Introduction
Osteosarcoma (OS), a mesenchymal tumor
histologically characterized by the presence of malignant
mesenchymal cells which produce osteoid, is the most common primary
malignancy of bone, particularly with a high incidence rate in
children and young adults (1).
After conventional treatment, consisting of the combination of pre-
and post-operative chemotherapy with surgical removal of the tumor,
the 5-year overall survival rate approaches 60–70% (2–4).
Nonetheless, ~30–35% patients, with successful resection and
adjuvant chemotherapy, still develop metastases (2,5). There
has been no substantial improvement in the long-term outcomes for
patients presenting with localized or disseminated disease over the
last three decades. Thus, identification and validation of novel
targetable agents as adjuvant to conventional chemotherapeutics to
provide better control of osteosarcoma is absolutely important and
urgent.
Osteosarcoma is a heterogeneous tumor with a
diversity of genetic changes. To date, the heterogeneity of
osteosarcoma has been reported through whole genome sequencing
approaches (6,7), while unfortunately, there has been no
significant improvement in molecularly targeted therapies that may
benefit osteosarcoma patients. Hence, identifying potential
targetable genetic aberrations or pathway alterations may lead to
the development of targeted therapies for osteosarcoma.
NSD3 (also known as WHSC1L1), a Su(var)3-9,
enhancer-of-zeste and trithorax (SET) domain containing histone
lysine methyltransferase, can dimethylate and trimethylate histone
H3 at lysine 36 (8–11). NSD3, the third member of the NSD
family, shares a C-terminal block of ~700 amino acids with the
other two family members, NSD1 (also known as SOTOS) and NSD2 (also
referred to as WHSC1 and MMSET) which also methylate H3K36. In
addition to the catalytic SET domain with pre- and post-SET
domains, NSD3 possesses two PWWP (the conserved sequence motif of
Pro-Trp-Trp-Pro) domains and five PHD (plant homeodomain) fingers,
both of which are often involved in chromatin-associated biological
processes through crosstalks with histone and DNA reader or
modifier proteins. Hence, NSD3 is involved in a variety of
biological processes such as chromatin modification,
transcriptional regulation, and DNA repair either through direct
regulation of histone methylation or through the protein-protein
interactions by these specific domains. There are two major
isoforms of NSD3, the long full-length isoform and the short
isoform lacking the SET domain and only possessing the first
N-terminal PWWP domain.
NSD3, as a nuclear protein, is located at chromosome
8p11.23, the locus that exhibits strong cancer relevance. Indeed,
NSD3 is likely involved in solid and hematological tumors (12–17),
and functions as an oncogene in the development and progression of
cancer (18–20). In midline carcinoma, a novel
NSD3-NUT fusion oncogene containing N-terminal region of NSD3 has
been demonstrated (21). Notably,
the NSD3 portion of the fusion protein lacks the catalytic SET
domain and contains only its first PWWP domain. In addition,
NSD-short, possessing only the N-terminal PWWP domain, also couples
BRD4 to the CHD8 chromatin remodeler and sustains acute myeloid
leukemia (AML) cell proliferation (22). These findings indicate the
importance of these specific nuclear domains in NSD3. NSD3 was
reported as a translocation partner of NUP98 in an AML patient
associated with t(8;11)(p11.2;p15) (23,24).
NSD3 is also essential for neural crest gene expression during
specification (25). Collectively,
all of these studies suggest the important role of NSD3 in human
carcinogenesis. However, the molecular mechanisms of NSD3 in human
carcinogenesis are still obscure and remain to be clarified.
In the present study, we demonstrated that
inhibition of NSD3 in osteosarcoma cells caused a marked reduction
in cancer cell viability and survival, with an increase in the
proportion of cells at the G2/M phase and the number of apoptotic
cells. Additionally, a set of NSD3-regulated genes involved in
multiple biological processes were identified by RNA-seq analysis.
Thus, these results demonstrated the oncogenic functions of NSD3 in
osteosarcoma, and furthermore, indicate that NSD3 could be a
promising target for the treatment of osteosarcoma.
Materials and methods
Cell lines and cell culture
Human osteosarcoma cell lines HOS, U2OS and MG-63
were purchased from the Shanghai Institute for Biological Sciences,
Chinese Academy of Cell Resource Center (Shanghai, China). HOS,
U2OS and MG-63 cells were grown in minimum essential medium (MEM;
Gibco, Shanghai, China), supplemented with 10% fetal bovine serum
(Beijing Yuanheng Shengma Research Institution of Biotechnology,
Beijing, China) and 1% antibiotic/antimycotic solution (Gibco) at
37°C in a humidified incubator with 5% CO2.
RNA extraction and real-time PCR
Total RNA was isolated from the cells using an
EASYspin Plus Tissue/Cell RNA extraction kit (Aidlab
Biotechnologies Co., Ltd., Beijing, China). RNA was reverse
transcribed to cDNA using the ThermoScript First Strand cDNA
Synthesis kit (Aidlab Biotechnologies Co., Ltd.) according to the
manufacturer's instructions. Semi-quantitative reverse
transcription-PCR was performed using KOD-Plus-Neo DNA polymerase
(Toyobo Biotech Co., Ltd., Shanghai, China) to investigate the
complete knockdown of NSD3. Each PCR regime consisted of initial
denaturation at 94°C for 2 min followed by 22 cycles (for ACTB), 32
cycles (for NSD3) at 98°C for 10 sec, 55°C for 30 sec and 68°C for
45 sec. The primer sequences were: 5′-TTGGCTTGACTCAGGATTTA-3′ and
reverse 5′-ATGCTATCACCTCCCCTGTG-3′ for β-actin (ACTB); and
5′-CCATGCAGAGAAAGCATTGA-3′ and 5′-TCTTCCTCTTCCGCACTTGT-3′ for
NSD3. qRT-PCR was conducted using SYBR Premix Ex Taq™
(Takara, Dalian, China) at 95°C for 30 sec, followed by 40 cycles
of 95°C for 5 sec and 60°C for 34 sec in the ABI StepOnePlus
Real-time PCR system. Relative gene expression was quantified
relative to the GAPDH level using the comparative cycle threshold
(Ct) method. Each sample was analyzed in duplicate.
Cell viability and colony formation
assays
Cells (104 cells/well) were plated in
96-well plates the day before treatment to allow cell attachment,
and transfected with NSD3-specific siRNA duplex (siNSD3#1,
5′-CUCACAAAUGGGUAUCCAU-3′; siNSD3#2, 5′-GUACUGAAAUUCGGAGACA-3′); or
EGFP siRNA duplex (5′-GCAGCACGACUUCUUCAAG-3′) as a negative
control, respectively, using Lipofectamin RNAiMAX (Invitrogen Life
Technologies, Shanghai, China) according to the manufacturer's
recommendations. Cell viability was measured using Cell-Counting
Kit-8 (Dojindo, Shanghai, China) 96 h after transfection. The
absorbance was measured at a wavelength of 450 nm, and the
percentage of cell viability was calculated as the percentage of
absorption. Each sample was assayed in triplicate. All experiments
were performed in triplicate and repeated two times independently.
For the colony formation assay, the cells were seeded into 6-well
plates (2×103 cells/well) and treated with the specific
siRNA for 4–8 h, respectively, followed by a medium refresh. After
10 days for HOS, U2OS and 14 days for MG-63, the cells in the
plates were stained with crystal violet solution.
Annexin V-FITC and propidium iodide
(PI) staining
Cells treated with the specific siRNA for 96 h were
centrifuged and collected. The cell pellets were washed with
phosphate-buffered saline (PBS) and resuspended in 1X
Annexin-binding buffer with 5 µl of FITC Annexin V and 5 µl PI
(both from BD Pharmingen, San Diego, CA, USA). After a 15-min
incubation at room temperature in the dark, the samples were
analyzed by BD FACSAria II. FITC+/PI− and
FITC+/PI+ cells were considered as early and
late apoptotic cells and analyzed. The percentage of apoptotic
cells in total cells was designated as the apoptotic index. For
cell cycle analysis, 96 h after the transfection, the cells were
fixed with 70% ethanol at 4°C, incubated with 500 µl of PBS
containing 0.5 mg of RNase at 37°C for 30 min, and finally, cells
stained with 50 µg/ml PI were analyzed by fluorescence-activated
cell sorting (FACS).
RNA-seq analysis
Indexed libraries from HOS cells incubated with
siEGFP or siNSD3 were subjected to RNA-seq on the Illumina HiSeq
2000 platform. Gene expression was quantified using the fragments
per kilobase of transcript per million mapped reads (FPKM)
normalization method (26), and
Cufflinks was used for FPKM quantification. Gene expression and
differential transcription between siEGFP- and siNSD3-treated cells
were evaluated using Cuffdiff. The lists of significantly
differentially expressed genes were obtained with the threshold of
p-value ≤0.05 and a fold-change ≥2. Gene ontology (GO) analysis was
performed in the standard enrichment computation method.
Results
Silencing of NSD3 attenuates
osteosarcoma cell viability
To investigate the biological significance of NSD3
in osteosarcoma cells, the HOS, U2OS and MG-63 cancer cell lines
were transfected with an NSD3-specific siRNA duplex (siNSD3#1,
siNSD3#2) or EGFP siRNA duplex (siEGFP) as a negative control,
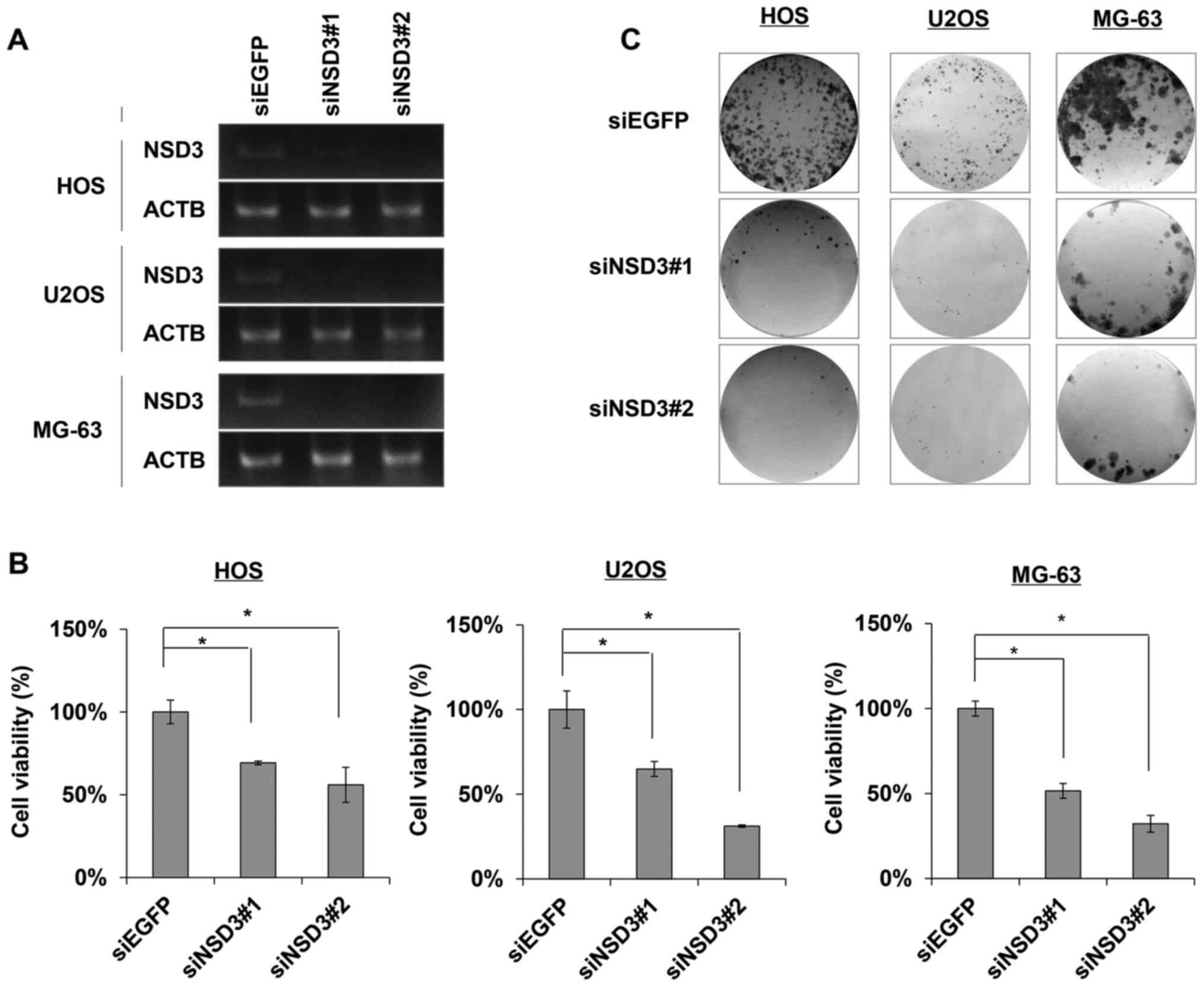
respectively. Results of semi-quantitative RT-PCR confirmed the
complete knockdown effects of NSD3 expression in the cells
transfected with siNSD3#1 or siNSD3#2 for 96 h compared with the
siEGFP control (Fig. 1A). Cell
viability assay data revealed a marked reduction in the number of
viable cells in the siNSD#1 or siNSD3#2-treated cancer cells as
compared to the siEGFP control, reaching a 30–50% reduction in the
HOS, U2OS and MG-63 osteosarcoma cells (Fig. 1B). The reduction in viable cells
upon NSD3 knockdown was investigated with two different siRNA
sequences specific for NSD3, indicating it was not due to
off-target effects of the siRNA. Colony formation assay was
conducted to assess the effect of NSD3 on the proliferation of
cancer cells, and as a result, siNSD3#1- or siNSD3#2-treated cancer
cells formed much fewer and smaller colonies than the
siEGFP-treated cancer cells (Fig.
1C), probably due to cell death caused by apoptosis and cell
cycle-related events. These findings elucidated the oncogenic
functions of NSD3 in the development and progression of
osteosarcoma.
Knockdown of NSD3 induces apoptosis of
cancer cells
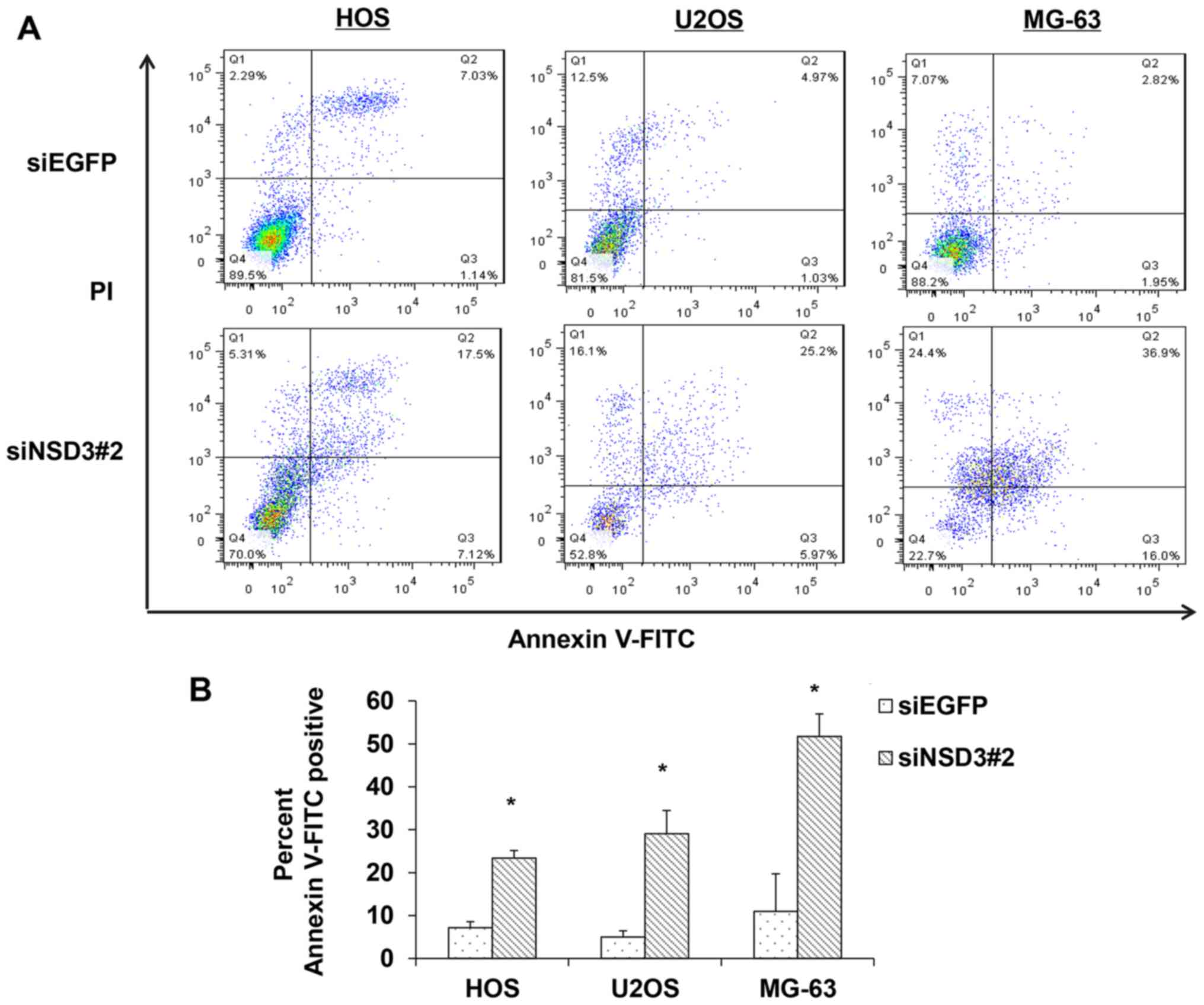
Given the drastic reduction in viable cells
following NSD3 silencing, we evaluated the effects of NSD3 on cell
apoptosis using Annexin V-FITC and PI staining followed by FACS
analysis. In the scatter plot of double variable events, cells in
quadrant Q4 (FITC−/PI−) were considered
viable, cells in quadrant Q2 (FITC+/PI+) were
in the late apoptotic stage, and cells in quadrant Q3
(FITC+/PI−) represented early apoptotic
cells. As a result, a striking increase was observed in both the
early and late stage of apoptosis in the NSD3-depleted HOS, U2OS
and MG-63 osteosarcoma cells compared with the control cells
(Fig. 2A). The number of apoptotic
cancer cells was increased 3.3-fold in the NSD3-depleted HOS cells,
5.8-fold in the U2OS cells, and 4.7-fold in the MG-63 osteosarcoma
cells (Fig. 2B), showing that
silencing of NSD3 induced apoptosis in all three measured
osteosarcoma cell lines. These data suggest that NSD3 may
contribute to cancer cell apoptosis.
Depletion of NSD3 increases the
proportion of cells in the G2/M phase
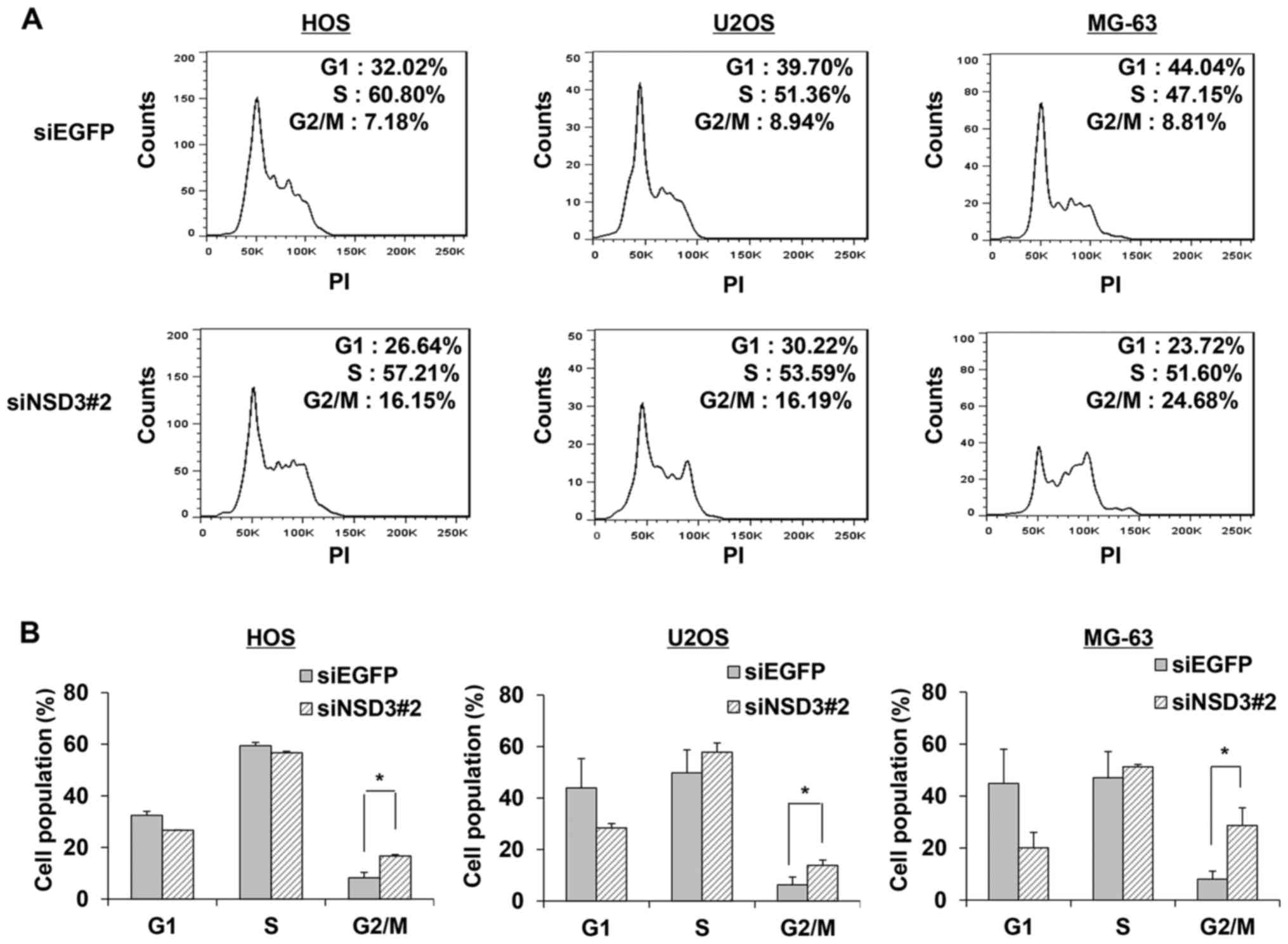
Since deletion of NSD3 decreased cell viability and
induced cell apoptosis, we hypothesized that NSD3 is also involved
in the cell cycle. To assess the roles of NSD3 in the progression
of the cell cycle, viable cells treated with siRNA specific to NSD3
or control were collected, fixed with ethanol and stained with PI
for flow cytometric analysis. A 2-fold increase in the population
of G2/M phase was found in the NSD3-depleted HOS cells (Fig. 3A). Concordant increases were
obtained in the other two osteosarcoma cancer cell lines U2OS and
MG-63. Furthermore, apart from the marked increases in the G2/M
population, slight increases in the S phase population were also
observed in the NSD3-depleted U2OS and MG-63 cells (Fig. 3A). Data from three independent
experiments were analyzed and the average population of each phase
was calculated (Fig. 3B).
Consistent alterations in G2/M populations upon NSD3 silencing were
observed. These findings imply that knockdown of NSD3 leads to G2/M
cell cycle arrest and NSD3 is likely to be involved in the
regulation of the G2/M checkpoint.
Identification of downstream genes of
NSD3 by RNA-seq analysis
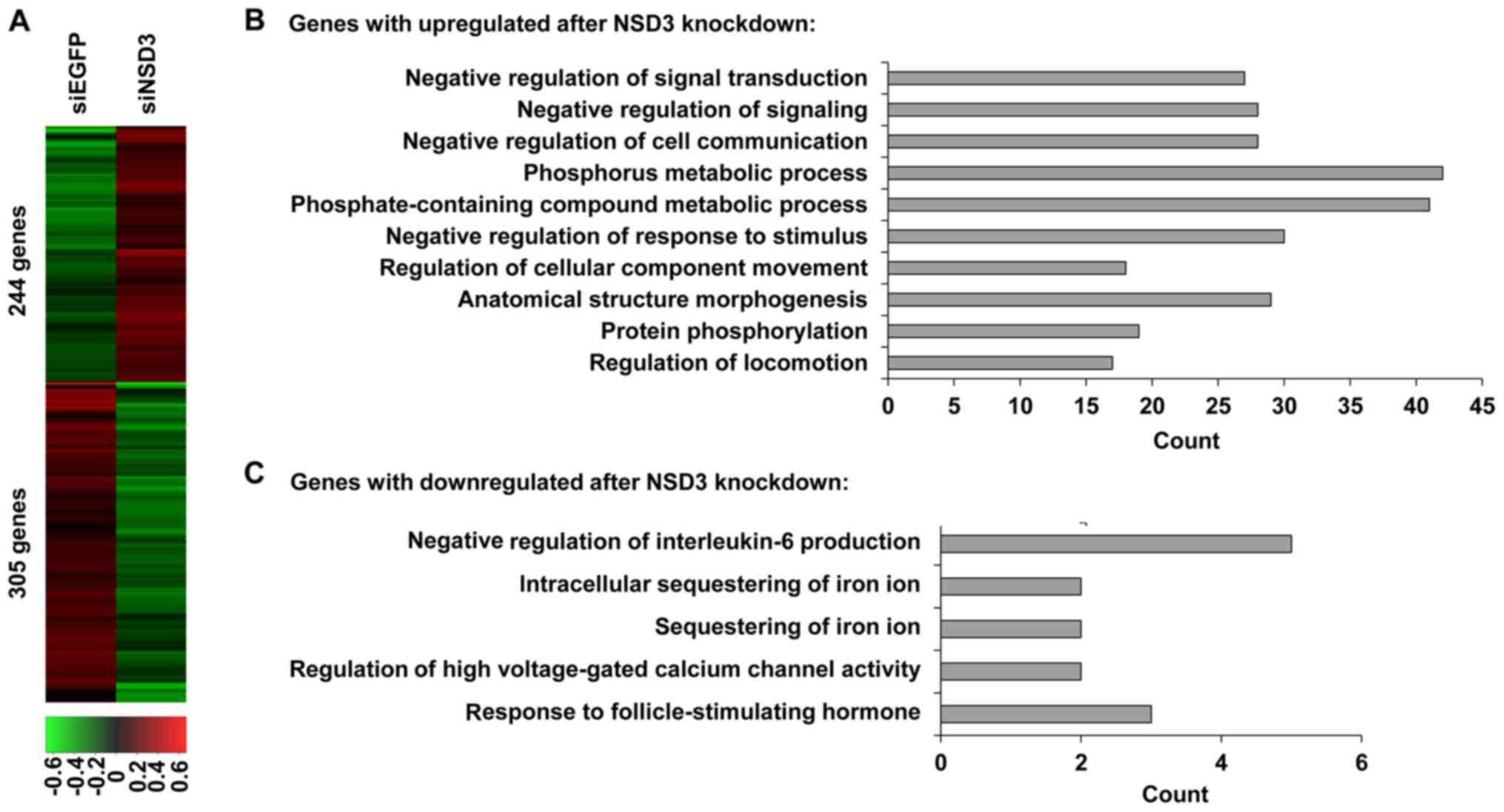
To identify how NSD3 contributes to the development
and progression of osteosarcoma, we searched NSD3-regulated genes
in HOS cells on a genome-wide scale. RNA sequencing gene expression
analysis was performed in HOS cells treated with NSD3-specific
siRNA (siNSD3#2) or EGFP-specific siRNA (siEGFP) as a control. A
variety of genes were identified, including 244 upregulated genes
and 305 downregulated genes in the NSD3-depleted HOS cells with the
threshold of p≤0.05 and a fold-change ≥2 as compared with the
control (Fig. 4A), indicating that
NSD3 is correlated with both gene activation and repression. GO
term enrichment analysis of the biological process category
revealed that genes with increased expression after NSD3 depletion
were involved in various biological processes, such as negative
regulation of signal transduction, negative regulation of
signaling, negative regulation of cell communication, phosphorus
metabolic process, negative regulation of response to stimulus
(Fig. 4B), whereas genes with
decreased expression after NSD3 depletion were involved in negative
regulation of interleukin-6 production and intracellular
sequestering of iron ion (Fig. 4C).
These findings, together with the effect of NSD3 on cancer cell
proliferation and cell apoptosis, provide evidence that NSD3 is an
oncogenic driver in osteosarcoma. To validate the findings of
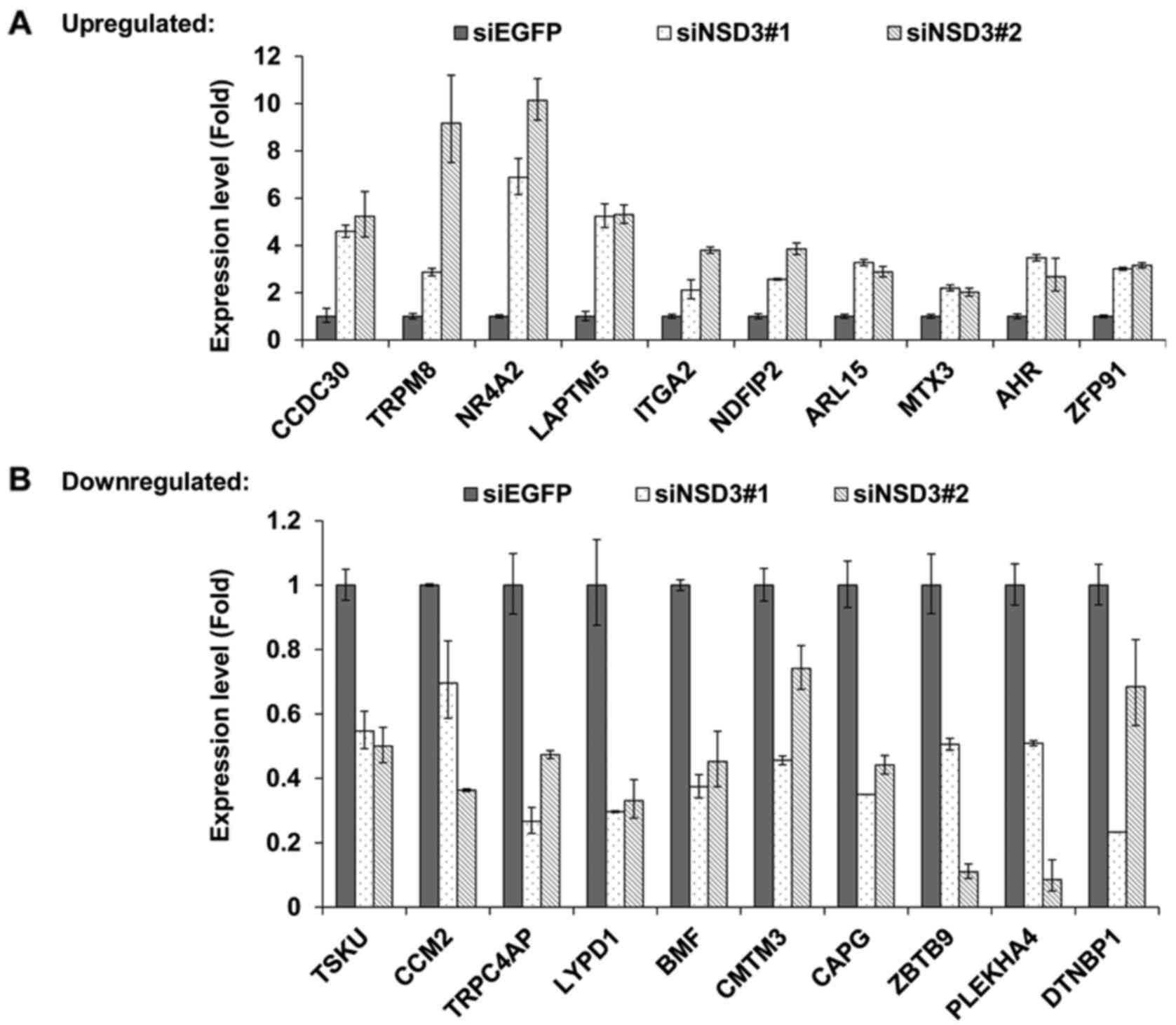
RNA-seq, the top 20 upregulated and downregulated genes were
selected to be analyzed by quantitative real-time PCR. Moreover, to
exclude the set of off-target genes, the results were confirmed
with two different NSD3-specific siRNAs, siNSD3#1 and siNSD3#2,
respectively (Fig. 5A). As a
result, 14 upregulated and 12 downregulated genes by depletion of
NSD3 were identified. Herein, the top 10 upregulated (Fig. 5A) and downregulated (Fig. 5B) genes are listed, including
tumor-related genes, such as TRPM8, LAPTM5, ITGA2, BMF, CAPG and
CMTM3.
Discussion
Epigenetic modifications, including DNA methylation,
covalent histone modifications and nucleosome positioning, play
critical roles in normal mammalian development and regulation of
gene expression, and are believed to be ubiquitous drivers of human
carcinogenesis. Histone methyltransferases, the key group in the
regulation of histone methylation, are commonly disrupted in cancer
and contribute to human carcinogenesis. Hence, histone
methyltransferases have emerged as crucial targets for the
treatment of cancer in both industrial and academic research
groups. Most notably, selective inhibitors of histone lysine
methyltransferase EZH2, DOT1L and histone demethylase LSD1 have
entered into human clinical trials for the treatment of non-Hodgkin
lymphoma, acute leukemia, acute myeloid leukemia and
relapsed/refractory small cell lung carcinoma, respectively
(27–32). Nevertheless, selective inhibitors
for many histone methyltranferases still remain unavailable.
As a member of the NSD histone lysine
methyltransferase family, NSD3 dimethylates and trimethylates
histone H3 at lysine 36, a conserved epigenetic marker regulating
gene transcription, DNA replication and repair, DNA methylation,
and alternative splicing that are dysregulated in cancer. NSD3,
also known as Wolf-Hirschhorn syndrome candidate 1-like 1
(WHSC1L1), is located within chromosome 8p11-12, a recurrent target
region of genetic alterations such as DNA amplification,
chromosomal breaks and loss. Abnormal expression of NSD3 in a
number of cancer types has been demonstrated (18–20).
Nonetheless, the biological roles of NSD3 in human carcinogenesis
are not clear and remain to be clarified.
In the present study, we elucidated the biological
importance of histone methyltransferase NSD3 in human osteosarcoma.
Our studies revealed that depletion of NSD3 in osteosarcoma cell
lines inhibited cell proliferation and survival, and induced cell
apoptosis. Furthermore, marked and consistent increases in the
population of G2/M phase after RNAi knockdown of NSD3 were observed
in all three analyzed osteosarcoma cell lines, probably due to G2/M
cell cycle arrest, indicating the involvement of NSD3 in the cell
cycle process. However, the detailed mechanisms of the role of NSD3
in cell proliferation, cell cycle, and apoptosis remain poorly
understood.
A key to understand the role of NSD3 in cancer may
be through its regulated genes. Hence, a series of NSD3-associated
genes were identified by RNA-seq, including 244 upregulated and 305
downregulated genes after NSD3 deletion, implying that NSD3
functions as either a transcriptional activator or a repressor.
Analysis of Gene ontology (GO) term enrichment revealed that NSD3
negatively regulated a variety of genes involved in the process of
negative regulation of signal transduction as well as negative
regulation of signaling, negative regulation of cell communication,
and negative regulation of response to stimulus, implying the
oncogenic functions of NSD3 in human osteosarcoma. GO categories
including regulation of cell cycle (GO:0051726), cell cycle arrest
(GO:0007050), regulation of cell cycle checkpoint (GO:1901976),
cell cycle G2/M phase transition (GO:0044839) and G2/M transition
of mitotic cell cycle (GO:0000086) were enriched in the
differentially expressed genes with depletion of NSD3, implying the
crucial functions of NSD3 in the G2/M transition of the cell cycle
process. Additionally, a set of selected candidate genes likely to
be involved in the development and progression of osteosarcoma was
validated by quantitative real-time PCR. Taking these findings into
account, NSD3 may function as an oncogenic driver in osteosarcoma.
Thus, NSD3 may be a novel target for the treatment of
NSD3-associated cancers, and it appears feasible to develop
inhibitors of NSD3 for cancer therapy.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bielack SS, Kempf-Bielack B, Delling G,
Exner GU, Flege S, Helmke K, Kotz R, Salzer-Kuntschik M, Werner M,
Winkelmann W, et al: Prognostic factors in high-grade osteosarcoma
of the extremities or trunk: An analysis of 1,702 patients treated
on neoadjuvant cooperative osteosarcoma study group protocols. J
Clin Oncol. 20:776–790. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Botter SM, Neri D and Fuchs B: Recent
advances in osteosarcoma. Curr Opin Pharmacol. 16:15–23. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gatta G, Botta L, Rossi S, Aareleid T,
Bielska-Lasota M, Clavel J, Dimitrova N, Jakab Z, Kaatsch P, Lacour
B, et al EUROCARE Working Group, : Childhood cancer survival in
Europe 1999–2007: Results of EUROCARE-5 - a population-based study.
Lancet Oncol. 15:35–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kempf-Bielack B, Bielack SS, Jürgens H,
Branscheid D, Berdel WE, Exner GU, Göbel U, Helmke K, Jundt G,
Kabisch H, et al: Osteosarcoma relapse after combined modality
therapy: An analysis of unselected patients in the Cooperative
Osteosarcoma Study Group (COSS). J Clin Oncol. 23:559–568. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Bahrami A, Pappo A, Easton J,
Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al:
St. Jude Children's Research Hospital-Washington University
Pediatric Cancer Genome Project: Recurrent somatic structural
variations contribute to tumorigenesis in pediatric osteosarcoma.
Cell Reports. 7:104–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuijjer ML, Hogendoorn PC and
Cleton-Jansen AM: Genome-wide analyses on high-grade osteosarcoma:
Making sense of a genomically most unstable tumor. Int J Cancer.
133:2512–2521. 2013.PubMed/NCBI
|
|
8
|
Morishita M, Mevius D and di Luccio E: In
vitro histone lysine methylation by NSD1, NSD2/MMSET/WHSC1 and
NSD3/WHSC1L. BMC Struct Biol. 14:252014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He C, Li F, Zhang J, Wu J and Shi Y: The
methyltransferase NSD3 has chromatin-binding motifs, PHD5-C5HCH,
that are distinct from other NSD (nuclear receptor SET domain)
family members in their histone H3 recognition. J Biol Chem.
288:4692–4703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Trojer P, Xu CF, Cheung P, Kuo A,
Drury WJ III, Qiao Q, Neubert TA, Xu RM, Gozani O, et al: The
target of the NSD family of histone lysine methyltransferases
depends on the nature of the substrate. J Biol Chem.
284:34283–34295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rahman S, Sowa ME, Ottinger M, Smith JA,
Shi Y, Harper JW and Howley PM: The Brd4 extraterminal domain
confers transcription activation independent of pTEFb by recruiting
multiple proteins, including NSD3. Mol Cell Biol. 31:2641–2652.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simon R, Richter J, Wagner U, Fijan A,
Bruderer J, Schmid U, Ackermann D, Maurer R, Alund G, Knönagel H,
et al: High-throughput tissue microarray analysis of 3p25 (RAF1)
and 8p12 (FGFR1) copy number alterations in urinary bladder cancer.
Cancer Res. 61:4514–4519. 2001.PubMed/NCBI
|
|
13
|
Balsara BR and Testa JR: Chromosomal
imbalances in human lung cancer. Oncogene. 21:6877–6883. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ray ME, Yang ZQ, Albertson D, Kleer CG,
Washburn JG, Macoska JA and Ethier SP: Genomic and expression
analysis of the 8p11-12 amplicon in human breast cancer cell lines.
Cancer Res. 64:40–47. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tonon G, Wong KK, Maulik G, Brennan C,
Feng B, Zhang Y, Khatry DB, Protopopov A, You MJ, Aguirre AJ, et
al: High-resolution genomic profiles of human lung cancer. Proc
Natl Acad Sci USA. 102:pp. 9625–9630. 2005; View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang ZQ, Liu G, Bollig-Fischer A, Giroux
CN and Ethier SP: Transforming properties of 8p11-12 amplified
genes in human breast cancer. Cancer Res. 70:8487–8497. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mahmood SF, Gruel N, Nicolle R,
Chapeaublanc E, Delattre O, Radvanyi F and Bernard-Pierrot I:
PPAPDC1B and WHSC1L1 are common drivers of the 8p11-12 amplicon,
not only in breast tumors but also in pancreatic adenocarcinomas
and lung tumors. Am J Pathol. 183:1634–1644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kang D, Cho HS, Toyokawa G, Kogure M,
Yamane Y, Iwai Y, Hayami S, Tsunoda T, Field HI, Matsuda K, et al:
The histone methyltransferase Wolf-Hirschhorn syndrome candidate
1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes
Chromosomes Cancer. 52:126–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Angrand PO, Apiou F, Stewart AF,
Dutrillaux B, Losson R and Chambon P: NSD3, a new SET
domain-containing gene, maps to 8p12 and is amplified in human
breast cancer cell lines. Genomics. 74:79–88. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saloura V, Vougiouklakis T, Zewde M,
Kiyotani K, Park JH, Gao G, Karrison T, Lingen M, Nakamura Y and
Hamamoto R: WHSC1L1 drives cell cycle progression through
transcriptional regulation of CDC6 and CDK2 in squamous cell
carcinoma of the head and neck. Oncotarget. 7:42527–42538. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
French CA, Rahman S, Walsh EM, Kühnle S,
Grayson AR, Lemieux ME, Grunfeld N, Rubin BP, Antonescu CR, Zhang
S, et al: NSD3-NUT fusion oncoprotein in NUT midline carcinoma:
Implications for a novel oncogenic mechanism. Cancer Discov.
4:928–941. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang
E, Roe JS, Suzuki Y, Pappin DJ, Joshua-Tor L and Vakoc CR:
NSD3-short is an adaptor protein that couples BRD4 to the CHD8
chromatin remodeler. Mol Cell. 60:847–859. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosati R, La Starza R, Veronese A, Aventin
A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF and Mecucci
C: NUP98 is fused to the NSD3 gene in acute myeloid leukemia
associated with t(8;11)(p11.2;p15). Blood. 99:3857–3860. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Taketani T, Taki T, Nakamura H, Taniwaki
M, Masuda J and Hayashi Y: NUP98-NSD3 fusion gene in
radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15)
and expression pattern of NSD family genes. Cancer Genet Cytogenet.
190:108–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jacques-Fricke BT and Gammill LS: Neural
crest specification and migration independently require
NSD3-related lysine methyltransferase activity. Mol Biol Cell.
25:4174–4186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Knutson SK, Warholic NM, Wigle TJ, Klaus
CR, Allain CJ, Raimondi A, Scott M Porter, Chesworth R, Moyer MP,
Copeland RA, et al: Durable tumor regression in genetically altered
malignant rhabdoid tumors by inhibition of methyltransferase EZH2.
Proc Natl Acad Sci USA. 110:pp. 7922–7927. 2013; View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Knutson SK, Kawano S, Minoshima Y,
Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G,
Kumar N, et al: Selective inhibition of EZH2 by EPZ-6438 leads to
potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol
Cancer Ther. 13:842–854. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Basavapathruni A, Olhava EJ, Daigle SR,
Therkelsen CA, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR,
Raimondi A, Scott MP, et al: Nonclinical pharmacokinetics and
metabolism of EPZ-5676, a novel DOT1L histone methyltransferase
inhibitor. Biopharm Drug Dispos. 35:237–252. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Klaus CR, Iwanowicz D, Johnston D,
Campbell CA, Smith JJ, Moyer MP, Copeland RA, Olhava EJ, Scott MP,
Pollock RM, et al: DOT1L inhibitor EPZ-5676 displays synergistic
antiproliferative activity in combination with standard of care
drugs and hypomethylating agents in MLL-rearranged leukemia cells.
J Pharmacol Exp Ther. 350:646–656. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Waters NJ, Smith SA, Olhava EJ, Duncan KW,
Burton RD, O'Neill J, Rodrigue ME, Pollock RM, Moyer MP and
Chesworth R: Metabolism and disposition of the DOT1L inhibitor,
pinometostat (EPZ-5676), in rat, dog and human. Cancer Chemother
Pharmacol. 77:43–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mohammad HP, Smitheman KN, Kamat CD, Soong
D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y,
Butticello M, et al: A DNA hypomethylation signature predicts
antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell.
28:57–69. 2015. View Article : Google Scholar : PubMed/NCBI
|