Introduction
Cervical cancer is the third most commonly diagnosed
cancer and the fourth in mortality among females worldwide
(1,2). The incidence is lower in developed
countries as a consequence of screening by cervical cytology and
human papillomavirus (HPV) (3).
However, the prognosis of patients with advanced-stage cervical
cancer remains unsatisfactory, especially for those in developing
countries where widespread screening is unavailable (4). What is more, cervical cytology and HPV
screening also have some shortcomings (5). Metastasis of cervical cancer is
responsible for ~90% of all poor prognosis (3). Therefore, it is urgent for cervical
cancer to identify causes and potential markers of metastasis in
order to find novel therapeutic strategies.
Inhibitor of β-catenin and T-cell factor (ICAT) is a
small protein of 81 amino acids that was first reported in 2000
(6).ICAT was demostrated as an
inhibitor of the Wnt/β-catenin signaling pathway at first (7). Subsequently, crystallographic analyses
of ICAT have proved its β-catenin binding sites which was a
β-sheet-like conformation and located in carboxy-terminal domain
(8–10). As research continued, ICAT was also
found to inhibit the intercellular adhesion function (11).
Epithelial-to-mesenchymal transition (EMT) has been
recognized as an important process in embryogenesis and
carcinogenesis (12,13). During EMT, epithelial cells convert
to mesenchymal cells, which involves profound phenotypic changes
including loss of cell-cell adhesion and cell polarity (14). Increasing evidence implicates that
EMT is a key process for metastasis in cervical cancer (15,16).
However, whether ICAT is associated with the process of EMT in
cervical cancer is still unknown.
In the present study, we demonstrated that ICAT was
highly expressed in cervical cancer tissues and had a role in
control of EMT in cervical cancer cells. Overexpression of ICAT
promoted cell proliferation, migration, invasion and resulted in
EMT by disrupting the stability of E-cadherin/β-catenin complex,
whereas the opposite effect was observed in downregulation of ICAT
in Caski cells. We thus, identified a novel role and regulatory
mechanism of ICAT in EMT in cervical cancer.
Materials and methods
Human tissue samples
Cervical cancer and normal cervical tissues were
collected from patients who undergone hysterectomy at the First
Affiliated Hospital, Chongqing Medical University. None of the
patients received any preoperative chemotherapy, immunotherapy, or
radiotherapy. Written informed consent was obtained from each
patient at the time of surgery, and all collections were approved
by the Clinical Ethics Committee of the hospital.
Cell culture
The human cervical cancer cell lines SiHa, HeLa and
Caski were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA) and cultured in Dulbecco's modified
Eagle's medium (DMEM; HyClone Laboratories, Inc., Logan, UT, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA,
USA), 1% penicillin and 1% streptomycin. All cells were cultured at
37°C in a 5% CO2 incubator under humidified
atmosphere.
Immunohistochemistory (IHC)
Cervical cancer tissues, normal cervical tissue and
paraffin-embedded nude mouse xenograft cervical tumors were sliced
into 4-µm consecutive sections. Sections were then dewaxed in
xylene, rehydrated in graded ethanol and heat-treated for antigen
retrieval with citric acid buffer. Sections were cooled and
incubated in a 0.3% hydrogen peroxide solution for 20 min, blocked
with normal goat serum for 30 min and then incubate with
corresponding antibody at 4°C overnight. Subsequently, the sections
were performed using an immunohistochemistry SP-9000 kit (Beijing
Zhongshan Golden Bridge Biotechnology, Co., Ltd., Beijing, China).
Staining procedures were performed under standardized conditions.
Subsequently, hematoxylin were used for counterstain. All slides
were scored by a pathologist in a blinded manner. The evaluation of
ICAT immunoreactivity was performed using the immunoreactivity
scores. The score was determined by multiplying the staining
intensity by the staining extent. The staining intensity for ICAT
was scored as 0, negative; 1, weak; 2, moderate; and 3 strong. The
staining extent was scored as 0, 0%; 1, 1–25%; 2, 26–50%; 3,
51–75%; and 4, 76–100% according to the percentage of positively
stained cells. The immunoreactivity was divided to two grades on
the basis of the final score: negative immunoreactivity was defined
as a total score of 0–4 and positive was defined as a total score
>4.
Western blot analysis
The cells (2×106) were washed three times
with cold phosphate-buffered saline (PBS) and lysed in RIPA lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China). Cell
lysis solution was centrifuged at 13,000 × g for 15 min at 4°C and
protein solution were collected. The BCA protein assay kit
(Beyotime Institute of Biotechnology) was used to measure the
protein concentration. Equivalent amounts of protein were loaded in
SDS-PAGE polyacrylamide gels and then transferred onto PVDF
membranes. The PVDF membranes were blocked with 5% bovine serum
albumin (BSA; Beijing Solarbio Science and Technology, Co., Ltd.,
Beijing, China) in TBST for 2 h at 37°C. Subsequently, the
membranes were incubated with the primary antibodies overnight at
4°C. The following primary antibodies were used in the present
study: monoclonal rabbit anti-ICAT, anti-MMP9 (1:5,000; Abcam,
Cambridge, MA, USA); monoclonal rabbit anti-β-catenin (1:1,000,
Abcam), polyclonal rabbit anti-E-cadherin, anti-vimentin and
anti-Snail (1:1,000; Bioworld Technology, Inc., Nanjing, China),
monoclonal mouse anti-β-actin (1:1,000; Beijing Zhongshan Golden
Bridge Biotechnology); polyclonal rabbit anti-PCNA, anti-cyclin D1
(1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The
membranes were washed with TBST 3 times and incubation with a
secondary antibody (1:5,000; Beijing Zhongshan Golden Bridge
Biotechnology) for 1 h at 37°C. The SuperSignal West Pico
Chemiluminescent substrate kit was used to quantifiy protein
levels.
RNA isolation and quantitative
real-time reverse transcription-PCR (qRT-PCR)
Cells were treated with AdICAT in FBS-free DMEM for
48 h. Total RNA was isolated using TRIzol reagent (Invitrogen,
Carlsbad, CA, USA) according to the RNA extraction protocol. Total
RNA (1.5 µg) was used for cDNA synthesis by reverse transcriptase
PCR. The cDNA was amplified by a real-time polymerase chain
reaction (qPCR) system (Bio-Rad Laboratories) using SYBR-Green PCR
Master Mix. β-actin was used as the endogenous control. Reaction
conditions were as follows: denaturation at 94°C for 10 sec,
annealing at 57°C for 20 sec and extension at 72°C for 10 sec. The
specific primers for ICAT were 5′-ATGAACCGCGAGGGAGCTCC-3′ (forward)
and 5′-CTACTGCCTCCGGTCTTCCG-3′ (reverse). Those for β-actin were
5′-GATGACCCAGATCATGTTTGAG-3 (forward) 5′-AGGGCATACCCCTCGTAGAT-3′
(reverse).
Adenoviral transfections and small
interfering RNA
Recombinant adenovirus AdICAT and negative control
AdRFP were kindly donated by Dr Tongchuan He (University of Chicago
Medical Center, Chicago, IL, USA). AdICAT or AdRFP was transfected
into the SiHa cells with polybrene (Sigma-Aldrich). After 8 h of
cultivation, the medium was replaced with a fresh medium without
FBS. The fluorescence was then observed 36 h later.
Three ICAT-siRNAs and negative control (NC) were
purchased from Shanghai GenePharma, Co., Ltd. (Shanghai, China).
The sequences are shown in Table I.
Cells were transfected using a Lipofectamine RNAiMAX kit
(Invitrogen) according to the manufacturers instructions.
 | Table I.The sequences of the siRNAs and
negative control. |
Table I.
The sequences of the siRNAs and
negative control.
| siRNAs | Sense | Antisense |
|---|
| siICAT |
#1:GGUGCUUUAGUUAGGUCAUTT |
AUGACCUAACUAAAGCACCTT |
|
|
#2:GGUCCACAGCUGGAAAGUUTT |
AACUUUCCAGCUGUGGACCTT |
|
|
#3:GGCUCCAUAUUUCAACUAATT |
UUAGUUGAAAUAUGGAGCCTT |
| siNC |
UUCUCCGAACGUGUCACGUTT |
ACGUGACACGUUCGGAGAATT |
MTT assay
Cell viability was detected by MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
assay. A total of 4×103 cells were seeded into each well
of 96-well plates in quintuplicate and cultured for 24, 48, 72 and
96 h. At the indicated time, 10 µl of MTT (5 mg/ml; Sigma-Aldrich)
was added into each well and then incubated for 4 h at 37°C. Next,
150 µl of dimethyl sulfoxide (DMSO) was added to the 96-well plates
to dissolve the formazan product. Finally, the absorbance was
measured at 492 nm on a microplate reader. the overall experiments
were repeated at least three times.
Colony-forming assay
Log-phase cells were collected and ~500 cells were
plated into each well of a 6-well plate. The cells were cultured in
10% FBS medium and the medium was repalaced every four days. When
clones were observed, the cells were fixed with 4%
paraformaldehyde, washed twice with PBS and stained with 0.1%
crystal violet. The visible colonies were counted. Each experiment
was repeated thrice.
Flow cytometric analysis
Cells (1×106) from each group were
harvested and washed thrice with cold PBS, followed by fixation
with 70% cold ethanol at 4°C overnight. After being washed in PBS,
the cells were incubated with propidium iodide (PI; Sigma-Aldrich)
and RNaseA for 30 min at room temperature. The cell cycle
distribution was measured by a FACSVantage SE flow cytometer
(Becton-Dickinson, San Jose, CA, USA).
Migration and invasion assay
Transwell chambers (24-well Transwell chambers, 8-µm
pore size; Corning, Inc., Corning, NY, USA) were utilized for
migration and invasion assays. For the migration assay,
~4×105/400 µl cells in serum-free media were seeded into
upper chambers after infection for 48 h. The lower chamber
contained medium with 20% FBS. Following 24-h incubation, the cells
which invaded to the lower surface of the chamber were fixed with
4% paraformaldehyde and stained with 0.1% crystal violet, and
counted from five random fields by bright field microscopy. The
Transwell invasion assay course was similar to the migration assay
except that the Transwell membrane was coated with 1:4 diluted
Matrigel beforehand. Each experiment was repeated thrice.
Immunofluorescence staining
Cells cultured on chamber coverslips were fixed with
4% paraformaldehyde for 15 min and blocked with 10% goat serum
(0.2% Triton X-100 in PBS) for 1 h at 37°C. Then, the sections were
incubated with anti-E-cadherin rabbit, anti-vimentin rabbit (1:100;
Bioworld Technology) and anti-β-catenin (1:100; Abcam) rabbit at
4°C overnight. Thereafter, cells were incubated with DyLight 488
AffiniPure goat anti-rabbit IgG (H+L) (1:800; EarthOx, LLC, San
Francisco, CA, USA) for 1 h at 37°C. Images were captured under a
fluorescence microscope (Eclipse Ti-S; Nikon, Tokyo, Japan).
Immunoprecipitation
Whole cells were washed three times with cold PBS
and resuspended in RIPA lysis buffer at 4°C for 30 min.
Anti-β-catenin antibody (5 µg; Santa Cruz Biotechnology) was
incubated with Protein A/G Magnetic Beads (Biotools Co., Ltd.,
Shanghai, China) at room temperature for 20 min. Cell lysis
solution was centrifuged at 14,000 × g for 10 min and protein
solution was added into the Protein A/G Magnetic Beads and
incubated overnight at 4°C. Then, the beads were washed three times
with wash buffer. Proteins were eluted by boiling with 20 µl 1X
SDS-PAGE loading buffer. Finally, the expression of the target
protein was evaluated by western blot analysis.
Xenograft mouse experiment
The in vivo experiments were approved by the
guidelines established by the Animal Care and Use Committee of
Chongqing Medical University Laboratory Animal Research. The
4–6-week old female nude mice were randomly divided into 3 groups
(n=5/group). Untreated SiHa cells (2×107/each nude
mouse), AdRFP-infected SiHa cells (2×107/each nude
mouse) and AdICAT-infected SiHa cells (2×107/each nude
mouse) were injected subcutaneously into the posterior flank
position of the nude mice. Untreated SiHa cells and SiHa/AdRFP
served as control groups, whereas SiHa/AdICAT served as the
treatment group. Tumor dimensions were recorded every week with
vernier calipers, and the volumes were calculated using the
following formula: π/6 × (lengh × width2). The mice were
sacrificed by cervical vertebra dislocation after 5 weeks, and
tumor tissues were collected, embedded in paraffin for H&E and
immunohistochemical analysis.
Results
ICAT is upregulated in human cervical
cancer tissues, and verification of recombinant SiHa/ICAT
To investigate the role of ICAT in human cervical
carcinogenesis, we first detected the endogenous expression of ICAT
in human normal cervix and cervical cancer by immunohistochemistry
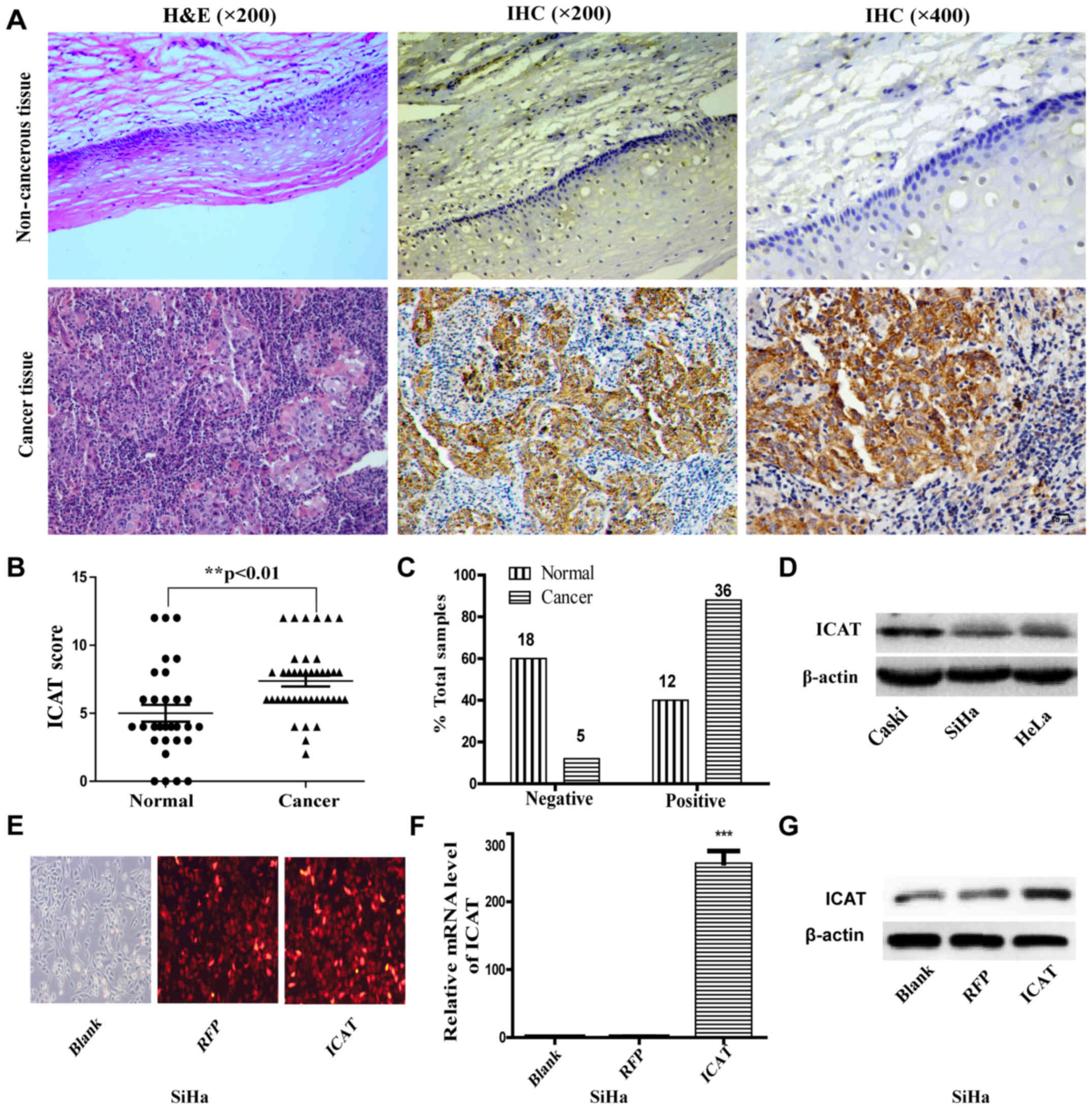
(IHC). The representative ICAT staining is shown in Fig. 1A. Samples were scored based on the
immunoreactivity scores: negative (1–4) and
positive (5–12) (17).
The average scores of IHC for ICAT were 5.000±0.6215 in normal
cervix samples and 7.366±0.3916 in cervical cancer (Fig. 1B). The positive ICAT expression
rates were 40.0% (12/30) in normal cervix and 87.8% (36/41) in
cervical cancer (Fig. 1C;
P<0.01). To further confirm the role of ICAT in human cervical
cancer, we detected the expression of ICAT by qRT-PCR and western
blot analysis in three human cervical cancer cell lines (HeLa, SiHa
and Caski) (Fig. 1C and D). The
results showed that ICAT mRNA and protein were detected in all
three cervical cancer cell lines and Caski cells showed higher
expression of ICAT; however, SiHa showed lower expression. These
data suggested that ICAT was upregulated in cervical cancer and it
may be involved in carcinogenesis. Thus, we used SiHa and Caski
cells as a model to investigate the function of ICAT on cell
proliferation, migration and invasion. SiHa cells were transfected
with ICAT-expressing adenoviruses (AdICAT) to generate recombinant
SiHa/ICAT. The transfection efficiency of SiHa cells at 36 h was
observed under a fluorescence microscope (Fig. 1E). qRT-PCR and western blot assay
showed that recombinant SiHa/ICAT cells were successfully
established and were appropriately prepared for the subsequent
experiments (Fig. 1F and G).
ICAT overexpression promotes
proliferation, migration and invasion in SiHa cells
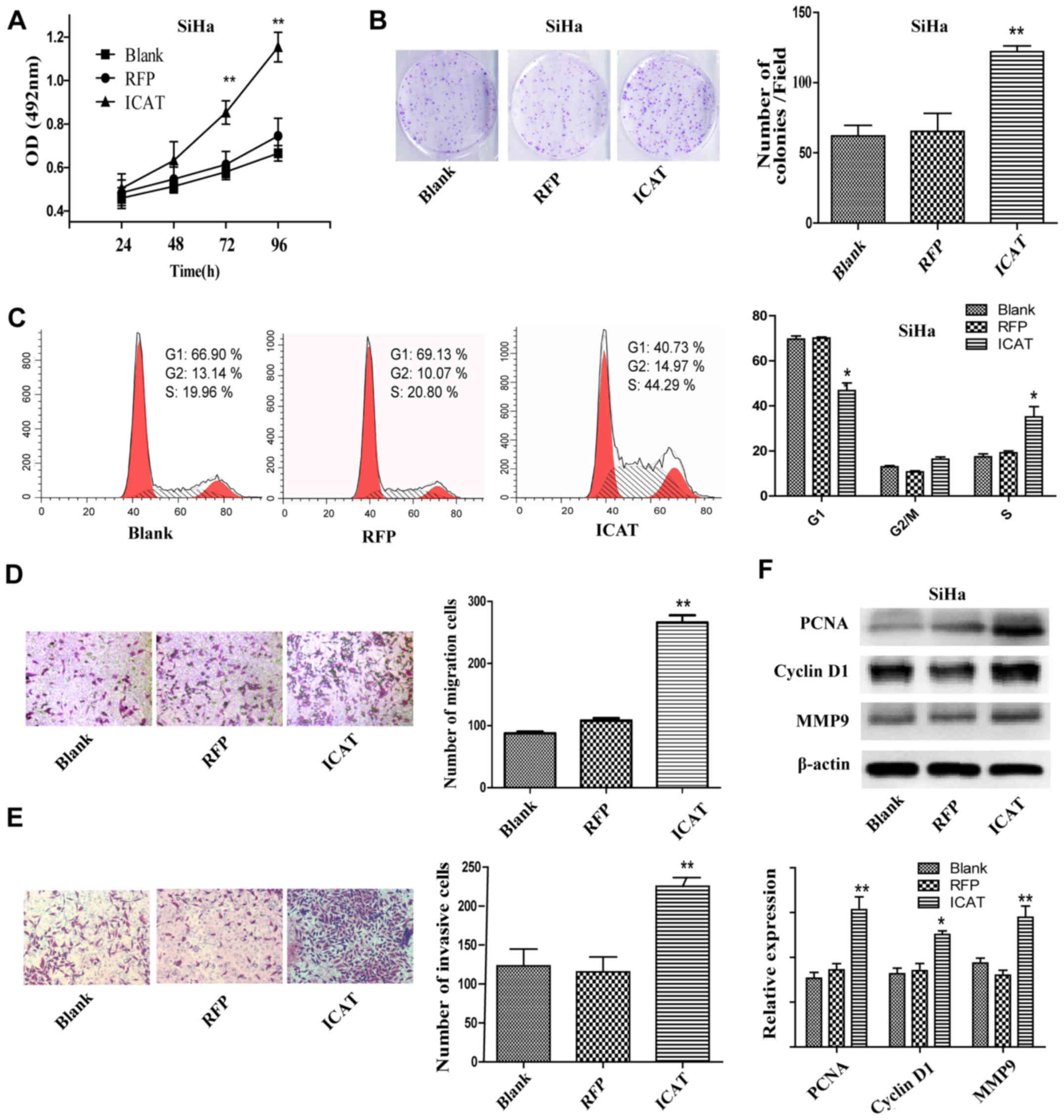
After AdICAT-mediated enforced ICAT expression in
SiHa cells, cell proliferation ability was assessed by MTT assay
and colony formation assay. The MTT assay showed that the
proliferation of the SiHa/ICAT cells was increased significantly
compared with the Blank and RFP groups (Fig. 2A). Colony formation assay results
showed that the number of colonies of SiHa/ICAT were approximately
double that of the controls (Fig.
2B). Flow cytometry revealed that ICAT overexpression led to an
increase in the percentage of cells in the S phase and a decrease
in the number of cells in the G1 phase compared with the controls
(Fig. 2C). Cell migration and
invasion abilities were detected by Transwell migration assays and
Transwell invasion assays, respectively. As shown in Fig. 2D and E, ICAT introduction caused a
remarkable increase of SiHa cells that invaded to the lower surface
of the chamber. Furthermore, we detected the protein levels of
migration-related factor 9 (MMP9), DNA replication factor (PCNA)
and cyclin D1 by western blotting. The results showed that PCNA,
cyclin D1 and MMP9 were upregulated after ICAT overexpression in
the SiHa cells (Fig. 2F).
Downregulation of ICAT inhibits the
proliferation, migration and invasion in Caski cells
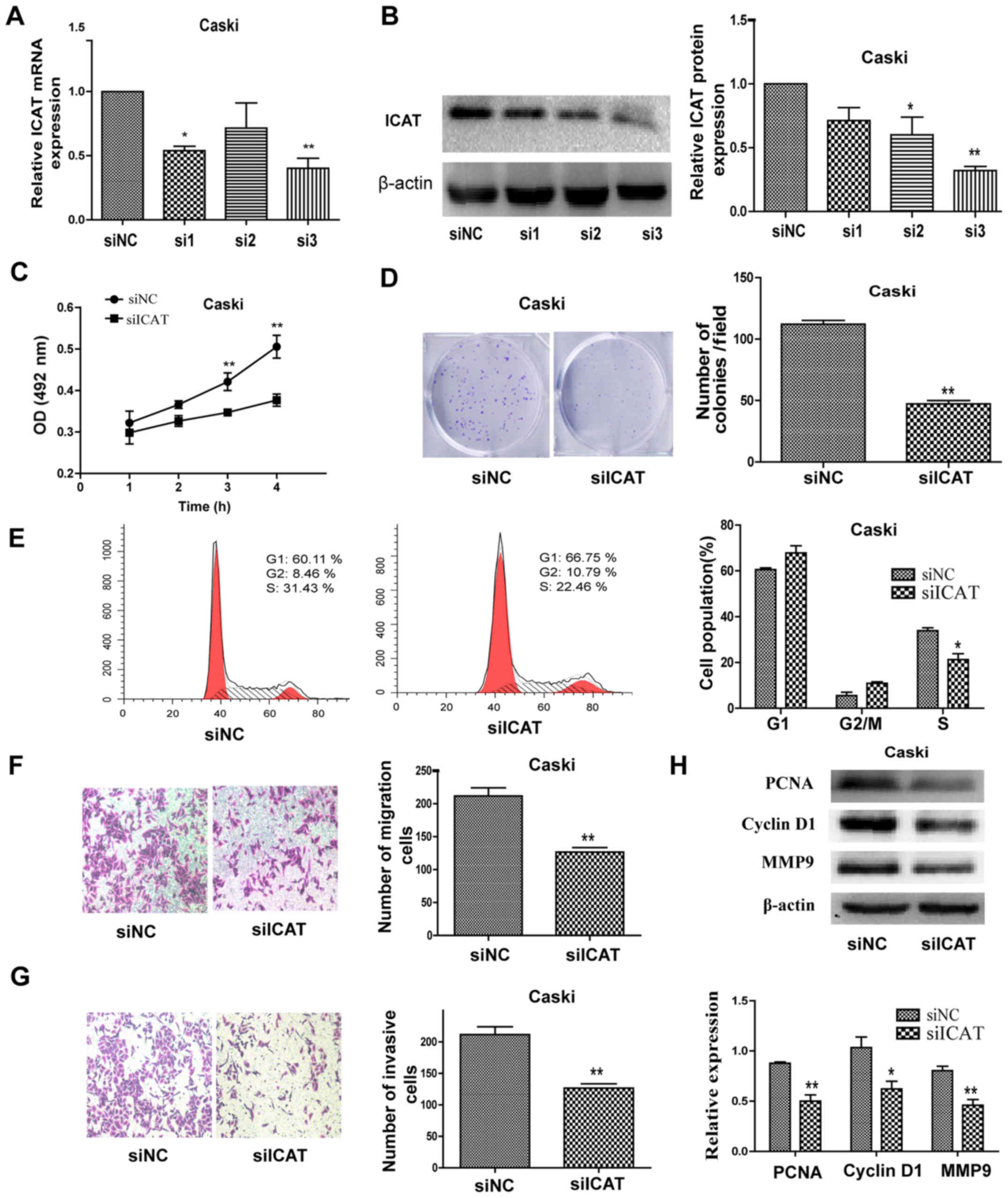
To further examine whether ICAT is involved in
cervical cancer progression, we synthesized three siRNAs
specifically targeting ICAT. The interference efficiency of the
three siRNAs targeting ICAT was verified by qRT-PCR and western
blotting (Fig. 3A and B). As shown
in Fig. 3A and B, ICAT-siRNA-3 had
a significant inhibitory effect on ICAT and were used in the
following assays. MTT assay and colony formation assay results
showed that knockdown of ICAT by RNAi significantly decreased cells
proliferation of Caski cells (Fig. 3C
and D). Furthermore, we compared the cell cycle profiles of
ICAT knockdown cells by flow cytometry. Suppression of ICAT led to
a decrease in the number of cells in the S-phase and an increase in
the percentage of cells in the G1 phase (Fig. 3E). The migration and invasion
effects in Caski cells by inhibiting ICAT were also investigated.
Following transfection with siICAT for 48 h, both the migratory and
invasive effects of Caski cells were significantly decreased
compared with the control groups (Fig.
3F and G). Quantification of invading cells revealed a
significant decrease in the number of migrating and invading cells
for Caski cells after ICAT knockdown. In addition, western blotting
showed that PCNA, cyclin D1 and MMP9 were downregulated after ICAT
knockdown in the Caski cells (Fig.
3H).
ICAT promotes EMT of cervical cancer
cells and disrupts E-cadherin/β-catenin complex
A significant morphological alteration was observed
by microscopy after ICAT-overexpressing in the course of
experiments. As seen in Fig. 4A,
AdICAT-mediated enforced ICAT expression in SiHa cells exhibited
more mesenchymal morphology with spindle-like shape, while control
cells displayed more cobblestone-shaped morphology (18,19).
Thus, we investigated whether ICAT have a regulatory effect on EMT
program in cervical cancer cells. The expression of EMT-related
markers were detected by western blot analsyis in SiHa and Caski
cells after transfection with AdICAT or siICAT. As expected, the
overexpression of ICAT induced marked upregulation of mesenchymal
marker vimentin and snail but downregulation of epithelial marker
E-cadherin and β-catenin in SiHa cells. Furthermore, ICAT knockdown
remarkably elevated the expression of E-cadherin and β-catenin and
at the same time significantly suppressed the expression of snail
and vimentin in Caski cells (Fig.
4B). In order to better demonstrate the ICAT regulated EMT in
cervical cancer cells, we tested the EMT-related marker changes by
immunofluorescence analysis. The results showed that the vimentin
staining was enhanced while E-cadherin staining was reduced in ICAT
groupe compared with the negative controls (Fig. 4C). Having identified ICAT as a
promoter of EMT, we were interested in identifying the mechanism
behind these effects. It has been reported that ICAT plays a role
in β-catenin-dependent nuclear signaling and E-cadherin/β-catenin
complex of the cell adhesion (11),
thus, we turned our attention on β-catenin in cervical cancer. Koay
et al (20) found that
β-catenin has aberrant localization in the cytoplasm and no
expression was found in the nucleus in 126 invasive carcinomas of
different histological types. We also illustrated the endogenous
expression of nuclear β-catenin and cytoplasm β-catenin in SiHa,
Caski, HeLa and MDA-MB-231 which is aberrantly activated in
Wnt/β-catenin signaling (22), and
used as postive control in western blot analysis. As seen in
Fig. 4D, β-catenin protein was
observed in the cytoplasm mainly and the expressiong of nuclear
β-catenin was very faint. Immunofluorescence staining analysis of
the cellular distribution of β-catenin proteins in SiHa (Fig. 4E) also proved β-catenin was mainly
localized in the cytoplasm. Thus, we hypothesized that ICAT
influence the E-cadherin/β-catenin complex and EMT mainly in
cervical cancer. As confirmation, we further verified that ICAT
competes with β-catenin binding to E-cadherin when overexpressed in
SiHa by immunoprecipitation (Fig.
4F).
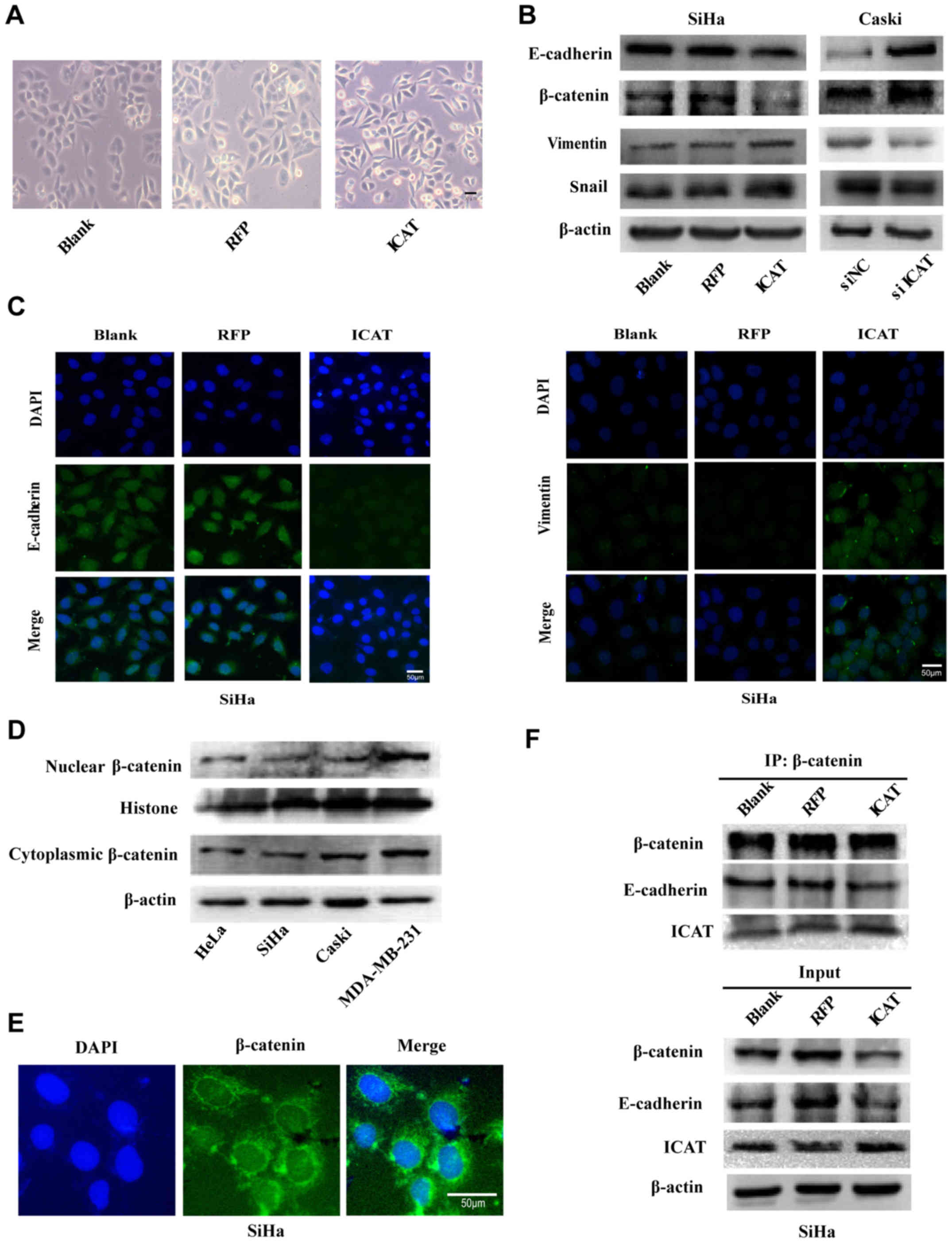 | Figure 4.ICAT regulates epithelial-mesenchymal
transition of cervical cancer cells and disrupts
E-cadherin/β-catenin complex. (A) The images of cell morphological
change caused by ICAT overexpression observed by microscopy; (B)
western blot analysis of the expression levels of EMT markers
(E-cadherin, β-catenin, vimentin and snail) in SiHa, SiHa/RFP,
SiHa/ICAT, Caski/NC and Caski/siICAT cells; (C) immunofluorescence
assay for E-cadherin and vimentin expression, proteins were stained
green, and the nuclei were stained with DAPI (blue). The images are
magnified ×400. Scale bar, 50 µm; (D) endogenous expression of
nuclear β-catenin and cytoplasm β-catenin in SiHa, Caski, HeLa and
MDA-MB-231; (E) immunofluorescence assay for cellular distribution
of β-catenin proteins in SiHa. Proteins were stained green, and the
nuclei were stained with DAPI (blue). Scale bar, 50 µm; (F)
immunoprecipitation (IP) of E-cadherin and ICAT proteins with
β-catenin in SiHa cells. |
Overexpression of ICAT in SiHa cells
promotes tumor growth and EMT in vivo
To investigate the effects of ICAT on the tumor
growth and EMT of cervical cancer cells in vivo, SiHa,
SiHa/RFP and SiHa/ICAT cells were subcutaneously implanted into
nude mice. The tumors were monitored weekly and dissected after
xenografting for 5 weeks. As shown in Fig. 5A the tumor volume of the SiHa/ICAT
cells was significantly increased relative to the controls after 3
weeks (Fig. 5B). Hence, ICAT
expression increased the proliferation of cervical cancer cells
relative to the control in nude mice and significantly increased
tumor burden over time. These results were consistent with those
in vitro. The immunohistochemical analysis of Ki-67 and MMP9
expression revealed that compared with the SiHa, SiHa/RFP groups
Ki-67 and MMP9 positive cell rates were increased in the SiHa/ICAT
group (Fig. 5C). The expression of
EMT-related markers also showed corresponding changes, the vimentin
staining was enhanced while E-cadherin staining was reduced in the
SiHa/ICAT group (Fig. 5D).
Furthermore, western blot results also showed that the protein
expression level of vimentin was increased in SiHa/ICAT group,
while the protein expression level of E-cadherin was significantly
decreased (Fig. 5E).
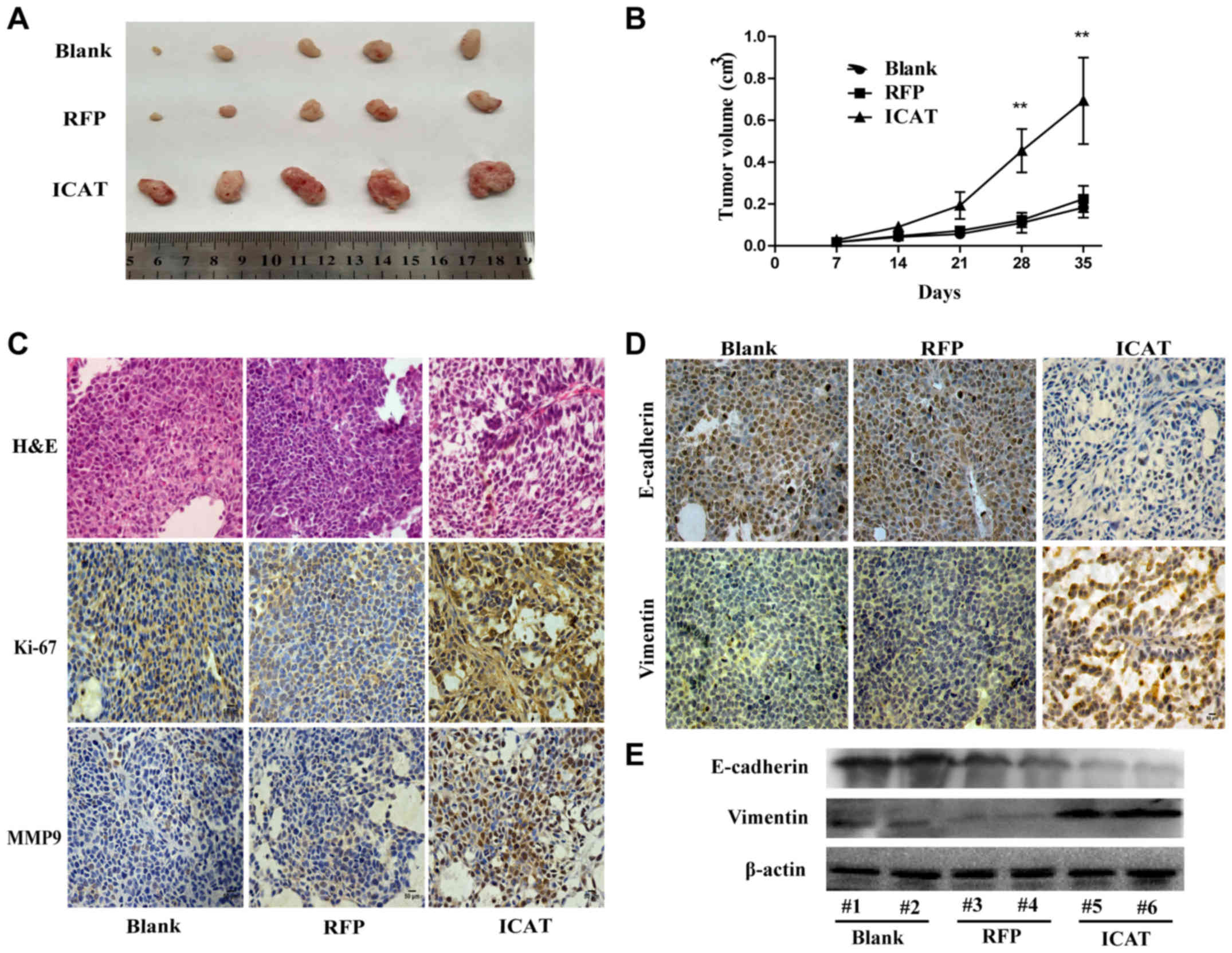 | Figure 5.Overexpression of ICAT in SiHa cells
promotes tumor growth in vivo. (A) The tumor sizes in the
SiHa, SiHa/RFP, SiHa/ICAT groups; (B) the tumor growth curves of
the SiHa, SiHa/RFP, SiHa/ICAT groups. (n=5 in each group); (C)
H&E staining of tumor tissues (×200 magnification), Ki-67 and
MMP9 staining of the SiHa, SiHa/RFP, SiHa/ICAT groups by
immunohistochemical staining (×200 magnification). Scale bar, 50
µm; (D) immunohistochemical staining of EMT markers (E-cadherin and
vimentin) in various groups; (E) western blot analysis of the
expression levels of EMT markers in various groups. Data are shown
as mean ± SD of three individual measurements (*P<0.05,
**P<0.01, as compared with Blank or RFP). |
Discussion
ICAT, a β-catenin interacting protein and
WNT/β-catenin pathway inhibitor, that was originally identified to
disrupts the formation of β-catenin and TCF complexes and inhibit
β-catenin-TCF-4-mediated transactivation. Dysregulation of WNT
signaling is associated with various human cancer including colon
(21), breast (22), prostate cancer (23) and glioma (24). As a negative regulator of WNT
signaling, ICAT was at first presumed to be a tumor suppressor gene
and its inactivation may result in carcinogenesis. Sekiya et
al (25) found that
overexpression of ICAT induces G2 arrest and cell death in
colorectal cancer cells carrying an APC or β-catenin mutation and
hepatocellular tumor cells with an Axin mutation (9). Other studies have demonstrated that
ICAT inhibited glioma cell proliferation, invasion and induced
apoptosis by suppressing Wnt/β-catenin activity in vitro and
in vivo (26,27). In addition, in accordance with two
other WNT/β-catenin pathway inhibitors, niclosamide and XAV939,
ICAT inhibited WNT/β-catenin pathway activation and exerted
antitumor effects in primary cultures of human leiomyoma cells
(28). All of these results seem to
indicate that ICAT may serve as a tumor suppressor gene. However,
some research showed different results. Ectopic overexpression of
ICAT increased melanoma cells motility and matrigel invasion which
was associated with conversion of an elongated/mesenchymal
phenotype to a round/amoeboid phenotype (29,30).
Zhang et al (31) found that
miR-424-5p could block EMT process of anchorage-independent
hepatocellular carcinoma cells by directly targeting ICAT, and
suppressed hepatocellular carcinoma progression. Therefore, the
effects of ICAT is different in various types of cancers. However,
its role in cervical cancer was unknown, and we investigated the
effects and its mechanism of ICAT on cervical cancer.
β-catenin as the major biochemical target of ICAT is
not only a downstream transcriptional activator of the Wnt
signaling pathway but also has a significant role in cell-cell
adhesion. It binds the cytoplasmic domain of E-cadherin and links
the actin cytoskeleton. This complex constitutes a key element in
intracellular adhesion (32).
β-catenin is a critical epithelial marker, the lack
of its expression could promote EMT of epithelial cells and the
cell morphology changed from epithelial morphology to a mesenchymal
phenotype characterized (33). This
function has been recognized as an important process in the
invasion and metastasis of cancer.
In this study, we first detected the expression of
ICAT by immunohistochemistry in 41 cervical cancer samples and 30
normal cervical tissues. The results indicated that ICAT was
upregulated in cervical cancer and it may play an important role in
cervical carcinogenesis and progression. Then, we enforced ICAT
expression by recombinant adenovirus to investigate the effects of
ICAT on SiHa cells. We used MTT, colony formation assay and flow
cytometry assays to analyze cells proliferation. The results showed
that ICAT overexpression promoted SiHa cells proliferation and the
percentage of cells in the S phase. As is known, metastasis of
cervical cancer is responsible for the vast majority of all poor
prognosis. Whether ICAT is involved in cervical cancer invasion and
metastasis has been proved. Transwell migration and Matrigel
invasion assay revealed that the migration and invasion of SiHa
cells were significantly promoted by ICAT, whereas ICAT silencing
by siRNAs targeting ICAT induced opposite effects in Caski cells.
The above suggested that ICAT promoted metastasis of cervical
cancer.
We then investigated how ICAT affects cell migration
and invasion of cervical cancer cells. Interestingly, a major cell
morphological change from a cobblestone-like morphology to a more
mesenchymal morphology with spindle-like shape was observed. The
findings indicated that ICAT may be involved in the cervical cancer
tumorigenesis and metastases through the regulation of EMT. To
further investigate the role of ICAT in the EMT of cervical cancer,
the expression of EMT related markers were detected by western blot
analysis in SiHa and Caski cells after ICAT overexpression or
downregulation. We proved that overexpression of ICAT induced
marked upregulation of mesenchymal marker but a downregulation of
epithelial marker in SiHa cells and ICAT knockdown induced opposite
effects in Caski cells. Immunofluorescence analysis also proved
that overexpression of ICAT promotes EMT in SiHa cells.
Furthermore, we illuminated that ICAT could disrupt the
E-cadherin/β-catenin complex and reduce the expression of
E-cadherin in SiHa cells by immunoprecipitation. The in vivo
function of ICAT on cervical cancer was also demonstrated in our
studies with a nude mouse xenograft model and were consistent with
previous studies. ICAT was found more than a decade ago, but most
of the studies only focus on its role in β-catenin-dependent
nuclear signaling while ignoring its role in EMT. We found ICAT
could disrupt the stability of E-cadherin/β-catenin complex and
result in EMT in cervical cancer. However, the precise mechanisms
underlying the feature of ICAT and its relation to EMT remain to be
defined in cervical cancer.
In summary, we revealed that ICAT expression was
upregulated in cervical cancer and participated in the EMT of
cervical cancer by inhibiting β-catenin binding to E-cadherin. The
in vivo function of ICAT on cervical cancer was also
demonstrated and was consistent with previous studies. These
results support the notion that inhibiting ICAT expression and
function may be an effective strategy for cervical cancer
therapy.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (no. 81172017) and the National
Basic Research Program of China (973 Program, 2011CB707906).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaccarella S, Lortet-Tieulent J, Plummer
M, Franceschi S and Bray F: Worldwide trends in cervical cancer
incidence: Impact of screening against changes in disease risk
factors. Eur J Cancer. 49:3262–3273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meijer CJ and Snijders PJ: Cervical cancer
in 2013: Screening comes of age and treatment progress continues.
Nat Rev Clin Oncol. 11:77–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Castle PE: Gynaecological cancer: New
standard of care-HPV testing for cervical cancer screening. Nat Rev
Clin Oncol. 12:194–196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tago K, Nakamura T, Nishita M, Hyodo J,
Nagai S, Murata Y, Adachi S, Ohwada S, Morishita Y, Shibuya H, et
al: Inhibition of Wnt signaling by ICAT, a novel
beta-catenin-interacting protein. Genes Dev. 14:1741–1749.
2000.PubMed/NCBI
|
|
7
|
Daniels DL and Weis WI: ICAT inhibits
beta-catenin binding to Tcf/Lef-family transcription factors and
the general coactivator p300 using independent structural modules.
Mol Cell. 10:573–584. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Graham TA, Clements WK, Kimelman D and Xu
W: The crystal structure of the beta-catenin/ICAT complex reveals
the inhibitory mechanism of ICAT. Mol Cell. 10:563–571. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koyama T, Tago K, Nakamura T, Ohwada S,
Morishita Y, Yokota J and Akiyama T: Mutation and expression of the
beta-catenin-interacting protein ICAT in human colorectal tumors.
Jpn J Clin Oncol. 32:358–362. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi HJ, Huber AH and Weis WI:
Thermodynamics of beta-catenin-ligand interactions: The roles of
the N- and C-terminal tails in modulating binding affinity. J Biol
Chem. 281:1027–1038. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gottardi CJ and Gumbiner BM: Role for ICAT
in beta-catenin-dependent nuclear signaling and cadherin functions.
Am J Physiol Cell Physiol. 286:C747–C756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: A role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995;
discussion 5995. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
An HT, Yoo S and Ko J: α-Actinin-4 induces
the epithelial-to-mesenchymal transition and tumorigenesis via
regulation of Snail expression and β-catenin stabilization in
cervical cancer. Oncogene. 35:5893–5904. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Q, Bao W, Fan Q, Shi WJ, Li ZN, Xu Y
and Wu D: Epidermal growth factor receptor kinase substrate 8
promotes the metastasis of cervical cancer via the
epithelial-mesenchymal transition. Mol Med Rep. 14:3220–3228. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun R, Jiang B, Qi H, Zhang X, Yang J,
Duan J, Li Y and Li G: SOX4 contributes to the progression of
cervical cancer and the resistance to the chemotherapeutic drug
through ABCG2. Cell Death Dis. 6:e19902015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He X, Qian Y, Cai H, Yang S, Cai J and
Wang Z: RhoC is essential in TGF-β1 induced epithelial-mesenchymal
transition in cervical cancer cells. Oncol Lett. 10:985–989.
2015.PubMed/NCBI
|
|
19
|
Zhang J, Zhu D, Lv Q, Yi Y, Li F and Zhang
W: The key role of astrocyte elevated gene-1 in CCR6-induced EMT in
cervical cancer. Tumour Biol. 36:9763–9767. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koay MH, Crook M and Stewart CJ: Cyclin
D1, E-cadherin and beta-catenin expression in FIGO stage IA
cervical squamous carcinoma: Diagnostic value and evidence for
epithelial-mesenchymal transition. Histopathology. 61:1125–1133.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ye GD, Sun GB, Jiao P, Chen C, Liu QF,
Huang XL, Zhang R, Cai WY, Li SN, Wu JF, et al: OVOL2, an inhibitor
of WNT signaling, reduces invasive activities of human and mouse
cancer cells and is down-regulated in human colorectal tumors.
Gastroenterology. 150:659–671.e616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Jin K, van Pelt GW, van Dam H, Yu X,
Mesker WE, Ten Dijke P, Zhou F and Zhang L: c-Myb enhances breast
cancer invasion and metastasis through the Wnt/beta-catenin/Axin2
pathway. Cancer Res. 76:3364–3375. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma F, Ye H, He HH, Gerrin SJ, Chen S,
Tanenbaum BA, Cai C, Sowalsky AG, He L, Wang H, et al: SOX9 drives
WNT pathway activation in prostate cancer. J Clin Invest.
126:1745–1758. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang K, Zhang J, Han L, Pu P and Kang C:
Wnt/beta-catenin signaling in glioma. J Neuroimmune Pharmacol.
7:740–749. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sekiya T, Nakamura T, Kazuki Y, Oshimura
M, Kohu K, Tago K, Ohwada S and Akiyama T: Overexpression of Icat
induces G2 arrest and cell death in tumor cell mutants
for adenomatous polyposis coli, beta-catenin, or Axin. Cancer Res.
62:3322–3326. 2002.PubMed/NCBI
|
|
26
|
Zhang K, Zhu S, Liu Y, Dong X, Shi Z,
Zhang A, Liu C, Chen L, Wei J, Pu P, et al: ICAT inhibits
glioblastoma cell proliferation by suppressing Wnt/β-catenin
activity. Cancer Lett. 357:404–411. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shi Z, Qian X, Li L, Zhang J, Zhu S, Zhu
J, Chen L, Zhang K, Han L, Yu S, et al: Nuclear translocation of
beta-catenin is essential for glioma cell survival. Pharmacology.
7:892–903. 2012.
|
|
28
|
Ono M, Yin P, Navarro A, Moravek MB, Coon
JSV, Druschitz SA, Gottardi CJ and Bulun SE: Inhibition of
canonical WNT signaling attenuates human leiomyoma cell growth.
Fertil Steril. 101:1441–1449. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Domingues MJ, Rambow F, Job B, Papon L,
Liu W, Larue L and Bonaventure J: β-catenin inhibitor ICAT
modulates the invasive motility of melanoma cells. Cancer Res.
74:1983–1995. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reifenberger J, Knobbe CB, Wolter M,
Blaschke B, Schulte KW, Pietsch T, Ruzicka T and Reifenberger G:
Molecular genetic analysis of malignant melanomas for aberrations
of the WNT signaling pathway genes CTNNB1, APC, ICAT and BTRC. Int
J Cancer. 100:549–556. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Y, Li T, Guo P, Kang J, Wei Q, Jia
X, Zhao W, Huai W, Qiu Y, Sun L, et al: MiR-424-5p reversed
epithelial-mesenchymal transition of anchorage-independent HCC
cells by directly targeting ICAT and suppressed HCC progression.
Sci Rep. 4:62482014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jamora C and Fuchs E: Intercellular
adhesion, signalling and the cytoskeleton. Nat Cell Biol.
4:E101–E108. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Angadi PV, Patil PV, Angadi V, Mane D,
Shekar SS, Kale AD and Kardesai SG: Immunoexpression of epithelial
mesenchymal transition proteins E-cadherin, β-catenin, and
N-cadherin in oral squamous cell carcinoma. Int J Surg Pathol.
24:696–703. 2016. View Article : Google Scholar : PubMed/NCBI
|