical studies (11,13,14).
For HNSCC, there are some studies using cancer cell lines, but
there is no report using clinical samples. Thus, the function of
NOTCH1 in tumors is unsettled (15). We previously reported that
NOTCH1 mutations in Japanese patients with OSCC frequently
occur near the ligand-binding region (9.5%, 8 of 84 patients with
OSCC). These mutations are thought to induce a conformational
change in NOTCH1 and its downregulation (16).
At present, there is no report on the function of
NOTCH1 mutations detected in OSCC clinical samples. In this
study, we examined the expression and the functional change of a
NOTCH1 mutant (p.A465T) detected in OSCC clinical samples using
stably transformed HEK293 cells.
Materials and methods
Construction of expression
vectors
The vectors contained the EBV promoter-cDNA of
NOTCH1 gene [wild-type (WT) NOTCH1, NOTCH1 mutant [G1393A
(p.A465T)], or empty vector (MOCK)]-IRES-anti-hygromycin. The
G1393A was introduced in NOTCH1 (NM_017617) human ORF cDNA
Clone (Origene Technologies, Inc., Rockville, MD, USA) by in
vitro mutagenesis. The cDNA was cloned into the pCEP4 vector
(Invitrogen, Carlsbad, CA, USA) between the SalI and
NruI restriction sites as previously described (17).
Cell culture and transfection
HEK293 cells and transformants were cultured in
Dulbecco's modified Eagle's medium with 10% fetal bovine serum
under 5% CO2 at 37°C. HEK293 cells were transfected
using the WT NOTCH1 and p.A465T NOTCH1 mutant expression vectors
with X-tremeGENE HP Reagent (Roche, Basel, Switzerland) in a 6-well
plate, and cells were cultured with hygromycin (400 µg/ml) over 21
days. Stable transformants were used for all assays.
Flow cytometry
Cells were collected by trypsinization followed by
centrifugation at 500 µg for 5 min. Cell pellets were then
resuspended in 100 µl of phosphate buffered saline (PBS, Life
Technologies, Carlsbad, CA, USA). The following antibodies were
used for fluorescence-activated cell sorting (FACS) analysis: PE
anti-human Notch1 antibody (MHN1-519) (BioLegend, San Diego, CA,
USA) and PE mouse IgG1 κ Isotype control (BioLegend; negative
control). After incubation of the cells with 1 µl of antibody for
20 min and washing in PBS, cells were incubated at 4°C in the dark
for 1 h. They were then analyzed using a FACS Aria cell sorter (BD
Biosciences, San Jose, CA, USA).
Western blotting
Cell extracts were prepared using an ultrasonic
disrupter, and protein concentration was determined by DC protein
assay (Bio-Rad, Hercules, CA, USA). T Protein (25 µg) were loaded
on 7.5% SDS/PAGE pre-casted gels (e-PAGEL, ATTO, Tokyo, Japan) and
transferred to polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). The primary antibody used was the NOTCH1
antibody (1:1,000, #3608, Cell Signaling Technology, Danvers, MA,
USA), which recognizes the C-terminal of NOTCH1, and the secondary
antibody was the peroxidase-conjugated rabbit anti-rabbit IgG
antibody (1:2,000, #7074, Cell Signaling Technology). Rabbit
anti-GAPDH antibody (1:3,000, G9545, Sigma-Aldrich, St. Louis, MO,
USA) was used for standardization. Detection was carried out by
chemiluminescence using Western Lightning Ultra (Perkin-Elmer,
Waltham, MA, USA).
Immunofluorescence imaging
WT and A465T cells cultured on glass slides were
washed three times with PBS, fixed in 4% paraformaldehyde (PFA) for
15 min at room temperature, washed 3 times with PBS, and incubated
with 10% goat serum (Rockland Immunochemicals, Inc., Pottstown, PA,
USA) for 10 min at room temperature. After washing, the cells were
incubated with a rabbit monoclonal anti-NOTCH1 antibody (1:100,
#3608, Cell Signaling Technology) diluted in 0.05% Triton X100/1%
BSA/0.01 M PBS at 4°C for 16 h. After being washed with PBS, the
cells were incubated with Alexa Fluor® 488
F(ab')2 fragment goat anti-rabbit IgG(H+L) (1:200,
Molecular Probes, Eugene, OR, USA) at room temperature for 1 h.
Cells were washed 3 times, and nuclei were stained with
4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI)
(Sigma-Aldrich). Cell fluorescence was analyzed by laser scanning
confocal microscopy (LSM700; Carl Zeiss, Jena, Germany).
Quantitative real-time PCR
Total RNA was extracted from WT cells, A465T cells,
and MOCK cells by using TRIzol reagent (Invitrogen). RNA was
reverse-transcribed to cDNA. Primers were as follows: HES1,
forward 5′-gaagcacctccggaa cct-3′, reverse
5′-gtcacctcttcatgcactc-3′; HEY1, forward 5′-cata
cggcaggagggaaag-3′, reverse 5′-gcatctagtccttcaatgatgct-3′. These
primers were designed by using primer3plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi).
Quantitative real-time PCR (qPCR) amplification was performed using
Fast SYBR-Green Master Mix (Thermo Scientific, Waltham, MA, USA)
according to the manufacturer's instructions. β-actin was used as
the housekeeping gene.
Cell growth assay
The Cell Counting Kit-8 (CCK-8) (Dojindo
Laboratories, Kumamoto, Japan) was used to determine cell
proliferation of WT, A465T, and MOCK cells. Two thousand five
hundred cells per well were cultured in 96-well culture plates in
Dulbecco's modified Eagle's medium with 10% fetal bovine serum for
96 h. CCK-8 solution (5 µl) were added to the cells for 2 h at
37°C, and optical density (OD) was examined at a wavelength of 450
nm using a spectra Max® i3 (Molecular Devices,
Sunnyvale, CA, USA) according to the manufacturer's
instructions.
Xenograft model
WT, A465T, and MOCK cells (5×106) were
suspended in a 1:1 mixture of 200 µl PBS and Matrigel (Corning,
Tewksbury, MA, USA) and injected subcutaneously into the flank of
five female BALB/C nude mice (5 weeks old) for each group (CLEA
Japan, Tokyo, Japan). At 8 weeks after transplantation, we
evaluated the cell implantation rate. All experiments were approved
by the Animal Ethics Committee of Tokai University School of
Medicine. These tumors were excised and preserved in 10% formalin.
Hematoxylin and eosin (H&E) staining was performed by using a
standard technique. Immunohistochemical staining (IHC) was
performed by using a rabbit monoclonal anti-NOTCH1 antibody (1:100,
#3608, Cell Signaling Technology).
Statistical analysis
Statistical analyses were performed by using the
SPSS version 23 software (SPSS, Chicago, IL, USA). The data shown
represent mean values ± SD. The statistical analyses were performed
using the Student's t-test, and P<0.01 was considered to
indicate a statistically significant difference.
Results
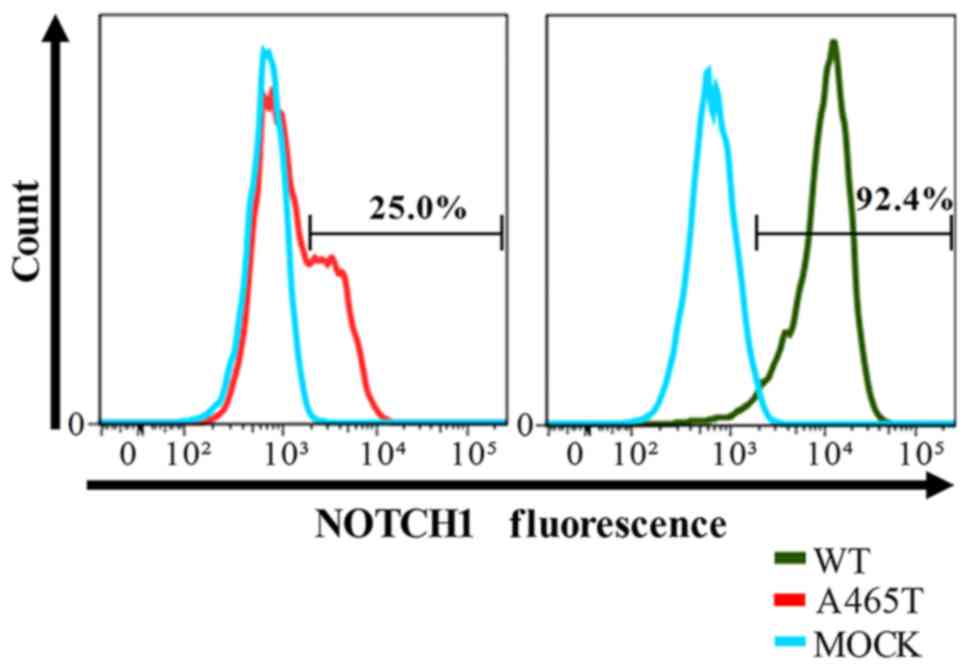
Flow cytometry
The percentage of NOTCH1 positive cells was 92.4%
and 25.0% in WT and p.A465T transformants, respectively. MOCK cells
were used as a negative control (NOTCH1 positive cells: 0.1%). The
histogram of A465T cells was diphasic, and the fluorescence
intensity of positive A465T transformed cells was lower than that
of WT transformed cells (Fig. 1).
We verified the mRNA expression level of each transfected vector.
The mRNA expression was standardized to that in MOCK cells. The
NOTCH1 mRNA level was 1.78 (WT/MOCK) in WT cells and that of A465T
cells was 0.48 (p.A465T/MOCK) consistent with the FACS
analysis.
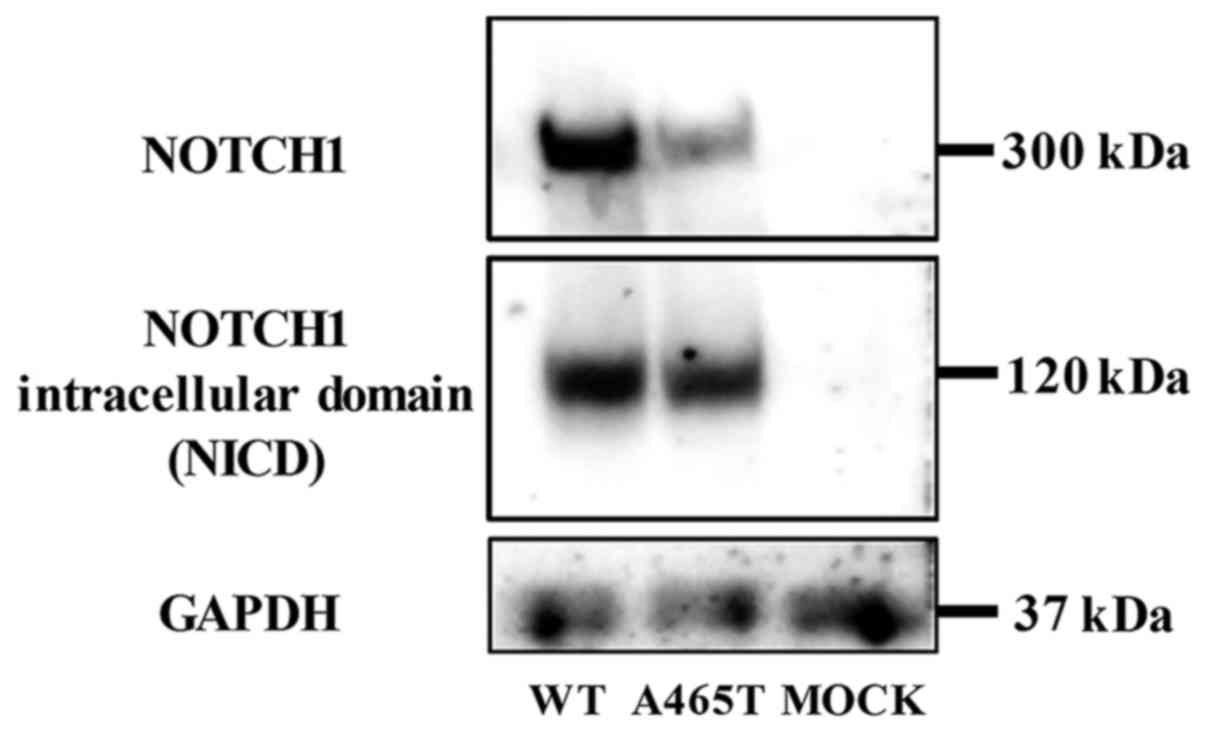
Western blotting
NOTCH1 and NICD were detected both in WT and A465T
cells. NOTCH1 and NICD expression levels were lower in A465T cells
than those in WT cells, but the NICD expression level, as compared
to that of NOTCH1, was relatively higher in A465T cells than that
in WT cells (Fig. 2).
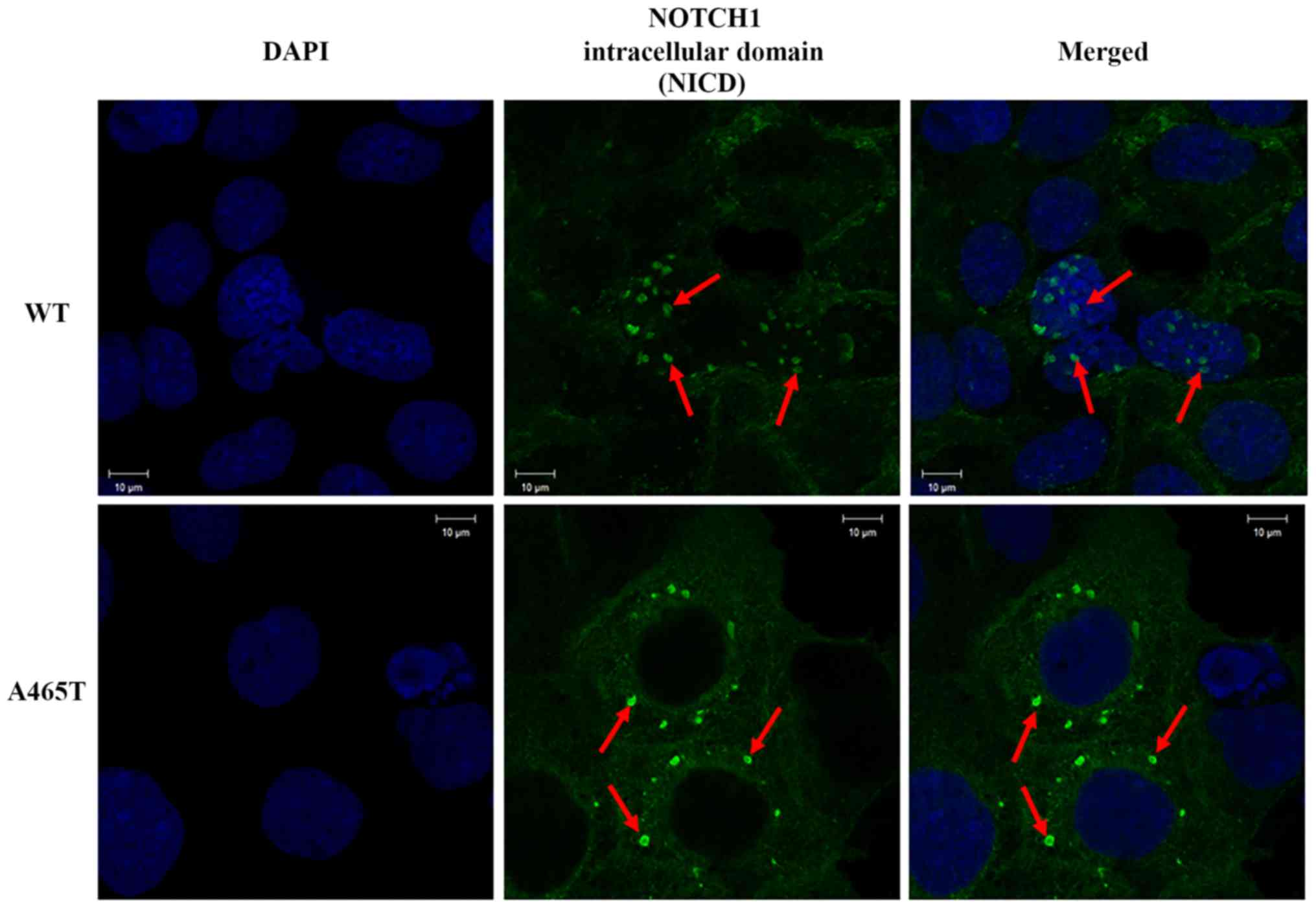
Immunofluorescence imaging
Consistent with previous studies, NICD was detected
in the nucleus in WT cells. However, we detected a strong NICD
signal in the cytoplasm of A465T cells and no signal in the
nucleus. This means that NICD was equally expressed in WT and A465T
cells, but the localization of the NICD in A465T cells was
different from that in WT cells (Fig.
3).
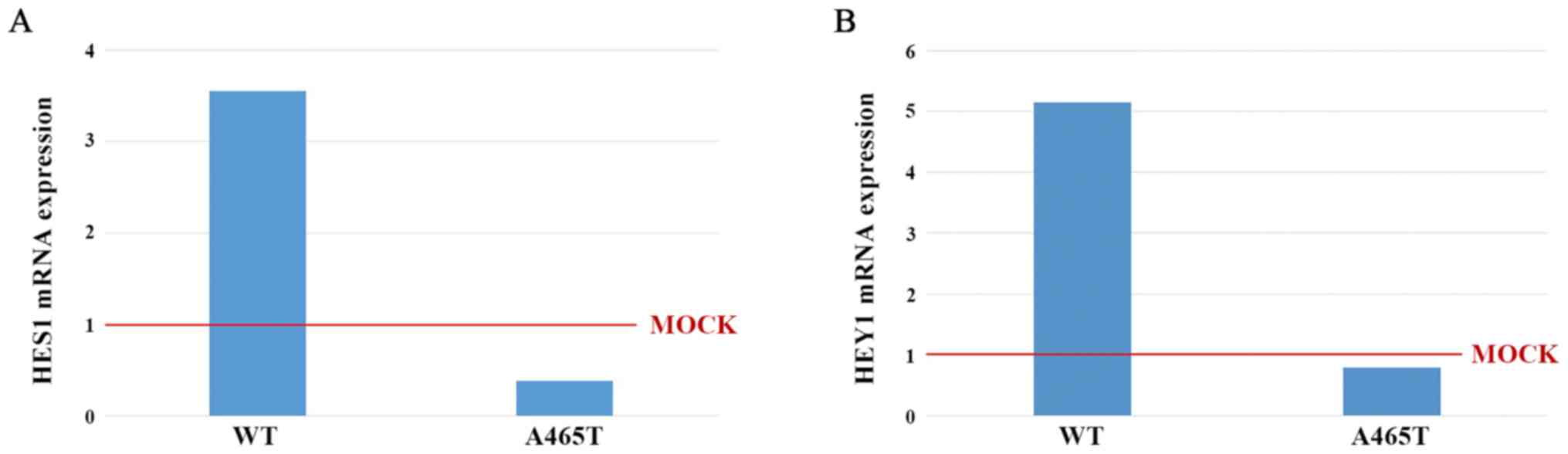
Quantitative real-time PCR
We measured HES1 and HEY1 mRNA
expression level in WT and A465T by qPCR. mRNA expression was
standardized to that in MOCK cells. The HES1 mRNA expression
levels in WT and A465T cells were 3.55 and 0.38, respectively
(Fig. 4A). The HEY1 mRNA
expression levels in WT and A465T cells were 5.15 and 079,
respectively (Fig. 4B).
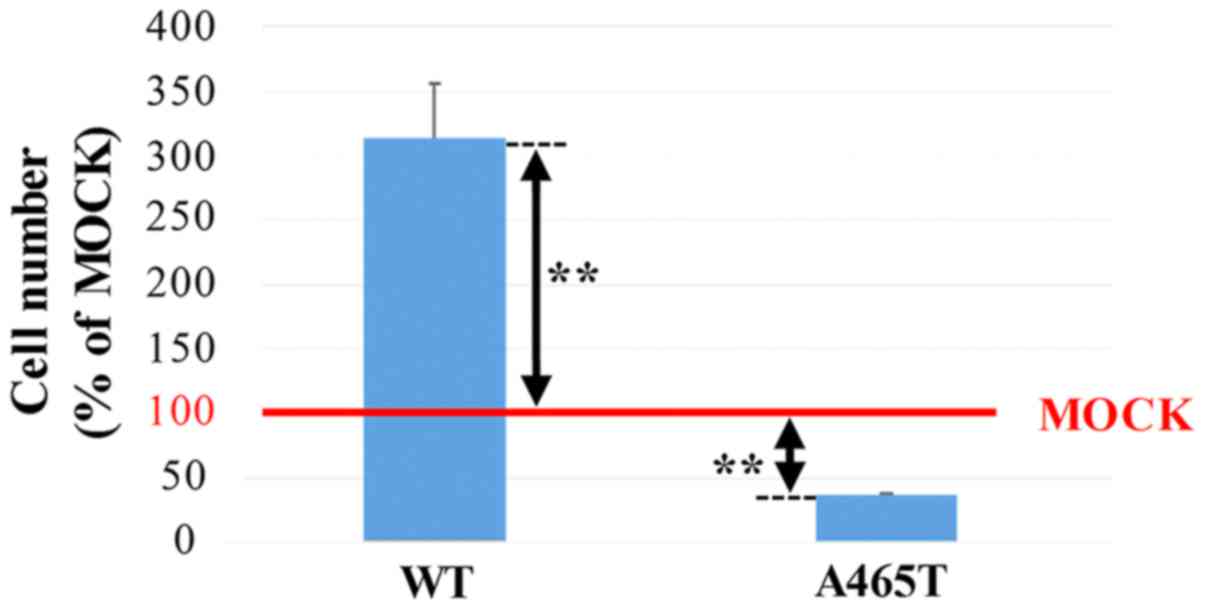
Cell growth
The cell growth rates of WT and A465T cells,
measured as a ratio of the cell number using MOCK cells as a
standard, were 313 and 37%, respectively. The A465T cell number was
significantly lower than the MOCK cell number (P<0.01), while
the WT cell number was significantly higher than the MOCK cell
number (P<0.01) (Fig. 5).
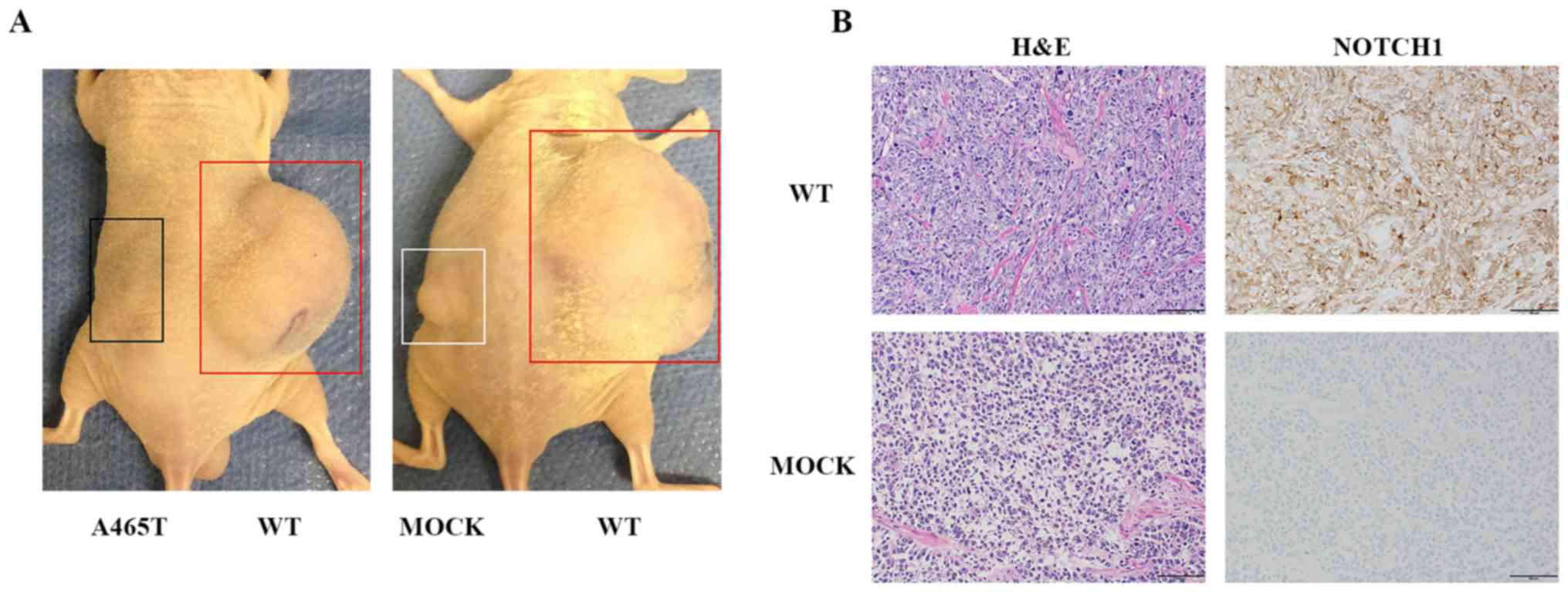
Xenograft model
WT, A465T, and MOCK cells were injected
subcutaneously in the flanks of five mice per group. WT cell
implantation was observed in the flank of four out of five mice
(implantation rate: 80%). MOCK cell implantation was detected in
the flank of three out of five mice (implantation rate: 60%).
However, no implantation was observed in mice injected with A465T
cells. Tumors generated from WT cells were obviously larger than
those generated from MOCK cells, consistent with the cell growth
assay results (Fig. 6A). We
performed H&E staining on tumors generated from each NOTCH1 WT
and MOCK cells in the flanks of nude mice at 8 weeks after
implantation. Macroscopically, nude mice presented a
well-circumscribed margin and slight fibrosis of the tumor masses.
Cell density of WT cells was higher than that of MOCK cells.
Immunohistochemical staining showed that all cells were NOTCH1
positive (Fig. 6B).
Discussion
NOTCH1 mutations are the most frequently
detected mutations in HNSCC (7).
These mutations are specifically detected in the vicinity of the
ligand-binding region of NOTCH1 in patients with OSCC and HNSCC
(7,16,18).
Among these mutations, the G1393A (p.A465T) mutant identified in
two Japanese OSCC patients is located in EGFr12, and the other six
mutations are located in EGFr10. A previous report showed that the
G1393A (p.A465T) mutation leads to a conformational change of
NOTCH1 ligand-binding domain by protein structure simulation
(16). However, recent studies
using cancer cell lines could not provide evidence about the
functional role of NOTCH1 mutations detected in clinical
samples (15,19,20).
Thus, to clarify the function of NOTCH1 in OSCC, we
established A465T cells expressing the NOTCH1 A465T mutant and
examined its characteristics.
Generally, the process of NOTCH1 activation requires
some key steps. The first step is ligand binding, which requires
NOTCH1 localization on the cell membrane. The second step is NOTCH1
cleavage between the NECD and NICD (21,22).
Finally, the translocation of NICD into the nucleus leads to the
activation of NOTCH1 signaling (23). To examine the NOTCH1 activity
between WT and A465T cells, we performed flow cytometry analysis to
quantify the amounts of NOTCH1 localized on the cell surface and
western blot analysis to evaluate NOTCH1 cleavage by detecting NICD
and NOTCH1 (24). Although NOTCH1
A465T expression was downregulated on the cell surface as shown by
flow cytometry analysis, both the cleaved NICD and full length
NOTCH1 were detected in the A465T cells by western blot analysis.
Consistent with our findings, a previous in silico report
suggested that p.A465T mutation affects the conformation of NOTCH1
ligand binding region and downregulates NOTCH1 function (16). Thus, we hypothesized that NOTCH1
A465T would be downregulated and the NICD would not be localized in
the nucleus. As mentioned above, NICD has to be cleaved from NOTCH1
in the cell membrane in order to migrate to the cytoplasm or
nucleus. To examine whether the NICD from NOTCH1 A465T participates
in the NOTCH1 signaling, we next analyzed NICD localization by
immunofluorescence and confocal microscopy. The NICD in A465T
mutant cells was localized in the cytoplasm instead of in the
nucleus by immunofluorescence analysis. Furthermore, HES1
and HEY1 mRNA expression levels were decreased in A465T
cells compared with that in MOCK cells. A recent report
demonstrated that a decrease in NOTCH1 mRNA expression
induces a decrease in HES1 and HEY1 mRNA expression
(25). Together with this study,
our findings suggest that NOTCH1 activity declined in A465T
cells.
O-fucosylation is an important factor in the
maturation of NOTCH1 because the chaperone activity must be
sufficient to support ligand binding (26). Furthermore, the disruption of a
single EGFr can dominantly perturb NOTCH activation (27). In this study, although NOTCH1 A465T
was cleaved, the NICD could not translocate to the nucleus.
According to these reports, we considered that the conformational
change in the NICD structure was associated with some
modifications, including O-fucosylation, or with a change in the
structure of the EGFrs induced by p.A465T mutation, which lead to
NOTCH1 downregulation.
We next performed cell growth assay and xenograft
implantation to evaluate the effect of WT and A465T NOTCH1 on
NOTCH1-related tumorigenesis. Cell growth assay showed that the
proliferation of WT cells increased, while that of A465T cells
decreased. The growth of NICD overexpressing cells increased and
that of NOTCH1 knocked down cells decreased in vitro
(20,28). In addition, recent reports
demonstrated that siRNA-mediated silencing of mRNA for both HEY1
and NOTCH1 causes lower levels of cell proliferation (29). In our study, HEY1 expression
levels were lower in A465T cells than in WT cells. The implantation
rate of cells in a xenograft model represents the resistance of
cell lines to immunosuppression and their adaptation to the host
microenvironment (30). NOTCH1
regulates the formation of the microenvironment (31). Our xenograft model showed that no
implantation was observed when A465T cells were injected to the
mice, while WT expressing cells were implanted at a higher rate
than MOCK cells. Thus, our cell growth assay and xenograft model
indicated that WT NOTCH1 leads to increases tumorigenicity, while
p.A465T mutation abolishes it. These data are consistent with our
previous report that median disease-free survival of OSCC patients
with NOTCH1 mutation in the vicinity of the ligand binding region
is significantly longer than that of patients presenting with WT
NOTCH1 (16). Together with
previous evidence, these findings suggest that the p.A465T mutation
mediates NOTCH1 downregulated tumorigenicity.
In this study, we expected a negative relationship
between p.A465T mutation and NOTCH1 activity. However, our results
did not directly demonstrate whether NOTCH1 activity was lost in
A465T cells. Further experiments are required to determine whether
p.A465T located in EGFr 12 leads to the instability of NOTCH1
within cell membrane. However, this mutation did not directly
contribute to the defect in NICD migration as shown in Fig. 3. Secondly, since NOTCH signaling
crosstalks with other pathways associated with tumorigenicity,
other molecular factors might abolish NOTCH1 tumorigenicity. Third,
ligand stimulation is followed by the cleavage step of NOTCH1.
Thus, we expect that the p.A465T mutant could lead to alternative
cleavage of NOTCH1, resulting in structural changes and/or unusual
migration of NICD.
In conclusion, our findings suggest that NOTCH1 acts
as an oncogene and that p.A465T NOTCH1 mutation in the
ligand-binding region affect cell growth and/or tumorigenicity of
OSCC. Furthermore, we consider that NOTCH1 mutations in the
ligand-binding region could be good prognostic factors and the
domain could be used as a new therapeutic molecule in OSCC. Further
studies are warranted to elucidate the direct relationship between
the p.A465T mutant and NOTCH1 activity.
Acknowledgements
We are grateful to the Support Center for Medical
Research and Education, Tokai University, for technical support.
This work was supported by Grants-in-Aid for Young scientists (B)
(KAKENHI Grant Number 16K20614) from Japan Society of the Promotion
of Science (JSPS), Grants-in-Aid for Scientific Research (C)
(KAKENHI Grant Number 15K11302) from JSPS and 2015 Tokai University
School of Medicine Research Aid. We thank Editage for language
editing.
References
|
1
|
Chaturvedi AK, Anderson WF,
Lortet-Tieulent J, Curado MP, Ferlay J, Franceschi S, Rosenberg PS,
Bray F and Gillison ML: Worldwide trends in incidence rates for
oral cavity and oropharyngeal cancers. J Clin Oncol. 31:4550–4559.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Warnakulasuriya S: Global epidemiology of
oral and oropharyngeal cancer. Oral Oncol. 45:309–316. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pfister DG, Spencer S, Brizel DM, Burtness
B, Busse PM, Caudell JJ, Cmelak AJ, Colevas AD, Dunphy F, Eisele
DW, et al: Head and Neck Cancers, Version 1.2015. J Natl Compr Canc
Netw. 13:847–856. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chi LM, Lee CW, Chang KP, Hao SP, Lee HM,
Liang Y, Hsueh C, Yu CJ, Lee IN, Chang YJ, et al: Enhanced
interferon signaling pathway in oral cancer revealed by
quantitative proteome analysis of microdissected specimens using
16O/18O labeling and integrated two-dimensional LC-ESI-MALDI tandem
MS. Mol Cell Proteomics. 8:1453–1474. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mazumdar A, Henderson YC, El-Naggar AK,
Sen S and Clayman GL: Aurora kinase A inhibition and paclitaxel as
targeted combination therapy for head and neck squamous cell
carcinoma. Head Neck. 31:625–634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Colella S, Richards KL, Bachinski LL,
Baggerly KA, Tsavachidis S, Lang JC, Schuller DE and Krahe R:
Molecular signatures of metastasis in head and neck cancer. Head
Neck. 30:1273–1283. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stransky N, Egloff AM, Tward AD, Kostic
AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C,
McKenna A, et al: The mutational landscape of head and neck
squamous cell carcinoma. Science. 333:1157–1160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vettore AL, Ramnarayanan K, Poore G, Lim
K, Ong CK, Huang KK, Leong HS, Chong FT, Lim TK, Lim WK, et al:
Mutational landscapes of tongue carcinoma reveal recurrent
mutations in genes of therapeutic and prognostic relevance. Genome
Med. 7:982015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
India Project Team of the International
Cancer Genome Consortium: Mutational landscape of gingivo-buccal
oral squamous cell carcinoma reveals new recurrently-mutated genes
and molecular subgroups. Nat Commun. 4:28732013.PubMed/NCBI
|
|
10
|
Ojesina AI, Lichtenstein L, Freeman SS,
Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio
L, Cibulskis K, Bertelsen B, et al: Landscape of genomic
alterations in cervical carcinomas. Nature. 506:371–375. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ranganathan P, Weaver KL and Capobianco
AJ: Notch signalling in solid tumours: A little bit of everything
but not all the time. Nat Rev Cancer. 11:338–351. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vicente C, Schwab C, Broux M, Geerdens E,
Degryse S, Demeyer S, Lahortiga I, Elliott A, Chilton L, La Starza
R, et al: Targeted sequencing identifies associations between
IL7R-JAK mutations and epigenetic modulators in T-cell acute
lymphoblastic leukemia. Haematologica. 100:1301–1310. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Radtke F and Raj K: The role of Notch in
tumorigenesis: Oncogene or tumour suppressor? Nat Rev Cancer.
3:756–767. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leong KG and Karsan A: Recent insights
into the role of Notch signaling in tumorigenesis. Blood.
107:2223–2233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yap LF, Lee D, Khairuddin A, Pairan MF,
Puspita B, Siar CH and Paterson IC: The opposing roles of NOTCH
signalling in head and neck cancer: A mini review. Oral Dis.
21:850–857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aoyama K, Ota Y, Kajiwara K, Hirayama N
and Kimura M: Frequent mutations in NOTCH1 ligand-binding regions
in Japanese oral squamous cell carcinoma. Biochem Biophys Res
Commun. 452:980–985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizutani A, Kikkawa E, Matsuno A,
Shigenari A, Okinaga H, Murakami M, Ishida H, Tanaka M and Inoko H:
Modified S/MAR episomal vectors for stably expressing fluorescent
protein-tagged transgenes with small cell-to-cell fluctuations.
Anal Biochem. 443:113–116. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Agrawal N, Frederick MJ, Pickering CR,
Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et
al: Exome sequencing of head and neck squamous cell carcinoma
reveals inactivating mutations in NOTCH1. Science. 333:1154–1157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun Y, Zhang R, Zhou S and Ji Y:
Overexpression of Notch1 is associated with the progression of
cervical cancer. Oncol Lett. 9:2750–2756. 2015.PubMed/NCBI
|
|
20
|
Su BH, Qu J, Song M, Huang XY, Hu XM, Xie
J, Zhao Y, Ding LC, She L, Chen J, et al: NOTCH1 signaling
contributes to cell growth, anti-apoptosis and metastasis in
salivary adenoid cystic carcinoma. Oncotarget. 5:6885–6895. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Yu S, Huang D, Cui M, Hu H, Zhang
L, Wang W, Parameswaran N, Jackson M, Osborne B, et al: Cellular
prion protein mediates pancreatic cancer cell survival and invasion
through association with and enhanced signaling of Notch1. Am J
Pathol. 186:2945–2956. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hsu KW, Fang WL, Huang KH, Huang TT, Lee
HC, Hsieh RH, Chi CW and Yeh TS: Notch1 pathway-mediated
microRNA-151-5p promotes gastric cancer progression. Oncotarget.
7:38036–38051. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Croquelois A, Domenighetti AA, Nemir M,
Lepore M, Rosenblatt-Velin N, Radtke F and Pedrazzini T: Control of
the adaptive response of the heart to stress via the Notch1
receptor pathway. J Exp Med. 205:3173–3185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pickering CR, Zhang J, Yoo SY, Bengtsson
L, Moorthy S, Neskey DM, Zhao M, Alves Ortega MV, Chang K, Drummond
J, et al: Integrative genomic characterization of oral squamous
cell carcinoma identifies frequent somatic drivers. Cancer Discov.
3:770–781. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shao S and Zhao X, Zhang X, Luo M, Zuo X,
Huang S, Wang Y, Gu S and Zhao X: Notch1 signaling regulates the
epithelial-mesenchymal transition and invasion of breast cancer in
a Slug-dependent manner. Mol Cancer. 14:282015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma L, Dong P, Liu L, Gao Q, Duan M, Zhang
S, Chen S, Xue R and Wang X: Overexpression of protein
O-fucosyltransferase 1 accelerates hepatocellular carcinoma
progression via the Notch signaling pathway. Biochem Biophys Res
Commun. 473:503–510. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okajima T, Xu A, Lei L and Irvine KD:
Chaperone activity of protein O-fucosyltransferase 1 promotes notch
receptor folding. Science. 307:1599–1603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yoshida R, Nagata M, Nakayama H,
Niimori-Kita K, Hassan W, Tanaka T, Shinohara M and Ito T: The
pathological significance of Notch1 in oral squamous cell
carcinoma. Lab Invest. 93:1068–1081. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun W, Gaykalova DA, Ochs MF, Mambo E,
Arnaoutakis D, Liu Y, Loyo M, Agrawal N, Howard J, Li R, et al:
Activation of the NOTCH pathway in head and neck cancer. Cancer
Res. 74:1091–1104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jung J: Human tumor xenograft models for
preclinical assessment of anticancer drug development. Toxicol Res.
30:1–5. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weber JM and Calvi LM: Notch signaling and
the bone marrow hematopoietic stem cell niche. Bone. 46:281–285.
2010. View Article : Google Scholar : PubMed/NCBI
|