Introduction
Breast cancer is one of the leading causes of death
among women worldwide due mainly to its ability to metastasize and
develop chemoresistance. It has been estimated that one-third of
breast cancer patients relapse at some point and that 25% of all
cases are resistant to therapy (1).
Chemoresistance is complex and involves several cellular and
molecular events including alterations in the cell cycle, apoptosis
or DNA damage repair pathways and a greater capacity to excrete
chemotherapeutic drugs (2). The
cell cycle regulator p21, which historically has been considered as
a suppressor protein in normal cells, was recently linked to cancer
progression and chemoresistance (3). For instance, the Nrf2-p21 axis has
been associated with an increase in the resistant tumor cell
population, activation of antioxidant mechanisms and
chemoresistance in MCF-7, MDA-MB-231 and T47D cells (4). Moreover, ErbB2-dependent
overexpression of p21 correlates with resistance to the
chemotherapeutic drug Taxol in breast cancer (5), suggesting that in pathological
conditions this cell arrest mechanism is triggered to protect the
tumor cells from toxic treatments commonly used to target DNA
division and/or induction of apoptosis. In recent years, drug
expulsion has been considered another key mechanism of
chemoresistance. The ATP-binding cassette (ABC) transporter family
with its 49 members present in the human genome is one of the
largest and oldest known protein families (6). One feature common to all members of
this family is that they are membrane transporters that, by
consuming ATP, are able to expel from cells a wide spectrum of
substrates, including vitamins, lipids, hormones, metabolic waste
products and xenobiotics such as toxins and drugs. Their expression
and activity, in fact, are correlated with a decrease in the
cytoplasmic concentration of drugs and consequent failure of
therapy (7). In addition, in an
analysis of cellular and population composition, the onset of
chemoresistance has been linked to cancer stem cells (CSCs). CSCs
are a cancer cell subpopulation that has been demonstrated to
possess tumor-initiating properties and metastatic potential, and
they are intrinsically chemoresistant (8). CSCs have been already described and
characterized in several hematologic and solid tumors including
breast cancer, where the CD44+/CD24− surface
marker profile has been considered a canonical CSC characteristic
(9); although emerging evidence
indicates that this profile is not exclusive to mammary cancer
cells with CSC properties (10).
Moreover, the origin of the CSC population is still controversial,
and some other cellular events are associated with their stem-like
profile as is the case for epithelial-mesenchymal transition (EMT).
EMT in cancer is well documented and is characterized by a
reversible conversion of cells with a polarized epithelial pattern
into cells with a mesenchymal profile (11). At the molecular level, during EMT,
epithelial cells lose adhesion molecules such as E-cadherin, lose
their epithelial differentiation markers and acquire high motility
by induction of vimentin and N-cadherin proteins. In fact, this
transformation highly correlates with the
CD44+/CD24− profile and chemoresistance
(12).
Several researchers have focused on improving
chemotherapeutic treatment using natural molecules to limit
chemoresistance and avoid significant increases in toxicity.
Molecular iodine (I2) is a chemical form of iodine that
exerts significant antineoplastic effects on several types of
cancer cells, and its actions could be mediated by multiple
mechanisms. At moderately high concentrations, iodine induces a
strong depolarization of mitochondrial membranes triggering
mitochondrion-mediated apoptosis (13). Furthermore, I2 is able to
react with lipids and proteins producing several iodinated
compounds. Among all the iodolipids
5-hydroxy-6-iodo-8,11,14,eicosatrienoic δ-lactone, also called
6-iodolactone (6-IL), has been confirmed to be an agonist of the
peroxisome proliferator-activated receptor type γ (PPARγ). IL-6
promotes differentiation by decreasing the expression of specific
markers associated with invasiveness and metastasis (14,15).
Moreover, previous studies from our laboratory showed that when
co-administered with doxorubicin (DOX), I2 significantly
improves conventional mammary cancer treatment in both women and
rodents, and it diminishes the chemoresistance response (16,17).
In the present study we developed a cell line resistant to low
doses of DOX as a model to analyze in-depth how the I2
supplement affects the chemoresistance response. DOX is an
anthracycline antibiotic and is the most widely used
chemotherapeutic drug in breast cancer treatment. Our results
showed that after 30 days of exposure to 10 nM of DOX, MCF-7/D
cells exhibited the same proliferation rate but higher expression
of the p21, Bcl-2 and MDR-1 proteins associated with
chemoresistance mechanisms in comparison with MCF-7/W. The
molecular iodine supplement maintained its apoptotic effect in both
types of cells, indicating that I2 and DOX exert
antineoplastic effects by different mechanisms. In addition,
I2 increased the intracellular retention of DOX and
exerted a differential down-selection of the highly tumorigenic
CD44+/CD24+ and
E-cad+/vim+ subpopulations. The I2
+ DOX-selected cells showed a reserved tumorigenic competence in
xenografts suggesting that the chemoresistance and invasive
mechanisms were defective. All these I2 actions were
associated with a significant increase in PPARγ expression.
Materials and methods
Cell culture and I2 + DOX
treatment
The MCF-7 cell line was obtained from the American
Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in
Dulbeccos modified Eagles medium (DMEM) (Gibco, Thermo Fisher
Scientific, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum (FBS) and maintained at 37°C in a 5% CO2
atmosphere. Adriblastin® (Pfizer Inc., New York, NY,
USA) was the source of DOX at a concentration of 35.4 ng/ml,
equivalent to 10 nM DOX. The MCF-7/D cell line was generated by
treating MCF-7/W cells for 30 days with 10 nM DOX. Both types of
cells were authenticated follow the ATCC protocol by short tandem
repeat analysis. Molecular iodine was prepared with 13 g of
crystalline iodine (Macron-Avantor, Center Valley, PA, USA) and 60
g of potassium iodide (Sigma-Aldrich, St. Louis, MO, USA) in one
liter of ddH2O. The iodine concentration was confirmed
by titration with a solution of 0.1 N sodium thiosulfate. A working
concentration of 200 µM I2 was employed in all
assays.
Proliferation assay
Cells (25,000) were seeded into 6-well plates and
left to recover for 24 h in DMEM before treatments. Medium and
treatments were replaced daily before counting. Cell counting was
performed using a Neubauer chamber. The coefficient of drug
interaction (CDI) was calculated as reported by Gong et al
(18) with the follow equation: CDI
= (I2 + DOX) × nt/(I2 × DOX), where
(I2 + DOX) is proliferation of the co-treated culture,
nt is proliferation of the non-treated cells, while I2
and DOX represent the proliferation of cultures treated with each
alone. Values <0.7 are considered as synergic interaction;
values in the range 0.7–1.3 indicate additive interaction, and
values >1.3 indicate an inhibitory effect.
RT-qPCR
Total RNA was extracted using TRIzol®
(Invitrogen, Carlsbad, CA, USA) as suggested by the manufacturer.
Reverse transcription (RT) was performed using M-MLV Reverse
Transcriptase (Promega Corp., Fitchburg, WI, USA) and antisense
specific primers according to the manufacturer's protocol.
Quantitative PCR was performed as follows: 0.5 µl of cDNA solution
was added together with 0.4 µl 10 µM-specific primer mix (forward
and reverse), 5 µl Maxima SYBR-Green/ROX qPCR Master Mix
(Fermentas, Burlington, ON, Canada) and 4.1 ddH2O. The
reaction was performed using a Corbett research 3,000 rotor-gene.
The thermal profile used was: 95°C for 10 min as hot-start step
followed by 35 repetitions of the amplification cycle (melting at
95°C for 15 sec, annealing at 60°C for 30 sec, elongation at 72°C
for 30 sec). Lastly, the melting curve was analyzed to check
amplification specificity. Absolute gene quantifications were
normalized to β-actin levels. Table
I summarizes the primers used in the present study.
 | Table I.Primer details. |
Table I.
Primer details.
| Gene name | Accession no. | Forward primer
sequences | Reverse primer
sequences | bp |
|---|
| ABCg2 | NM_001257386.1 |
AGTGTTTCAGCCGTGGAACT |
GCATCTGCCTTTGGCTTCAA | 194 |
| BAX | NM_001291428.1 |
AAGCTGAGCGAGTGTCTCAAGCGC |
TCCCGCCACAAAGATGGTCACG | 327 |
| Bcl-2 | NM_000633.2 |
GTGGAGGAGCTCTTCAGGGA |
AGGCACCCAGGGTGATGCAA | 306 |
| Survivin
(Birc5) | NM_001168.2 |
TTCTCAAGGACCACCGCATC |
CCAAGTCTGGCTCGTTCTCA | 126 |
| E-cadherin | NM_004360.3 |
TGCCCAGAAAATGAAAAAGG |
GTGTATGTGGCAATGCGTTC | 200 |
| MDR-1 | NM_000927.4 |
GAGAGATCCTCACCAAGCGG |
ATCATTGGCGAGCCTGGTAG | 122 |
| p21 | NM_000389.4 |
GACCATGTGGACCTGTCACT |
GCGGATTAGGGCTTCCTCTT | 176 |
| Vimentin | NM_003380.3 |
GAGAACTTTGCCGTTGAAGC |
GCTTCCTGTAGGTGGCAATC | 163 |
| β-actin | NM_001101.3 |
CCATCATGAAGTGTGACGTTG |
ACAGAGTACTTGCGCTCAGGA | 175 |
Flow cytometry
CD44 and CD24 staining was performed as follows.
After a 72-h treatment, cells were washed with phosphate-buffered
saline (PBS) and detached with 0.05% EDTA/PBS. Cells
(1–2×106) were incubated in PBS containing 0.05% EDTA +
0.05% BSA, and then for 1 h in ice with antibodies against CD24
(coupled to PE; diluted 1:50; Abcam, Cambridge, UK) and CD44
(coupled to FITC; diluted 1:50; BD Biosciences, San Jose, CA, USA).
After a wash with PBS, cells were fixed using 2% formaldehyde in
PBS for 10 min. After washing again with PBS, the cells were
re-suspended with 1 ml PBS and analyzed.
Due to the cytoplasmic location of their epitope,
E-cadherin (E-cad) and vimentin (vim) were stained as follows.
After detaching using trypsin + 0.05% EDTA solution and washing
with 0.05% EDTA/PBS, the cells were fixed with 2% formaldehyde in
PBS for 10 min on ice. Cells were permeabilized using a 1:1
methanol/acetone solution at −20°C for 1 min. After a PBS wash, the
cells were incubated for 1 h on ice with antibodies to E-cad
coupled to Alexa 647 (diluted 1:2,000) and to vim coupled to PE
(diluted 1:20) (both from BD Biosciences). After a last PBS wash,
cells were re-suspended in 1 ml PBS. A BD Biosciences Accuri C6
flow cytometer was used to analyze the population.
VirtualGain® was applied to normalize background
fluorescence among treatments. Data were acquired and visualized
using BD Biosciences Accuri C6 software.
DOX retention assay
After a 72-h pretreatment with 200 µM I2,
cells were incubated with 20 or 500 nM DOX as follows. The medium
was replaced with fresh DMEM, and the cells were incubated for 1 h.
An appropriate volume of concentrated DOX was added directly to the
culture, which was incubated for another 1.5 h. At this point the
cells were detached with trypsin + 0.05% EDTA solution. A sample
containing 1–2×106 cells was fixed with 2% formaldehyde
in PBS. DOX fluorescence was detected by BD Biosciences Accuri C6
cytometer with excitation at 488 nM; emission filter 585/40. Data
were acquired and visualized using BD Biosciences Accuri C6
software.
Tumorogenic capacity
Female athymic homozygotic
(Foxn1nu/nu, Harlan, Indianapolis, IN, USA) mice
were housed in a temperature-controlled room (21±1°C) with a
12-h/12-h light/dark schedule. They were given food (Purina
certified rodent chow; Ralston Purina Co., St. Louis, MO, USA) and
water ad libitum. All of the procedures followed the Animal
Care and Use Program of the National Institutes of Health (NIH)
(Bethesda, MD USA), and were approved by the Committee on Ethics in
Investigation from INB (Protocol #035). When homozygotic animals
were 6-weeks old, each animal was injected subcutaneously with
2×106 MCF-7/D cells in 50 µl PBS and 50 µl Matrigel. All
animals were monitored daily for 20 days; any xenografts were
detected and measured with an automatic Vernier, and their volume
was calculated using the ellipsoid formula (19). On day 20, the presence of a tumor
mass was corroborated by the use of a thermograph camera FLIR E40
(parameters are summarized in Table
II), and digital processing software was implemented in MATLAB
and FLIR Tools to calculate tumor temperature (MathWorks, Natick,
MA, USA).
| Table II.Camera parameters. |
Table II.
Camera parameters.
| Temperature
range | –20 to 650°C |
| Thermal
sensitivity | <0.07 to
30°C |
| Detector type | Focal plane array
(FPA); uncooled microbolometer 160×120 pixels |
| Field of view | Focus 25° ×
19° |
| Spectral range | 7.5–13 µm |
Statistical analyses
One- or two-way ANOVA was performed to determine the
significance of differences between groups, followed by Tukey's
test for the significance of differences among multiple
experimental groups. DOX retention data were analyzed by Student's
t-test. Tumor progression was calculated by linear regression
analysis.
Results
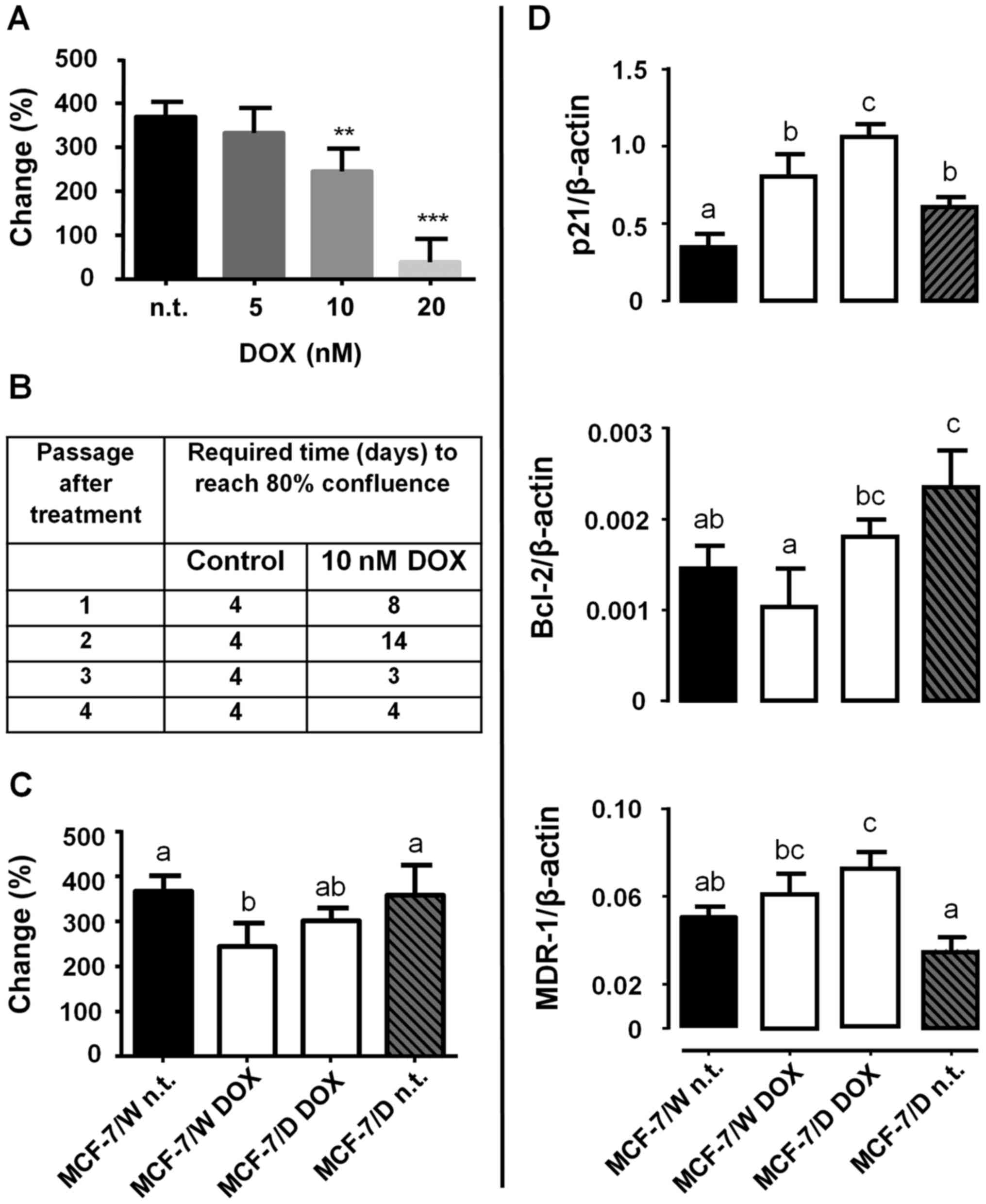
Initial characterization of the low-dose
DOX-resistant model showed that at 10 nM DOX, the MCF-7/W cell
culture maintained its proliferation rate at 60% of the untreated
control, whereas 20 nM DOX induced a total block of proliferation
at 96 h (Fig. 1A). The established
DOX-resistant model required two and four times longer (8 and 14
days) to reach 80% confluence after the first and second
subcultures (passages), but within 30 days, the duplication rate
had returned to the control value (Fig.
1B). The acute treatment (96 h) with 10 nM DOX decreased the
proliferation rate (% change) only in MCF-7/W cells (Fig. 1C). DOX adaptation was accompanied by
significant increases in the expression of the chemoresistance
markers p21, Bcl-2 and MDR-1 (Fig.
1D, MCF-7/D DOX). Removal of chronic DOX treatment from MCF-7/D
cells decreased p21 and MDR-1 expression (MCF-7/D n.t.).
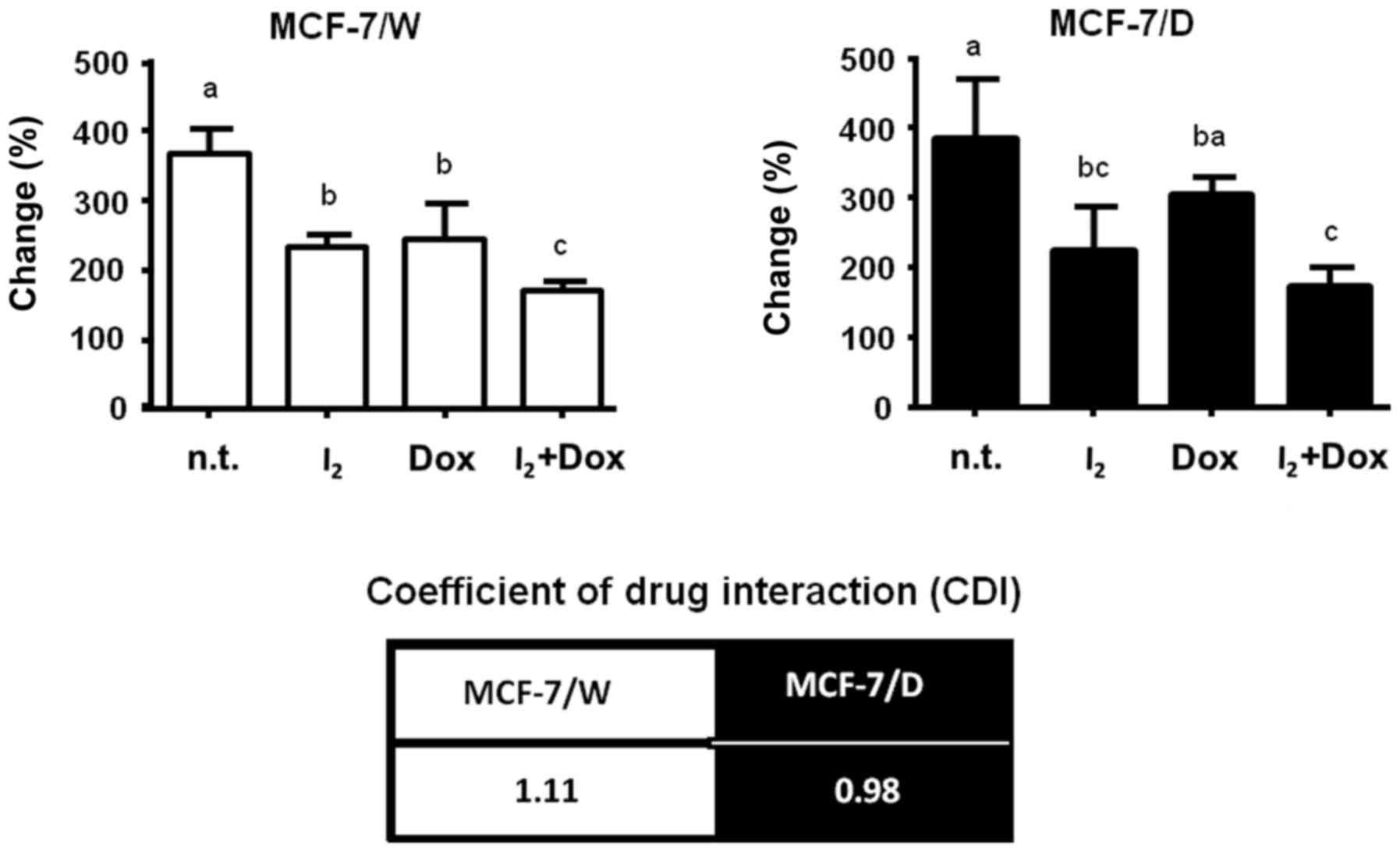
Fig. 2 shows the
effect of 200 µM I2 alone or co-administered with 10 nM
DOX. Iodine alone inhibited proliferation similarly in both types
of cells, and the magnitude of this inhibition was also similar to
that observed in the MCF-7/W cells treated with 10 nM DOX.
Co-administration of I2 + DOX exerted an additive effect
on both cellular populations, as indicated by the coefficients of
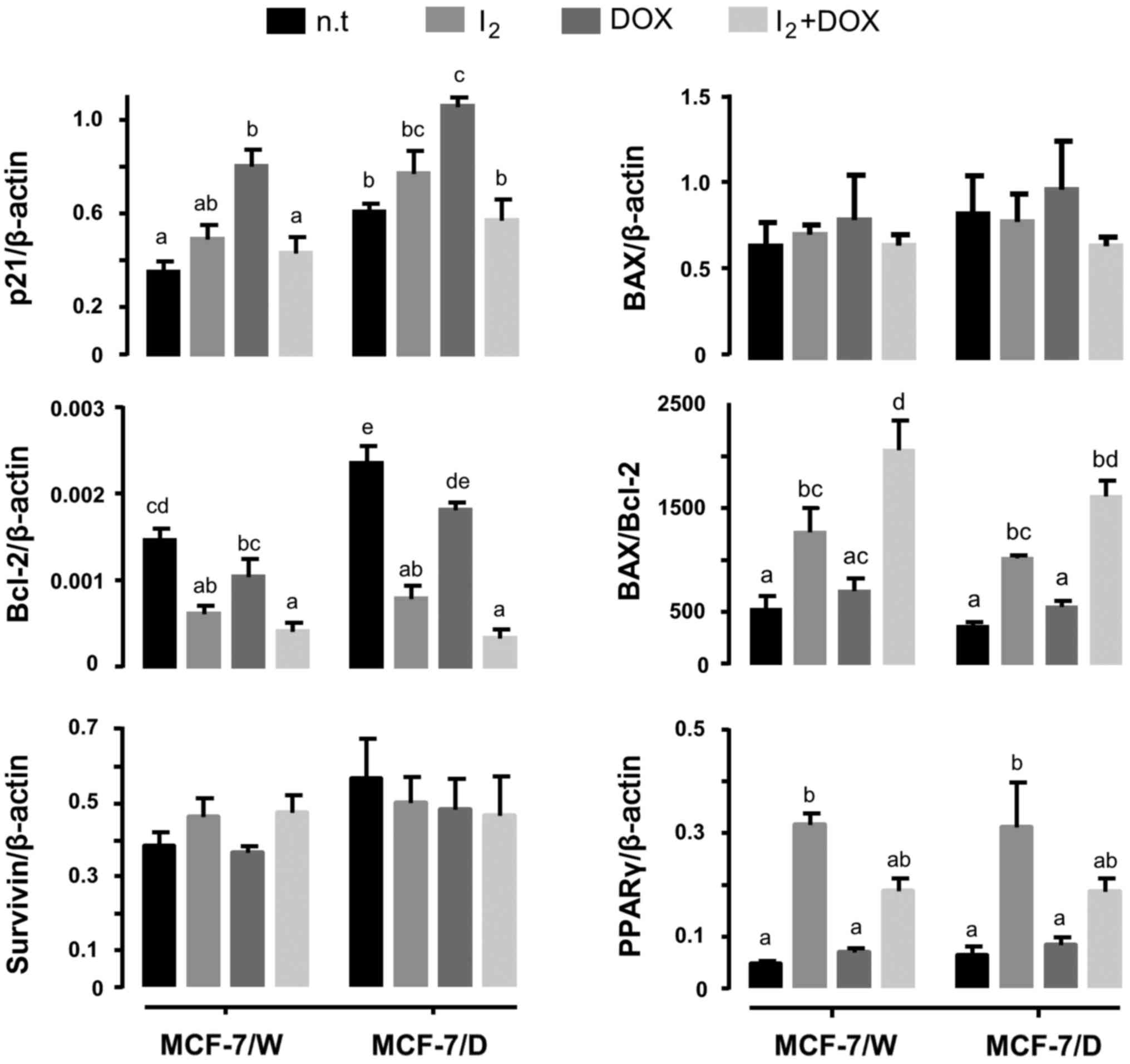
drug interaction (CDI). Gene analysis (Fig. 3) showed that the antineoplastic
effect of I2 per se was associated with a
decrease in Bcl-2 and an increase in PPARγ expression in both the
MCF-7/W and MCF-7/D cells. These effects were also observed with
I2 + DOX, but in this case I2 also impaired
cell cycle arrest (canceled the increase caused by DOX) and
intensified the decrease in Bcl-2 expression, thereby enhancing
apoptosis induction (BAX/Bcl-2 index). Survivin expression did not
show any change.
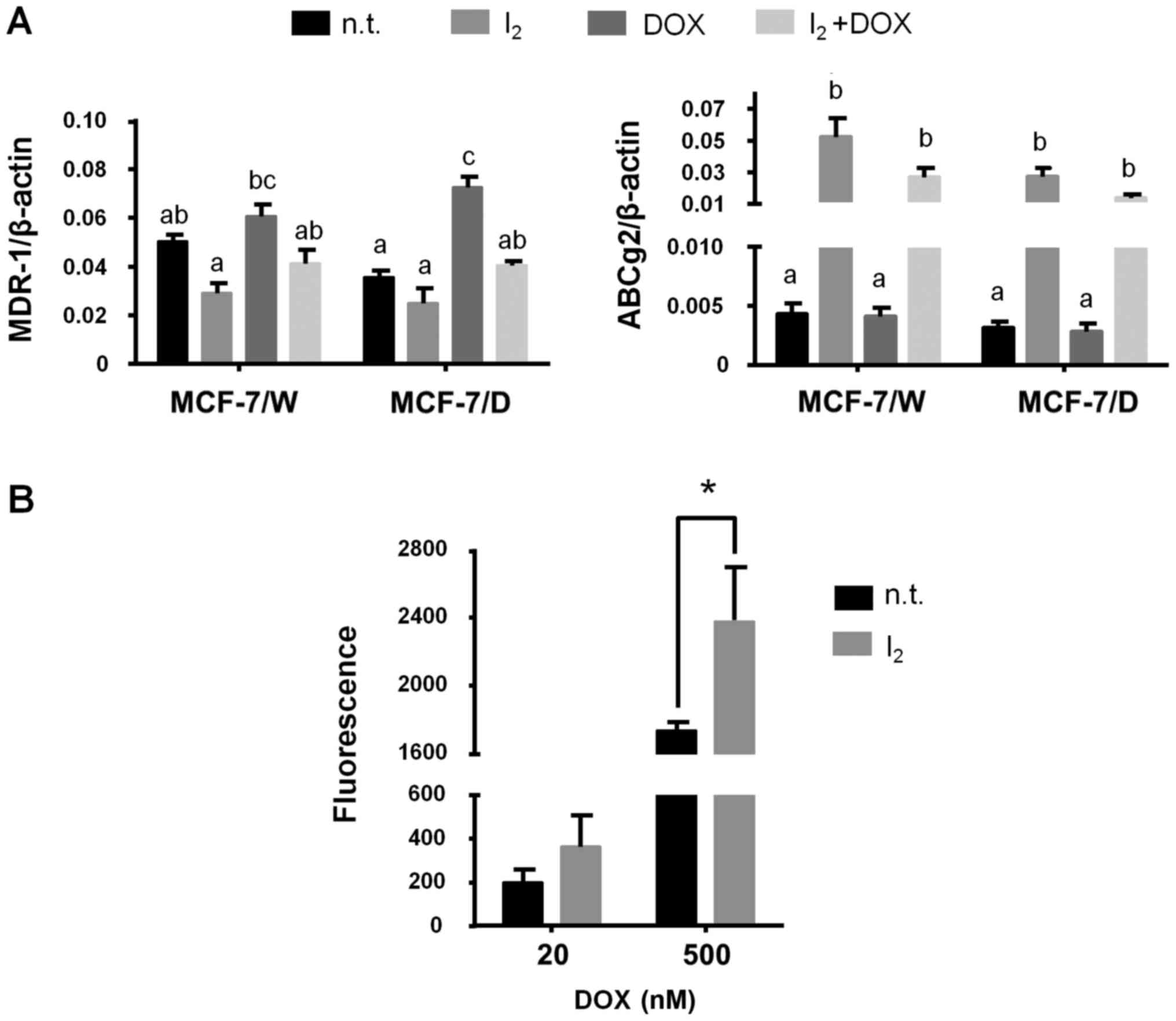
Fig. 4 shows the
effect of I2 on the expression of two of the most
important drug expulsion membrane transporters. Iodine did not
modify the expression of MDR-1 in the MCF-7/W cells but blocked its
induction by DOX in the MCF-7/D cells. In contrast, the
I2 supplement showed significant induction of ABCg2
transporter expression in all conditions (Fig. 4A). The DOX functional retention
assay showed an increase in the intracellular concentration of DOX
(fluorescence) when I2 was administered for 72 h, with a
tendency observed at low concentrations, but a clear and
significant increase at 500 nM DOX (Fig. 4B).
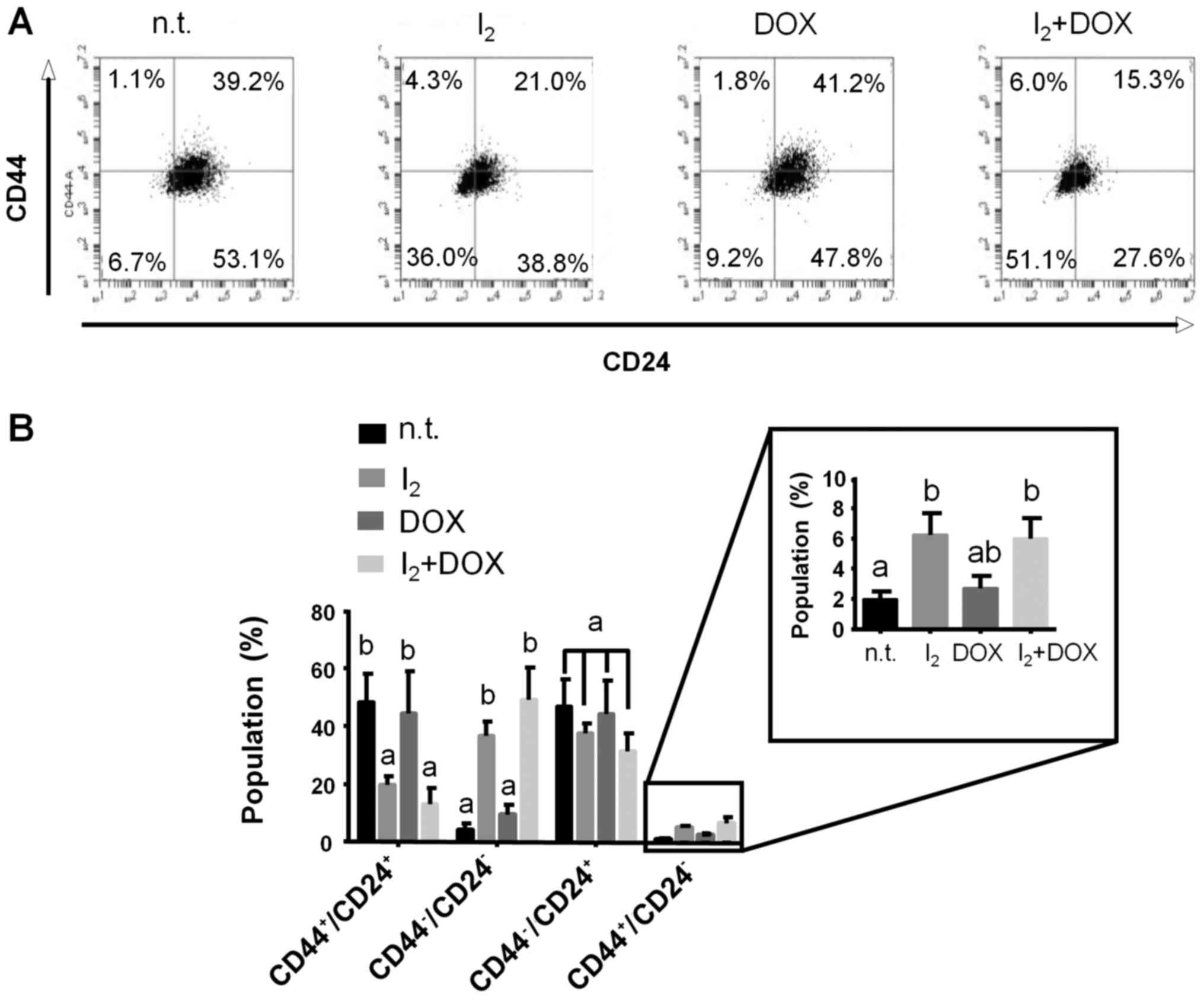
Phenotypes of mammary CSCs (CD44/CD24) and the EMT
process (E-cad/vim) were analyzed in MCF-7/D cells. Fig. 5 shows a significant decrease in the
CD44+/CD24+ population in favor of the
double-negative cell population in I2-treated cells with
and without DOX. The CD44+/CD24− phenotype,
which is the scarcest subtype (<4%) observed in these resistant
cells, showed a modest but significant increase (6%) after
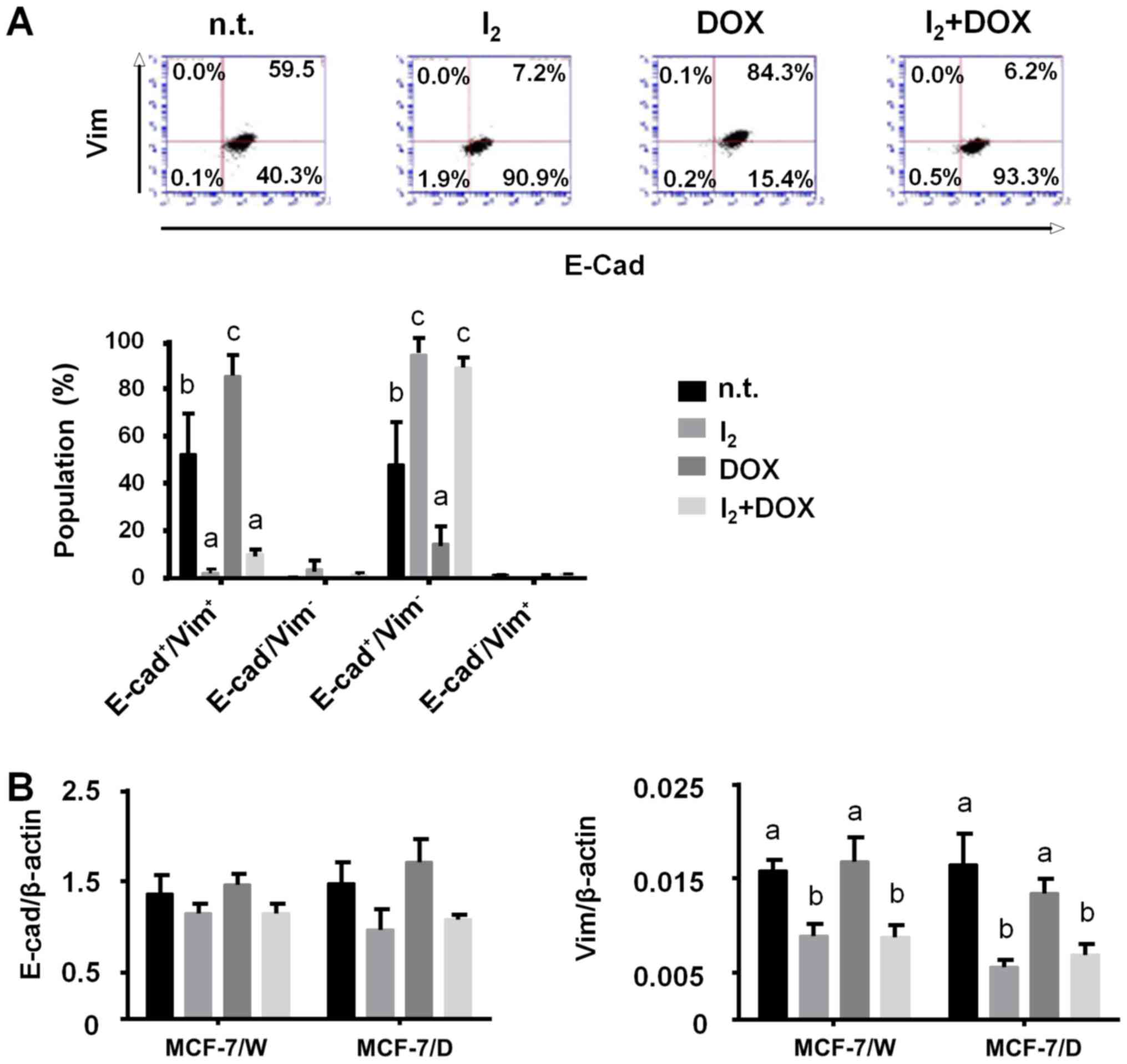
I2 treatment. Fig. 6
shows that in terms of EMT classification, the most abundant
population in the MCF-7/D cells corresponded to E-cad+.
Iodine treatment was accompanied by a significant decrease in
E-cad+/vim+ in favor of
E-cad+/vim−, and again, this was independent
of DOX presence. RT-PCR analysis show that the I2
supplement diminished vimentin expression (Fig. 6B).
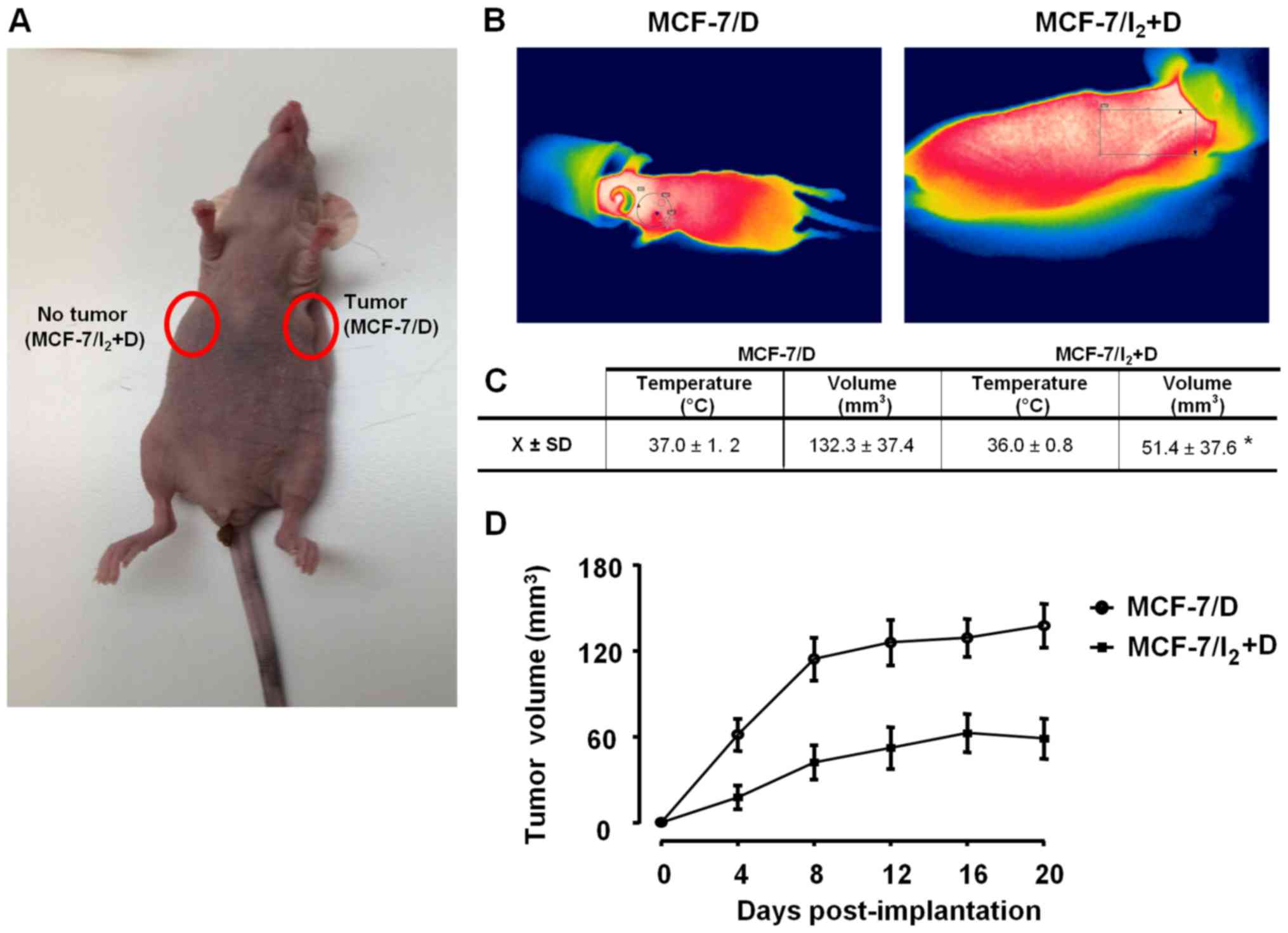
To analyze the in vivo tumorigenic capacity
of MCF-7/D subpopulations, athymic mice were inoculated with
DOX-resistant cells pre-incubated for 96 h with 10 nM DOX (MCF-7/D)
or 200 µM I2 + 10 nM DOX (MCF-7/I2 + D). Each
animal was inoculated with both subpopulations on the left or right
side, respectively. Fig. 7 shows
that MCF-7/D cells induced xenograft beginning on day 4 and
maintained a rapid growth until day 12, whereas with
MCF-7/I2 + D, its growth rate and tumor size were
significantly less.
Discussion
The multistep protocol has been commonly used to
establish in vitro models to study chemoresistance. Based on
this method, resistant cells are selected by treating with a
sequence of increasing DOX concentrations starting from low to over
1 mM depending on the author. However, in some cases this
multi-step selection is accompanied by a loss of identity of
cellular origin (20,21) or has only a weak correlation with
clinical reality, where tumors undergo neither such step-by-step
exposure nor such high DOX concentrations (22,23).
For that reason, we used a single-step treatment extended for one
month, a period that resembles the interval that separates one
treatment cycle from the next. In our experience, DOX at a low
concentration (10 nM) favors drug adaptation, as suggested by cell
proliferation to a normal rate and decreasing sensitivity to DOX.
At the molecular level, the antineoplastic effect of DOX results
from a variety of actions; the best known are its ability to
intercalate into DNA and to form a complex with topoisomerase II
and DNA which triggers apoptosis, apparently via the p53-caspase
pathway (24). In agreement,
numerous studies indicate that DOX-resistant cells respond by
decreasing topoisomerase II expression and increasing the
expression of membrane drug transporters and the anti-apoptotic
signal (22,25,26).
Some of these mechanisms were observed in our MCF-7/D cells, such
as cell cycle arrest (p21 upregulation), efficient drug expulsion
(upregulated MDR-1 expression), and apoptosis evasion (BAX/Bcl-2
ratio decrease), indicating that these DOX-adapted cells can be
considered as a chemoresistant cell model. The apparently
paradoxical increase in p21 expression in response to DOX in both
cell types agrees with recent studies showing that p21 can exert
both anti- and pro-apoptotic effects in response to antitumor
drugs, depending on cell type and cellular context (3). Cytotoxic drugs commonly act in
mitotically active cells where they trigger apoptosis by inducing
DNA damage (27). From this, it is
reasonable to assume that early cellular alterations in reaction to
such drugs may include apoptosis evasion and quiescence. Although
the general observation that MCF-7/D cells return to the same
proliferative rate as the wild-type, careful analysis reveals that
these DOX-resistant cells include several subpopulations that could
have different proliferation rates. Studies in our laboratory
designed to confirm this hypothesis are now in progress.
The primary objective of the present study was to
analyze for the first time the effect of iodine on the
chemoresistance acquisition to DOX. Previous studies from our
laboratory and others have shown that I2 exerts
antiproliferative and apoptotic effects in different models of
cancer (16,17,28,29).
Specifically, in mammary cell lines it has been demonstrated that
cancerous (MCF-7, MDA-MB134, MDA-MB157 and MDA-MB436) and normal
(MCF-10, MCF-12F) lines exhibit different sensitivity to
I2, but they all have a lower rate of proliferation when
iodine is present (28,29). The most sensitive cell line is
MCF-7, which is the focus of the present study as it represents the
most frequent breast cancer in women (luminal, estrogen-positive)
(30). Molecular iodine exerts a
direct apoptotic effect by mitochondrial membrane depolarization
and/or an indirect action via 6-iodolactone (6-IL). This iodolipid
is generated by iodination of arachidonic acid; by activating
PPARγ, 6-IL induces apoptosis and differentiation effects in MCF-7
cells (16,17). In the present study, I2
maintained its apoptotic effect independent of the DOX-resistance
mechanisms acquired by the cells. Several studies have shown that
DOX-adaptation confers resistance to other drugs, due mainly to the
overexpression of ABC transporters (22). It is possible that these membrane
transporters cannot expel iodine. Indeed, our results showed that
I2 alone inhibited MDR-1 expression only weakly, but it
significantly impaired MDR-1 upregulation by DOX treatment in
MFC-7/D cells, suggesting that the changes associated with
I2 treatment were capable of interfering with the
installation of DOX-resistance. One interesting observation is the
significant increase in ABCg2 expression in both types of cells
treated with I2. It is well documented that PPARγ
activation inversely modulates MDR-1 and ABCg2. Although the MDR-1
gene does not contain response elements to PPARs, these receptors
can inhibit the Wnt/β-catenin pathway, which is directly involved
in MDR-1 regulation (31). In
contrast, ABCg2 is directly stimulated by PPARγ agonists (32) and although these transporters are
overexpressed in some tumor types, they have also been detected in
several normal tissues such as intestine, liver, brain, placenta
and mammary glands (7). Moreover,
this breast cancer resistance protein (ABCg2) is strongly induced
in the mammary gland during pregnancy and lactation and is
responsible for pumping vitamin B2 into milk, suggesting a
physiological role in differentiated mammary cells (33). These facts, along with the
observation that I2 treatment is accompanied by
significantly higher intracellular retention of DOX, suggest that
the antineoplastic effect of iodine could be related to PPARγ
activation resulting in maintaining drug sensitivity
(downregulation of MDR-1 and, therefore, lower drug expulsion) and
the induction of cell differentiation. It is well established that
MCF-7 cells can respond to synthetic agonists of PPARγ by
increasing lipid accumulation, terminating cell growth and
undergoing changes characteristic of a less malignant state
(14,34,35).
These re-differentiation responses were also described by our group
in mammary (MCF-12 and MCF-7), prostate (RWPE-1, LNCaP and DU-145)
and neuroblastoma (SKN-AS and SKN-SH5Y) cell lines after
I2 or 6-IL administration (28,36,37).
In this context, it is possible that the significant increase in
the ABCg2 transporter corresponds more to an induction of
differentiation than of chemoresistance. The analysis of CSC and
EMT markers showed that the canonic CSC profile
CD44+/CD24− expected to be enriched in
drug-resistant cells was poorly represented (<4%) in MCF-7/D
cells, whereas the most abundant populations were the
CD44+/CD24+ and
CD44−/CD24+ subtypes (~40% each). The
supplementation with I2 showed a discrete increase in
CD44+/CD24− (~6%), no change in
CD44−/CD24+ and a clear differential
selection against CD44+/CD24+ with a
significant increase in the double-negative population
(CD44−/CD24−). Previous studies have
described that the canonic CD44+/CD24−
profile is not the only profile that corresponds to an invasive
phenotype. Indeed, in a recent study, using sphere-promoting
(Mammocult; Stem Cell Technologies, Vancouver, BC, Canada)
conditions, this double-positive subpopulation was found to be the
most representative group in the MCF-7 CSC culture (30). Increases in the double-positive
population were found to be associated with a worse outcome in
salivary gland (38), pancreatic
carcinomas (39), and in colorectal
cancer this double-positive population represents the specific
marker for CSCs (40).
Controversial results have been reported in relation to the
CD44−/CD24+ profile. In various studies,
increases in CD24+ cells were found to be correlated
with the most aggressive phenotype (41–43),
whereas in others there was no correlation with prognosis (44,45).
In contrast, the double-negative phenotype had no prognostic
significance in breast cancer patients (45), and in preclinical studies these
cells showed reduced capacity to induce tumor growth in soft agar
and xenografts in mouse models (46), suggesting that these cells are less
invasive. This less-aggressive profile found in I2 + DOX
cells was confirmed by the enrichment of
E-cad+/vim− cells. Indeed, the expected EMT
profile (E-cad−/vim+) was absent in MCF-7/D
cells, and the double-positive population was significantly
diminished in favor of the E-cad+/vim−
subpopulation when these cells were treated with I2.
E-cadherin is a transmembrane glycoprotein involved in epithelial
adherens junctions, and its loss could be sufficient to promote the
invasion-metastasis cascade, activating specific downstream signal
transduction pathways that bestow high motility on the cells by
inducing vimentin and N-cadherin proteins (47). In contrast, vimentin which is the
most commonly expressed and highly conserved member of the type III
intermediate filament protein family is considered the main EMT
marker. High vimentin expression is observed in several aggressive
breast cancer cell lines. In MCF-7 cells, vimentin overexpression
is accompanied by increases in motility and invasiveness. These
characteristics were reduced by vimentin antisense oligos in
MDA-MB-231 cells, which constitutively express this protein
(48). Congruently, our results
showed that I2-treated cells exhibited the lowest
vimentin expression and that the I2 + DOX-treated
subpopulation was powerless to initiate tumor xenografts,
corroborating its weak invasive potential. The EMT process, which
is triggered by factors such as transforming growth factor-β
(TGF-β), SNAIL and TWIST, is reverted by PPARγ activation (49,50).
Studies have shown the antineoplastic effects of PPARγ ligands in
various preclinical models (51);
however, agonists of these receptors used as monotherapy failed to
exert therapeutic benefits in advanced stage breast patients
(52). Notably, PPARγ agonists in
combination with the conventional antineoplastic drugs, such as
carboplatin or tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL), showed synergistic effects, indicating that
differentiation induced by PPARγ activation restored sensitivity to
the cytotoxic drug (53,54). These synergistic effects were
replicated in cells treated with DOX + I2 in both
preclinical (16) and clinical
studies (17), supporting the
notion that some I2 effects are mediated by PPARγ
activation.
In conclusion, the use of molecular iodine at a
moderately high concentration restored the sensitivity of mammary
cancer cells MCF-7/D to DOX. Impaired DOX expulsion and decreased
expression of the chemoresistance markers p21, Bcl-2 and MDR-1
resulted in the selection of a less aggressive population,
suggesting the potential of I2 as a clinically useful
anti-chemoresistance agent.
Acknowledgements
We thank M. Juana Cárdenas-Luna, Felipe Ortiz,
Adriana González and Michael Jeziorski for technical assistance,
Leonor Casanova and Lourdes Lara for academic support, Dorothy
Pless for proofreading and Martin Garcia-Servín and Alejandra
Castillo for animal care advice. We extend special thanks to Mario
Nava-Villalba and Silvia Angulo Barbosa for their contributions to
scientific discussions and to Guadalupe Delgado, who will live
forever in our memories, for technical and academic assistance. The
present study was partially supported by grants: PAPIIT-UNAM,
IN201516. Alexander Bontempo is a graduate student of UNAM in the
PhD Program in Biomedical Sciences of the National Autonomous
University of Mexico (Programa de Doctorado en Ciencias Biomédicas,
Universidad Nacional Autónoma de México) and received fellowship
262489 from CONACYT.
References
|
1
|
Martin HL, Smith L and Tomlinson DC:
Multidrug-resistant breast cancer: Current perspectives. Breast
Cancer. 6:1–13. 2014.PubMed/NCBI
|
|
2
|
Gong J, Jaiswal R, Mathys JM, Combes V,
Grau GE and Bebawy M: Microparticles and their emerging role in
cancer multidrug resistance. Cancer Treat Rev. 38:226–234. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Achuthan S, Santhoshkumar TR, Prabhakar J,
Nair SA and Pillai MR: Drug-induced senescence generates
chemoresistant stemlike cells with low reactive oxygen species. J
Biol Chem. 286:37813–37829. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hawthorne VS, Huang WC, Neal CL, Tseng LM,
Hung MC and Yu D: ErbB2-mediated Src and signal transducer and
activator of transcription 3 activation leads to transcriptional
up-regulation of p21Cip1 and chemoresistance in breast
cancer cells. Mol Cancer Res. 7:592–600. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vasiliou V, Vasiliou K and Nebert DW:
Human ATP-binding cassette (ABC) transporter family. Hum Genomics.
3:281–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liang Y, Li S and Chen L: The
physiological role of drug transporters. Protein Cell. 6:334–350.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abdullah LN and Chow EK: Mechanisms of
chemoresistance in cancer stem cells. Clin Transl Med. 2:3–12.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meyer MJ, Fleming JM, Lin AF, Hussnain SA,
Ginsburg E and Vonderhaar BK:
CD44posCD49fhiCD133/2hi defines
xenograft-initiating cells in estrogen receptor-negative breast
cancer. Cancer Res. 70:4624–4633. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bhat-Nakshatri P, Goswami CP, Badve S,
Sledge GW Jr and Nakshatri H: Identification of FDA-approved drugs
targeting breast cancer stem cells along with biomarkers of
sensitivity. Sci Rep. 3:2530–2542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
May CD, Sphyris N, Evans KW, Werden SJ,
Guo W and Mani SA: Epithelial-mesenchymal transition and cancer
stem cells: A dangerously dynamic duo in breast cancer progression.
Breast Cancer Res. 13:202–212. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sarrio D, Franklin CK, Mackay A,
Reis-Filho JS and Isacke CM: Epithelial and mesenchymal
subpopulations within normal basal breast cell lines exhibit
distinct stem cell/progenitor properties. Stem Cells. 30:292–303.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shrivastava A, Tiwari M, Sinha RA, Kumar
A, Balapure AK, Bajpai VK, Sharma R, Mitra K, Tandon A and Godbole
MM: Molecular iodine induces caspase-independent apoptosis in human
breast carcinoma cells involving the mitochondria-mediated pathway.
J Biol Chem. 281:19762–19771. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nuñez-Anita RE, Arroyo-Helguera O,
Cajero-Juárez M, López-Bojorquez L and Aceves C: A complex between
6-iodolactone and the peroxisome proliferator-activated receptor
type gamma may mediate the antineoplastic effect of iodine in
mammary cancer. Prostaglandins Other Lipid Mediat. 89:34–42. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nava-Villalba M, Nuñez-Anita RE, Bontempo
A and Aceves C: Activation of peroxisome proliferator-activated
receptor gamma is crucial for antitumoral effects of 6-iodolactone.
Mol Cancer. 14:168–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alfaro Y, Delgado G, Cárabez A, Anguiano B
and Aceves C: Iodine and doxorubicin, a good combination for
mammary cancer treatment: Antineoplastic adjuvancy, chemoresistance
inhibition, and cardioprotection. Mol Cancer. 12:45–55. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peralta G, Torres JM, Delgado G, Dominguez
A, De Obaldía R, Duarte L, Paredes E, Avecilla C, Hernández S,
Vega-Riverol L, et al: Iodine exhibits dual effects on breast
cancer as a co-treatment with anthracyclines: Anti-neoplastic
synergy and cardioprotector. In 102nd Annual Meeting, AACR,
Orlando, FL, 2011. (adstract 3509). Cancer Res. 71(Suppl 8): pp.
35092011; doi: 10.1158/1538-7445.AM2011-3509.
|
|
18
|
Gong JH, Liu XJ, Shang BY, Chen SZ and
Zhen YS: HERG K+ channel related chemosensitivity to
sparfloxacin in colon cancer cells. Oncol Rep. 23:1747–1756.
2010.PubMed/NCBI
|
|
19
|
Thompson HJ: Methods for the induction of
mammary carcinogenesis in the rat using either
7,12-dimethylbenz[a]antracene or 1-methyl-1-nitrosoureaMethods in
Mammary Gland Biology and Breast Cancer Research. Ip M and Aschz
BB: 8th edition. Kluwer Academic/Plenum Publishers; NY: pp. 19–29.
2000, View Article : Google Scholar
|
|
20
|
Mehta K, Devarajan E, Chen J, Multani A
and Pathak S: Multidrug-resistant MCF-7 cells: An identity crisis?
J Natl Cancer Inst. 94:1652–1654. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pirnia F, Breuleux M, Schneider E,
Hochmeister M, Bates SE, Marti A, Hotz MA, Betticher DC and Borner
MM: Uncertain identity of doxorubicin-resistant MCF-7 cell lines
expressing mutated p53. J Natl Cancer Inst. 92:1535–1536. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Calcagno AM, Fostel JM, To KKW, Salcido
CD, Martin SE, Chewning KJ, Wu CP, Varticovski L, Bates SE, Caplen
NJ, et al: Single-step doxorubicin-selected cancer cells
overexpress the ABCG2 drug transporter through epigenetic changes.
Br J Cancer. 98:1515–1524. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kars MD, Iseri OD, Gündüz U, Ural AU,
Arpaci F and Molnár J: Development of rational in vitro models for
drug resistance in breast cancer and modulation of MDR by selected
compounds. Anticancer Res. 26:4559–4568. 2006.PubMed/NCBI
|
|
24
|
Mehta K: High levels of transglutaminase
expression in doxorubicin-resistant human breast carcinoma cells.
Int J Cancer. 58:400–406. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Calcagno AM, Salcido CD, Gillet JP, Wu CP,
Fostel JM, Mumau MD, Gottesman MM, Varticovski L and Ambudkar SV:
Prolonged drug selection of breast cancer cells and enrichment of
cancer stem cell characteristics. J Natl Cancer Inst.
102:1637–1652. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Järvinen TA, Tanner M, Rantanen V, Bärlund
M, Borg A, Grénman S and Isola J: Amplification and deletion of
topoisomerase IIalpha associate with ErbB-2 amplification and
affect sensitivity to topoisomerase II inhibitor doxorubicin in
breast cancer. Am J Pathol. 156:839–847. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
AbuHammad S and Zihlif M: Gene expression
alterations in doxorubicin resistant MCF7 breast cancer cell line.
Genomics. 101:213–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arroyo-Helguera O, Rojas E, Delgado G and
Aceves C: Signaling pathways involved in the antiproliferative
effect of molecular iodine in normal and tumoral breast cells:
Evidence that 6-iodolactone mediates apoptotic effects. Endocr
Relat Cancer. 15:1003–1011. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rösner H, Torremante P, Möller W and
Gärtner R: Antiproliferative/cytotoxic activity of molecular iodine
and iodolactones in various human carcinoma cell lines. No
interfering with EGF-signaling, but evidence for apoptosis. Exp
Clin Endocrinol Diabetes. 118:410–419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smart CE, Morrison BJ, Saunus JM, Vargas
AC, Keith P, Reid L, Wockner L, Askarian-Amiri M, Sarkar D, Simpson
PT, et al: In vitro analysis of breast cancer cell line
tumourspheres and primary human breast epithelia mammospheres
demonstrates inter- and intrasphere heterogeneity. PLoS One.
8:e643882013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang H, Jing X, Wu X, Hu J, Zhang X, Wang
X, Su P, Li W and Zhou G: Suppression of multidrug resistance by
rosiglitazone treatment in human ovarian cancer cells through
downregulation of FZD1 and MDR1 genes. Anticancer Drugs.
26:706–715. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weiss J, Sauer A, Herzog M, Böger RH,
Haefeli WE and Benndorf RA: Interaction of thiazolidinediones
(glitazones) with the ATP-binding cassette transporters
P-glycoprotein and breast cancer resistance protein. Pharmacology.
84:264–270. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van Herwaarden AE, Wagenaar E, Merino G,
Jonker JW, Rosing H, Beijnen JH and Schinkel AH: Multidrug
transporter ABCG2/breast cancer resistance protein secretes
riboflavin (vitamin B2) into milk. Mol Cell Biol.
27:1247–1253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ruiz-Vela A, Aguilar-Gallardo C,
Martínez-Arroyo AM, Soriano-Navarro M, Ruiz V and Simón C: Specific
unsaturated fatty acids enforce the transdifferentiation of human
cancer cells toward adipocyte-like cells. Stem Cell Rev. 7:898–909.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Davies GF, Juurlink BH and Harkness TA:
Troglitazone reverses the multiple drug resistance phenotype in
cancer cells. Drug Des Devel Ther. 3:79–88. 2009.PubMed/NCBI
|
|
36
|
Aranda N, Sosa S, Delgado G, Aceves C and
Anguiano B: Uptake and antitumoral effects of iodine and
6-iodolactone in differentiated and undifferentiated human prostate
cancer cell lines. Prostate. 73:31–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Núñez-Anita RE, Nava-Villalba M and Aceves
C: Dose-dependent apoptotic effect of molecular iodine in two
neuroblastoma cell lines. Possible Participation of Retinoic Acid
Receptor. 14th International Thyroid Congress, Paris, France, Sept
2010. https://www.thyroid.org/professionals/meetings/past-meetings/14th-international-thyroid-congress/
|
|
38
|
Soave DF, da Costa JP Oliveira, da
Silveira GG, Ianez RC, de Oliveira LR, Lourenço SV and
Ribeiro-Silva A: CD44/CD24 immunophenotypes on clinicopathologic
features of salivary glands malignant neoplasms. Diagn Pathol.
8:29–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ohara Y, Oda T, Sugano M, Hashimoto S,
Enomoto T, Yamada K, Akashi Y, Miyamoto R, Kobayashi A, Fukunaga K,
et al: Histological and prognostic importance of
CD44+/CD24+/EpCAM+ expression in
clinical pancreatic cancer. Cancer Sci. 104:1127–1134. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yeung TM, Gandhi SC, Wilding JL, Muschel R
and Bodmer WF: Cancer stem cells from colorectal cancer-derived
cell lines. Proc Natl Acad Sci USA. 107:pp. 3722–3727. 2010;
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kristiansen G, Winzer KJ, Mayordomo E,
Bellach J, Schlüns K, Denkert C, Dahl E, Pilarsky C, Altevogt P,
Guski H, et al: CD24 expression is a new prognostic marker in
breast cancer. Clin Cancer Res. 9:4906–4913. 2003.PubMed/NCBI
|
|
42
|
Bretz N, Noske A, Keller S, Erbe-Hofmann
N, Schlange T, Salnikov AV, Moldenhauer G, Kristiansen G and
Altevogt P: CD24 promotes tumor cell invasion by suppressing tissue
factor pathway inhibitor-2 (TFPI-2) in a c-Src-dependent fashion.
Clin Exp Metastasis. 29:27–38. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma ZL, Chen YP, Song JL and Wang YQ:
Knockdown of CD24 inhibits proliferation, invasion and sensitizes
breast cancer MCF-7 cells to tamoxifen in vitro. Eur Rev Med
Pharmacol Sci. 19:2394–2399. 2015.PubMed/NCBI
|
|
44
|
Mylona E, Giannopoulou I, Fasomytakis E,
Nomikos A, Magkou C, Bakarakos P and Nakopoulou L: The
clinicopathologic and prognostic significance of
CD44+/CD24−/low and CD44/CD24+
tumor cells in invasive breast carcinomas. Hum Pathol.
39:1096–1102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen Y, Song J, Jiang Y, Yu C and Ma Z:
Predictive value of CD44 and CD24 for prognosis and chemotherapy
response in invasive breast ductal carcinoma. Int J Clin Exp
Pathol. 8:11287–11295. 2015.PubMed/NCBI
|
|
46
|
Shen YA, Wei YH and Chen YJ: High
CD44/CD24 expressive cells presented cancer stem cell
characteristics and undergo mitochondrial resetting and metabolic
shift in nasopharyngeal carcinoma. Cancer Res. 71 Suppl 8:Abstract
nr 4822011. View Article : Google Scholar
|
|
47
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tan X, Dagher H, Hutton CA and Bourke JE:
Effects of PPARγ ligands on TGF-β1-induced epithelial-mesenchymal
transition in alveolar epithelial cells. Respir Res. 11:21–33.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Reka AK, Kurapati H, Narala VR, Bommer G,
Chen J, Standiford TJ and Keshamouni VG: Peroxisome
proliferator-activated receptor-γ activation inhibits tumor
metastasis by antagonizing Smad3-mediated epithelial-mesenchymal
transition. Mol Cancer Ther. 9:3221–3232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Elrod HA and Sun SY: PPARgamma and
apoptosis in cancer. PPAR Res. 2008:7041652008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Burstein HJ, Demetri GD, Mueller E, Sarraf
P, Spiegelman BM and Winer EP: Use of the peroxisome
proliferator-activated receptor (PPAR) gamma ligand troglitazone as
treatment for refractory breast cancer: A phase II study. Breast
Cancer Res Treat. 79:391–397. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Girnun GD, Chen L, Silvaggi J, Drapkin R,
Chirieac LR, Padera RF, Upadhyay R, Vafai SB, Weissleder R, Mahmood
U, et al: Regression of drug-resistant lung cancer by the
combination of rosiglitazone and carboplatin. Clin Cancer Res.
14:6478–6486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bräutigam K, Biernath-Wüpping J,
Bauerschlag DO, von Kaisenberg CS, Jonat W, Maass N, Arnold N and
Meinhold-Heerlein I: Combined treatment with TRAIL and PPARγ
ligands overcomes chemoresistance of ovarian cancer cell lines. J
Cancer Res Clin Oncol. 137:875–886. 2011. View Article : Google Scholar : PubMed/NCBI
|