Introduction
Breast cancer is one of the leading causes of
cancer-related deaths in women (1).
Despite the great advance in the treatment of breast cancer, the
prognosis of these patients still remains poor to date (2,3).
Abnormal activation of the RAF/MEK/ERK signaling pathway has been
observed in breast cancer and was proposed as a candidate for
cancer therapy (4,5). Thus, a number of MEK inhibitors have
been designed to test their antiproliferative activity against
breast cancer cells (6,7). Although some studies have found that
these MEK inhibitors inhibit the growth of a variety of human
cancer cells, some trials have shown that the use of MEK inhibitors
as a treatment for breast cancer does not adequately improve
survival for unknown reasons (8–10).
Therefore, understanding the reasons for the poor outcome in such
treatments implies a better knowledge of the function of the MEK
pathway.
Previous studies have suggested that the treatment
of breast cancer cells with MEK inhibitors produced an increase in
the phosphorylation of the epidermal growth factor receptor (EGFR)
through a negative feedback loop (11,12).
Activated EGFR phosphorylates the Y593 residue of the protein known
as family with sequence similarity 129, member B (FAM129B),
resulting in an increased PKM2-dependent β-catenin transactivation
and tumor cell invasion (13).
β-catenin is an important intermediate in several signal
transduction pathways including the Wnt pathway (14–16).
In the canonical Wnt pathway, β-catenin accumulates and
translocates to the nucleus where it acts as a key transcriptional
co-activator to activate a series of genes that are associated with
cell proliferation and metastasis in cancer. In addition, EGFR
signaling also stimulates the phosphorylation of LRP6, increases
the active β-catenin level, and induces its nuclear translocation
(17). Recent studies of breast
cancer suggested that β-catenin nuclear accumulation is usually
correlated with poor outcome (18,19).
Aberrant upregulation of β-catenin was observed in various mammary
carcinoma cell lines which conferred resistance to PI3K inhibitors
(18,20). Therefore, we aimed to ascertain
whether EGFR-mediated β-catenin nuclear accumulation acts as an
alternative pathway for the poor outcome related to MEK inhibitors
in breast cancer.
In the present study, we used in vitro
approaches to investigate the effects of the MEK inhibitor PD98059
on MCF-7 and MDA-MB-231 breast cancer cells. Our results revealed
that MEK inhibitor PD98059 exhibited antiproliferative effects in a
dose- and time-dependent manner in MCF-7 and MDA-MB-231 breast
cancer cells. Conversely, incubation of MCF-7 and MDA-MB-231 cells
with PD98059 promoted their migration. Further investigation
disclosed that enhanced ability of migration by PD98059 was
dependent on β-catenin nuclear translocation in MCF-7 and
MDA-MB-231 cells. In addition, we also demonstrated that β-catenin
nuclear accumulation depended on the activation of EGFR induced by
PD98059 in the MCF-7 and MDA-MB-231 cells. Taken together, our
findings may elucidate a possible mechanism explaining the
ineffectiveness of MEK inhibitors in breast cancer treatment.
Materials and methods
Materials
PD98059 was purchased from Cell Signaling Technology
(Beverly, MA, USA). XAV-939, gefitinib and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma (St. Louis, MO, USA). RPMI-1640 medium
and fetal bovine serum (FBS) were purchased from Gibco (Grand
Island, NY, USA). The sources of primary antibodies used for
western blotting: polyclonal rabbit anti-human β-catenin (sc-7199;
1:500), monoclonal mouse anti-human p-EGFR (sc-81490; 1:200),
monoclonal mouse anti-human β-actin (sc-47778; 1:1,000) and
polyclonal rabbit anti-human lamin B1 antibodies (sc-20682; 1:500)
were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). Polyclonal rabbit anti-human phospho-MEK1 (Thr292) antibody
(#51265; 1:1,000) was purchased from Cell Signaling Technology. The
horseradish peroxidase-conjugated secondary antibodies including
goat anti-rabbit IgG (sc-2054; 1:1,000) and goat anti-mouse IgG
(sc-2973; 1:1,000) were also purchased from Santa Cruz
Biotechnology. All other chemicals used in the present study were
commercial products of reagent grade.
Cell lines
Cell lines, derived from human breast cancer (MCF-7
and MDA-MB-231) were used in the present study. All of the cell
lines were purchased from the Cell Bank of the Chinese Academy of
Science (Shanghai, China). These cells were maintained in RPMI-1640
medium containing 10% FBS, 100 U/ml penicillin, and 100 µg/ml
streptomycin, at 37°C in a humidified incubator containing 5%
CO2.
MTT assay
Cells were seeded in 96-well plates at an initial
density of 4×103 cells/well in 90 µl of medium and
allowed to grow overnight. After cells grew to 30% of the bottom of
cell culture plates, various concentrations of PD980589 (1, 5, 10,
20 and 50 µM) were added to the cells and incubation was carried
out for 24 h. Then, 50 µl of MTT (1 mg/ml) was added to each well
for 4 h of incubation at 37°C and 100 µl dimethyl sulfoxide (DMSO)
was added to solubilize the crystal products at room temperature
for 10 min subsequently. The optical density (OD) was measured at a
wavelength of 490 nm with a microplate reader (BioTek, Winooski,
VT, USA). Growth inhibition ratio was calculated as follows: Growth
inhibition ratio (%) = (ODcontrol -
ODdrug)/ODcontrol × 100. The experiments were
repeated at least three times.
Wound scratch assay
The wound scratch assay was performed as previously
described (21,22). Cells (2×104) were seeded
in a 24-well plate and cultured overnight prior to serum
starvation. After incubation, a linear wound in the cellular
monolayer was created by scratching a confluent cell monolayer. The
monolayer of the scratched cells was washed by phosphate-buffered
saline (PBS) to remove debris. After incubation for 24 h, the area
of migration was photographed under a microscope. The width of the
wound was measured and recorded as t=0. The cells were then allowed
to migrate back into the wounded area. Twenty-four hours later, the
width of the open area was measured. Cell migration was expressed
as the percentage of the gap (t=24 h) relative to the primary width
of the open area (t=0 h). All experiments were performed in
triplicate.
Cell migration assay
The migration assays were performed in a 24-well
Boyden chamber with an 8-µm pore size polycarbonate membrane
(Corning, Corning, NY, USA) as previously described (21–23).
For the migration assay, 200 µl of serum-free medium (containing
1×105 cells) was added to the upper compartment of the
chamber, while the lower compartment was filled with 600 µl of
RPMI-1640 supplemented with 10% FBS. After incubation at 37°C for
24 h, the tumor cells remaining inside the upper chamber were
removed with cotton swabs. The cells on the lower surface of the
membrane were stained with 0.1% crystal violet after fixation with
methanol, and then counted under a light microscope.
Western blotting
The total proteins were isolated from cancer cell
lines using RIPA lysis buffer. The nuclear proteins were isolated
from the cancer cell lines using Nuclear and Cytoplasmic Protein
Extraction kit (Beyotime, Shanghai, China) following the
manufacturer's instructions. The protein concentration was
determined using a BCA assay kit (Pierce, Rockford, IL, USA).
Samples were denatured in 5X SDS sample buffer at 95°C for 5 min.
Equal amounts of total proteins were separated using 10% SDS-PAGE,
and then transferred onto polyvinylidene difluoride (PVDF)
membranes. The membranes were then blocked with 5% dried skimmed
milk in Tris-buffered saline with Tween-20 (TBST) at room
temperature for 1 h. After blocking, the membranes were incubated
with corresponding primary antibodies overnight at 4°C. After being
washed three times with TBST, the membranes were incubated with the
appropriate HRP-conjugated secondary antibody, and then washed
three times with TBST. Proteins were detected using the Enhanced
Chemiluminescence (ECL) Plus reagents (Beyotime).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5 for Windows (GraphPad Software, Inc., La Jolla, CA, USA).
All data are expressed as mean ± SEM. A two-tailed unpaired t-test
was used for the comparison of the mean values between two groups.
One-way analysis of variance (ANOVA) followed by Dunnett's multiple
comparison test or two-way ANOVA followed by Bonferroni post hoc
test was used for multiple comparison. Differences with P<0.05
were considered statistically significant.
Results
Antiproliferative effects of PD98059
on MCF-7 and MDA-MB-231 cells
MCF-7 cells are estrogen receptor-positive breast
cancer cells and MDA-MB-231 is a triple-negative breast cancer.
Therefore, we selected these two cell lines with different genetic
backgrounds for our studies. PD98059, one of the first selective
MEK inhibitors, is a potent and selective inhibitor of MEK with
anticancer activity in vitro. To determine the
antiproliferative effects of PD98059 on breast cancer cells, we
first treated MCF-7 and MDA-MB-231 breast cancer cells with 1, 5,
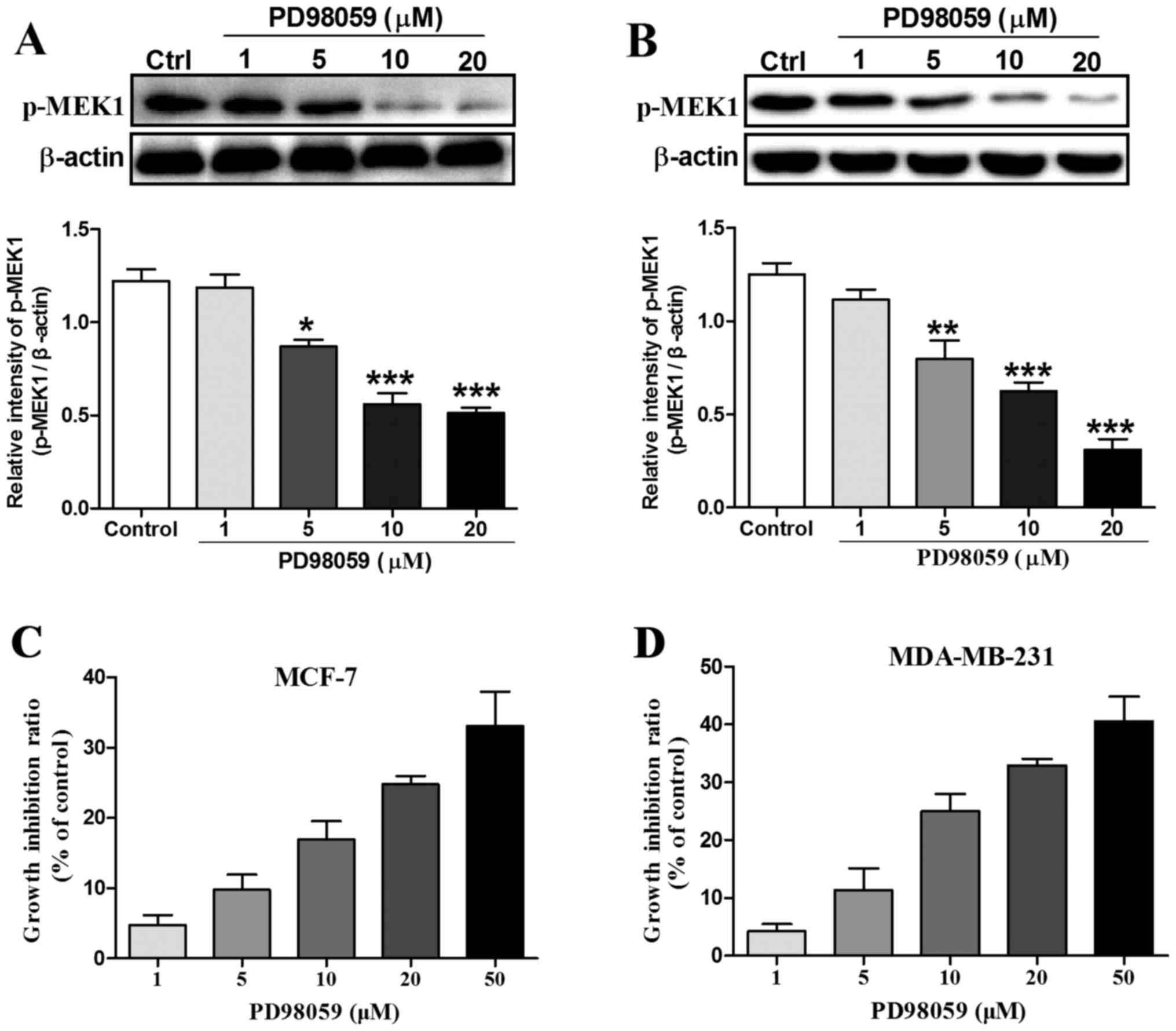
10 and 20 µM PD98059 for 24 h. As shown in Fig. 1A and B, after incubation of the
MCF-7 and MDA-MB-231 cells with 1, 5, 10 and 20 µM PD98059 for 24
h, the expression of phosphorylated MEK1 which is an indicator of
MEK1 activation was markedly decreased from 5 to 20 µM compared
with the control group. Then, we observed that PD98059 inhibited
MCF-7 and MDA-MB-231 cell proliferation in a dose-dependent manner
using MTT assay (Fig. 1C and D).
For example, the cell growth inhibition ratio was increased from
4.7% at the dose of 1 µM to 33.1% at the dose of 50 µM in MCF-7
breast cancer cells. Similarly, the cell growth inhibition ratio
was increased from 4.2% at the dose of 1 µM to 40.5% at the dose of
50 µM in the MDA-MB-231 breast cancer cells. In addition, we
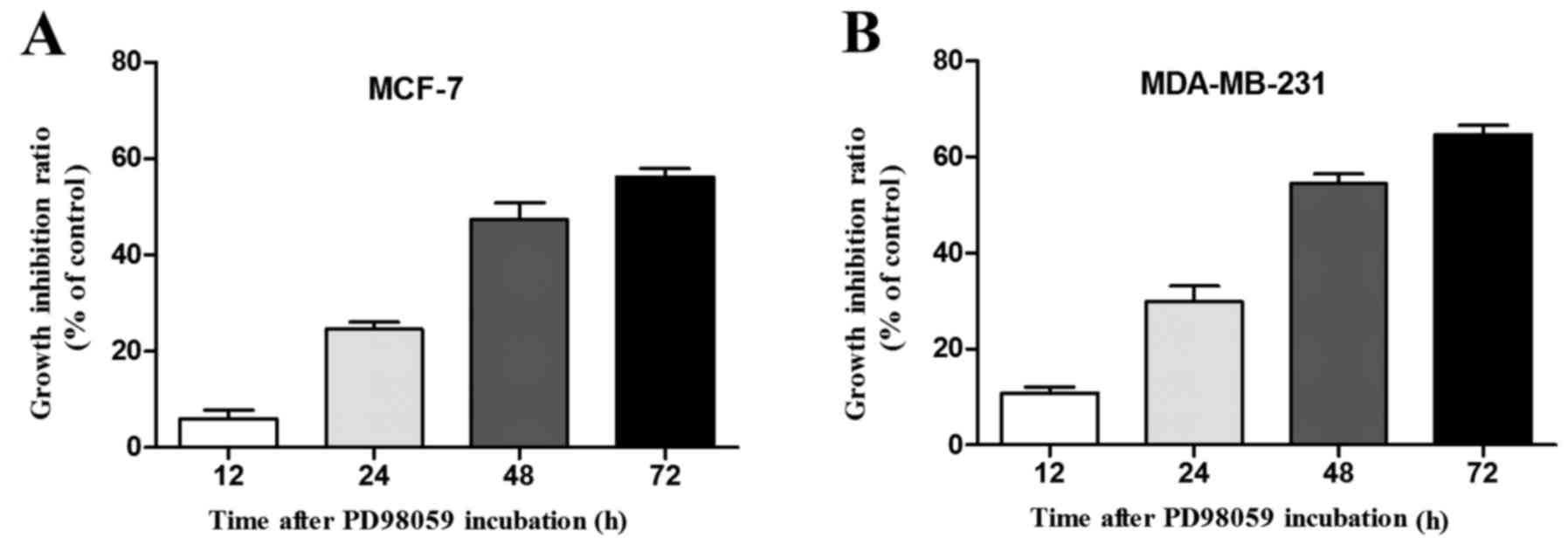
further evaluated whether PD98059 exerts antiproliferative activity
in a time-dependent manner in the breast cancer cells. As our
results showed, the growth inhibition ratio in the MCF-7 cells
incubated with 20 µM PD98059 was markedly increased from 6.1% at 12
h to 56.2% at 72 h (Fig. 2A).
Accordingly, the growth inhibition ratio in the MDA-MB-231 cells
incubated with 20 µM PD98059 was also increased from 10.7% at 12 h
to 64.6% at 72 h (Fig. 2B).
PD98059 promotes MCF-7 and MDA-MB-231
cell migration
Given that PD98059 not only influences tumor cell
proliferation but also cell motility in several types of cancer
cells, we investigated whether PD98059 regulates MCF-7 and
MDA-MB-231 cell migration, which is one of the most vital features
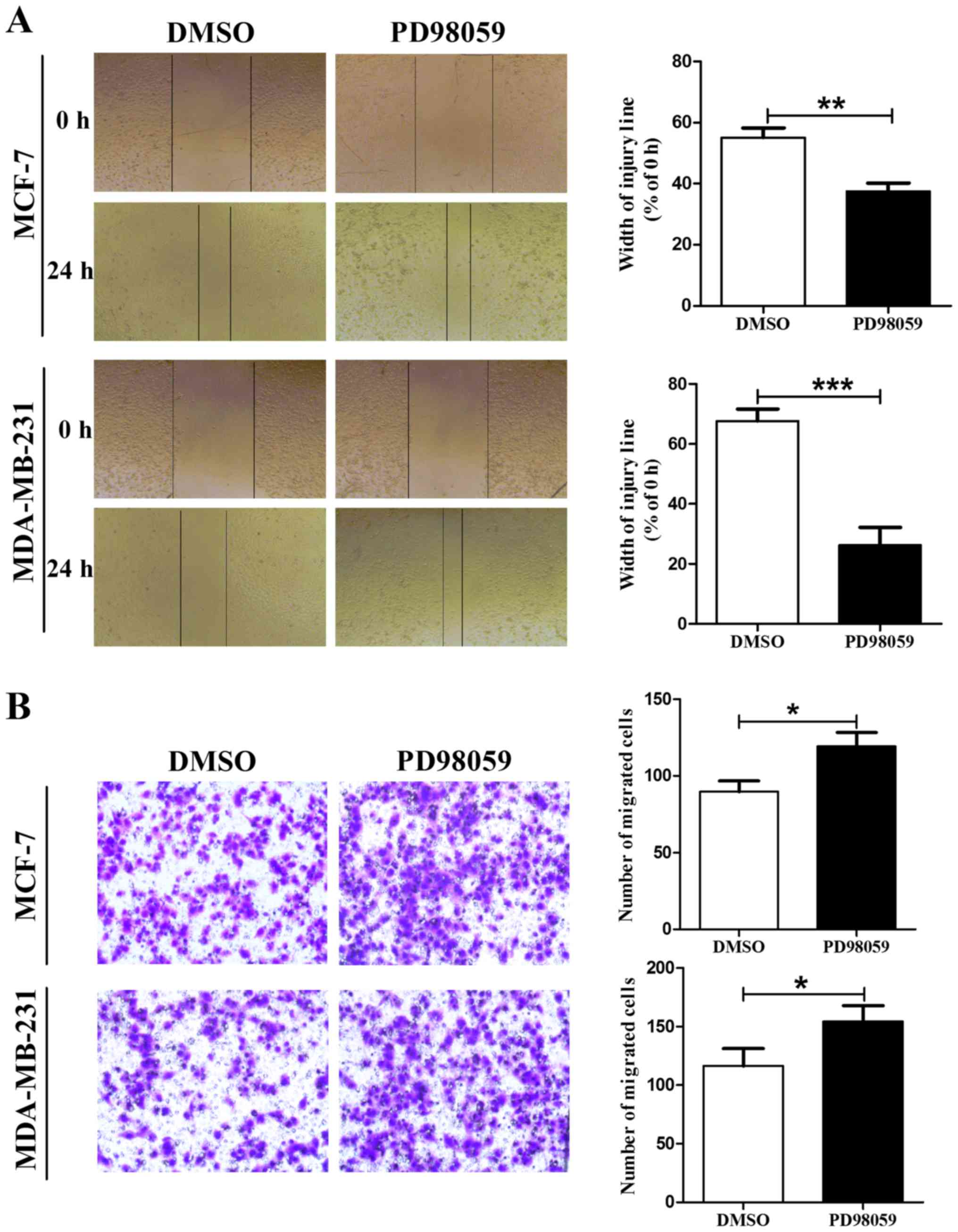
associated with malignant cell behavior. First, we examined the
role of PD98059 in MCF-7 and MDA-MB-231 cell migration using a
wound scratch assay. Since 20 µM is a commonly used dosage for
inhibiting MEK activity (24,25),
we selected this concentration for our subsequent experiments. As
shown in Fig. 3A, the cells
incubated with 20 µM PD98059 for 24 h displayed a higher ability of
migration compared with the cells treated with vehicle (DMSO). To
further identify these results, we examined the effects of PD98059
on cell migration using the Boyden chamber Transwell assay without
Martrigel. Consistent with the wound scratch assay, MCF-7 and
MDA-MB-231 cells treated with PD98059 also displayed an increased
ability of migration compared with the control group cells
(Fig. 3B). These results indicated
that PD98059 promoted cell migration in the MCF-7 and MDA-MB-231
cells.
β-catenin expression in the MCF-7 and
MDA-MB-231 cells is increased after treatment with PD98059
Given that β-catenin is a key mediator for cell
proliferation, migration and differentiation, we aimed to ascertain
whether β-catenin is involved in the regulation of cell
proliferation and migration in MCF-7 and MDA-MB-231 cells by
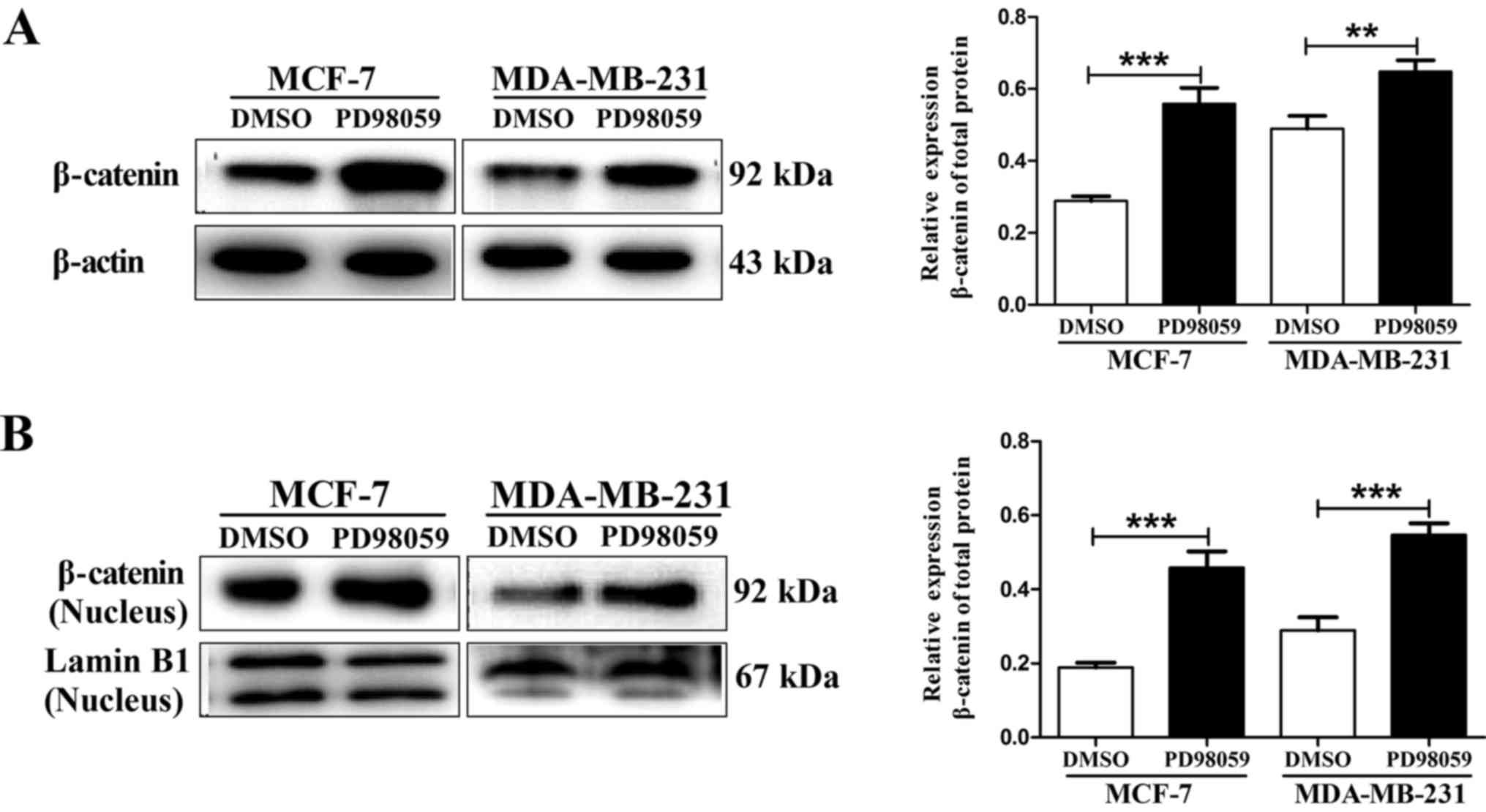
PD98059. Therefore, we first examined the total protein expression
of β-catenin using western blot assay. As shown in Fig. 4A, the expression of β-catenin total
protein in the MCF-7 and MDA-MB-231 cells was statistically
increased at 24 h after incubation with 20 µM PD98059 as compared
to β-catenin expression following incubation with vehicle.
Considering that β-catenin nuclear accumulation often induces
efficient metastasis formation by enhancing metastasis-related gene
transcription, we analyzed nuclear β-catenin levels as the most
direct way to assess the effects of PD98059 on β-catenin
transcriptional activity. Similarly, we also found that PD98059
significantly increased β-catenin nuclear accumulation in the MCF-7
and MDA-MB-231 cells (Fig. 4B).
Inhibition of β-catenin nuclear
accumulation reverses PD98059-induced MCF-7 and MDA-MB-231 cell
migration
In order to determine whether β-catenin nuclear
accumulation contributes to PD98059-mediated enhanced ability of
cell migration, the AXIN stabilizer XAV939 was used in the
following study. As a stabilizer of AXIN, XAV939 promotes the
degradation of β-catenin, thus leading to decreased β-catenin
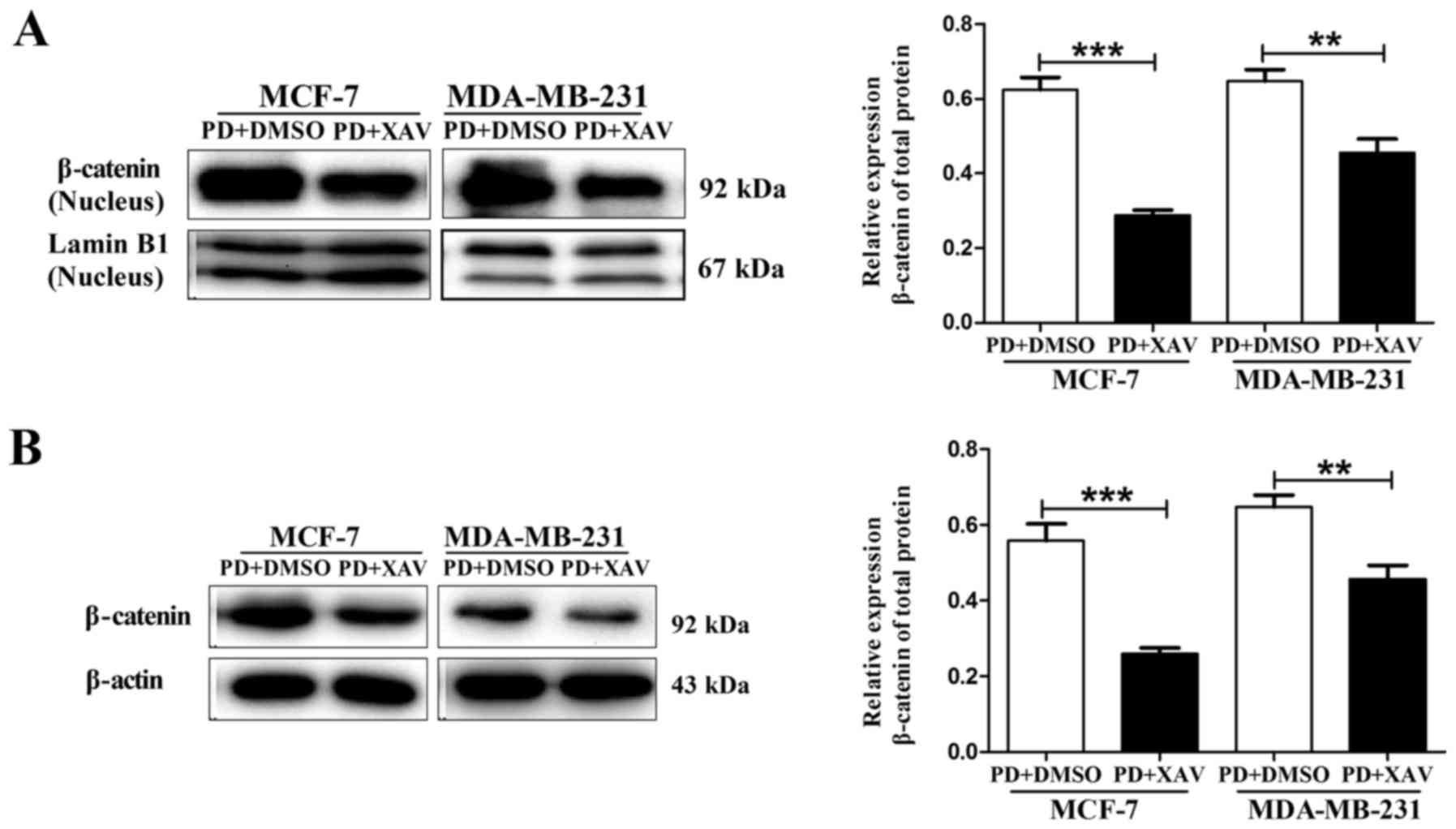
nuclear translocation (26,27). Our results showed that when MCF-7
and MDA-MB-231 cells were pre-treated with 5 µM XAV-939 for 30 min
followed by co-treatment with PD98059 (20 µM) for 24 h, the
PD98059-induced increase in β-catenin nuclear accumulation was
markedly blocked by XAV-939 (Fig.
5A). Similarly, PD98059-induced increase in β-catenin total
protein expression was also blocked by XAV-939 in the MCF-7 and
MDA-MB-231 cells (Fig. 5B). Then,
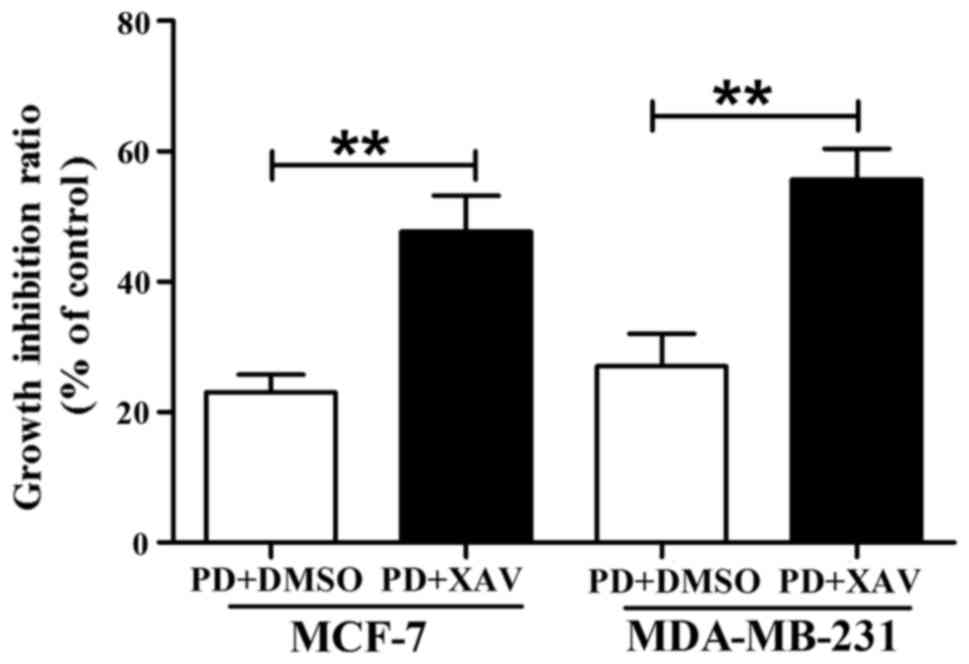
we examined whether XAV-939 reverses PD98059-mediated changes in
cancer cell behaviors. As shown in Fig.
6, XAV-939 did not reverse PD98059-mediated ability to decrease
MCF-7 and MDA-MB-231 cell growth, but produced a cooperative
inhibitory effect with PD98059 on cell proliferation. Finally, we
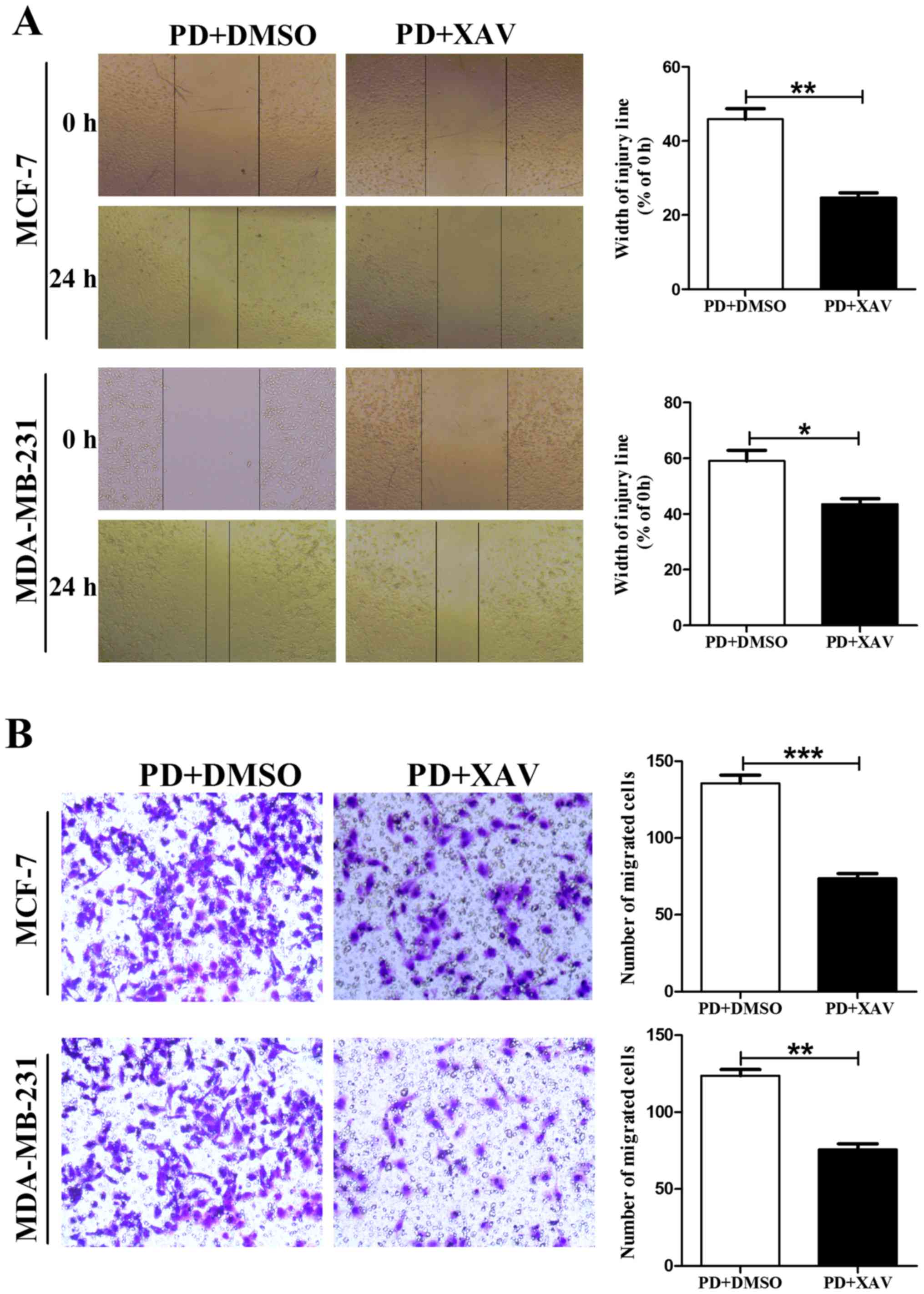
assessed the effects of XAV939 on cell migration using a wound
scratch assay and Boyden chamber Transwell assay without Martrigel.
Our results showed that XAV-939 reversed PD98059-induced MCF-7 and
MDA-MB-231 cell migration (Fig.
7).
β-catenin nuclear accumulation induced
by PD98059 depends on the activation of EGFR in breast cancer
cells
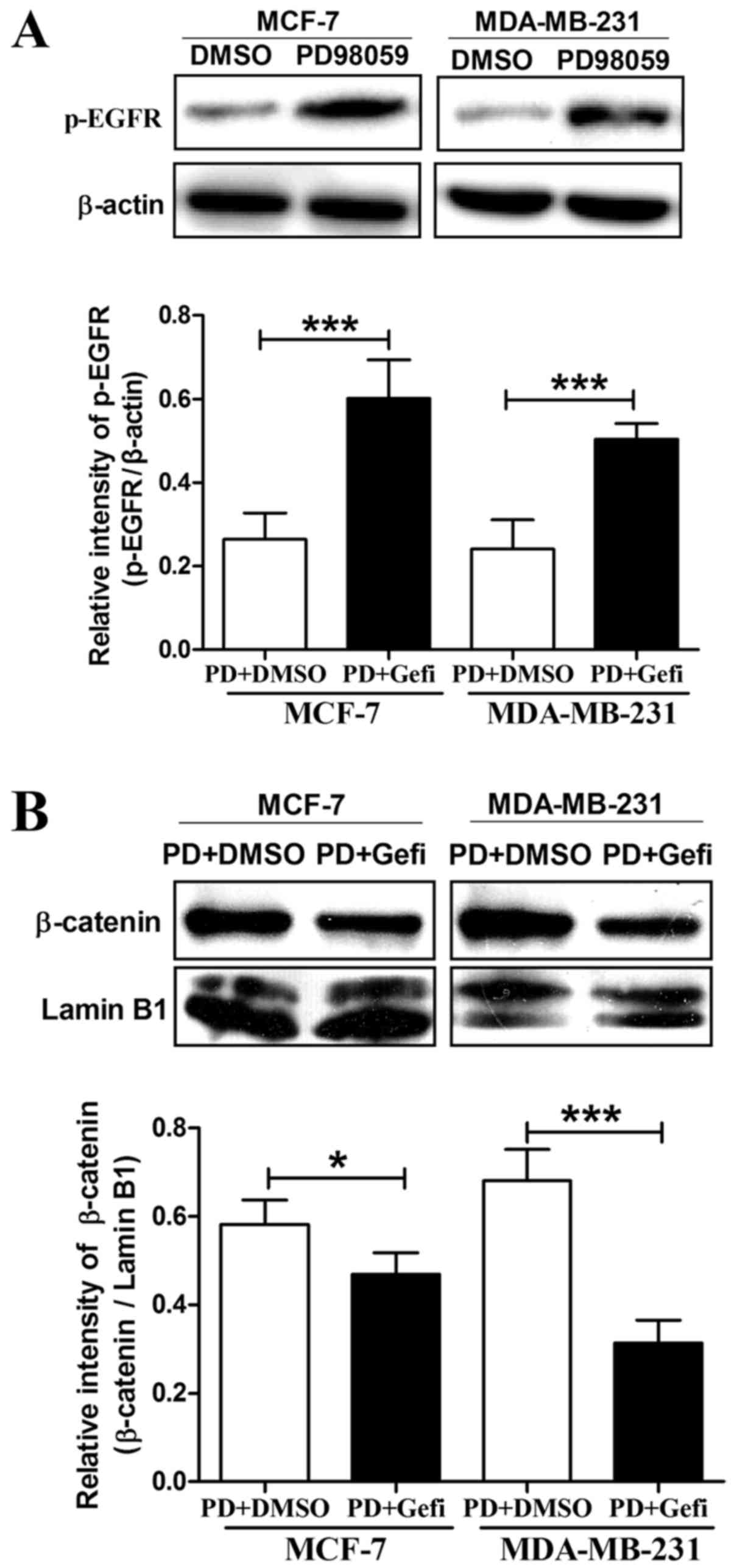
MEK inhibition is able to induce EGFR activation in
breast cancer cells (12), and
nuclear translocation of β-catenin sometimes depends on the
activation of EGFR (28,29). Therefore, we investigated whether
activation of EGFR can be induced by MEK inhibitor PD98059 in the
MCF-7 and MDA-MB-231 cells. In agreement with previous findings, we
also observed an increase in the phosphorylation of EGFR following
treatment with 20 µM PD98059 for 24 h in the MCF-7 and MDA-MB-231
cells (Fig. 8A). To elucidate the
underlying interaction between EGFR phosphorylation and β-catenin
nuclear accumulation, we then used PD98059 combined with the EGFR
inhibitor gefitinib to treat MCF-7 and MDA-MB-231 cells. As shown
in Fig. 8B, EGFR kinase inhibitor
gefitinib was able to abolish the PD98059-induced β-catenin nuclear
translocation. Hence, we conclude that β-catenin nuclear
accumulation induced by PD98059 depends on the activation of EGFR
in breast cancer cells.
Discussion
The Ras/MEK/ERK pathway plays a central role in
cancer cell biology, including proliferation, survival, migration,
invasion and angiogenesis (5,6).
Therefore, MEK has been proposed as a suitable target for
therapeutic intervention in cancer. To our disappointment, numerous
studies have found that these inhibitors may showed variable
effects on cell growth depending on tumor type (6,7,30). For
example, MEK inhibitors showed growth inhibition effects on various
types of neuroblastoma, colon cancer and hepatocellular carcinoma
cells via inducing apoptosis or G1 phase arrest (30–32),
while some breast cancer cell lines were found to be resistant to
MEK inhibitors in pre-clinical studies and early clinical trials
(10). In the present study, we
observed that PD98059 suppressed cell growth in a dose-dependent
manner in breast cancer MCF-7 and MDA-MB-231 cells. This is
consistent with previous studies by Zhou et al and Ye et
al. In their studies, Zhou et al found that the MEK
inhibitor suppressed cell growth via induction of apoptosis and G1
phase arrest in the breast cancer MDA-MB-231 and HCC1937 cell lines
(33). Ye et al observed
that inhibition of MEK/ERK with the pharmacological inhibitor
PD98059 resulted in a significant enhancement of growth inhibition
in breast cancer MCF-7 cells (34).
In the present study, the expression of β-catenin
was increased in the MCF-7 and MDA-MB-231 cells after treatment
with MEK inhibitor PD98059. It is well known that β-catenin usually
acts as a key transcriptional factor and widely participates in
promoting cancer cell proliferation and migration (35,36).
Why β-catenin nuclear accumulation induced by PD98059 was
associated with decreased cell growth in the present study is
unclear. We hypothesize that other proliferation-related signaling
pathways were inhibited by the MEK inhibitor. For example, the
Raf/MEK/ERK pathway can control cell survival and proliferation by
induction of cell cycle regulatory proteins such as BCL-2, CDKs and
cyclins. Conversely, MEK inhibition causes reduction in this
signaling in cancer cells concomitant with apoptosis (37,38).
Although β-catenin nuclear accumulation occurred after MEK
inhibition in the MCF-7 and MDA-MB-231 cells, it could not overcome
the anti-proliferative signaling pathway which was also induced by
the MEK inhibitor. Finally, PD98059 showed antiproliferative
effects in the MCF-7 and MDA-MB-231 cells in this case. Of course,
further studies are needed to ascertain whether our assumption is
correct.
β-catenin nuclear accumulation often induces
efficient metastasis formation by enhancing metastasis-related gene
transcription including matrix metalloproteinases (MMPs) (39–41).
In the present study, we also demonstrated that β-catenin nuclear
translocation was vital to cell migration in breast cancer MCF-7
and MDA-MB-231 cells, since the MEK inhibitor PD98059 promoted
β-catenin into the nucleus, and inhibition of β-catenin nuclear
accumulation with XAV-939 markedly reversed the cell migration
ability induced by PD98059. Subsequent experiments demonstrated
that β-catenin nuclear entry relied on the activation of EGFR in
MCF-7 and MDA-MB-231 cells which is in line with previous studies.
The epidermal growth factor receptor (EGFR) is a tyrosine kinase
receptor that participates in the regulation of cell proliferation
and migration. It has been shown that the activation of EGFR
signaling stimulates β-catenin nuclear translocation and
contributes to the acquisition of a motile phenotype by
upregulating the expression of MMPs (13,42).
Although MCF-7 and MDA-MB-231 are cell lines with two totally
different genetic backgrounds, EGFR is expressed in these cells
(43,44). Therefore, it is not surprising that
β-catenin nuclear translocation induced by PD98059 depends on the
activation of EGFR in these breast cancer cell lines.
Contrary to our results in breast cancer cells,
previous studies suggest that inhibition of ERK activity in lung
cancer activates glycogen synthase kinase 3β (GSK3β) potentially
leading to β-catenin degradation, which in turn inhibits cell
growth and metastasis (45,46). We hypothesized that these
conflicting results may be due to the different tumor types.
However, the mechanisms of EGFR overexpression induced by MEK
inhibition in breast cancer cells remain unresolved. Further
studies are needed to demonstrate this novel pathway and its role
in breast cancer progression in vivo. In addition, our
results also demonstrated that inhibition of β-catenin nuclear
translocation with XAV-939 inhibited the expression of β-catenin
total protein. This may occur as β-catenin is retained in the
cytoplasm where it is recognized by a destruction complex
containing GSK3β, casein kinase 1 (CK1), axin and adenomatous
polyposis coli (APC), thus leading to ubiquitin proteasome-mediated
degradation of β-catenin (45,46).
Taken together, our results demonstrated that MEK
inhibitor PD98059 promoted cell migration in MCF-7 and MDA-MB-231
cells by promoting β-catenin nuclear translocation. These results
may elucidate a possible mechanism explaining the ineffectiveness
of MEK inhibitors in breast cancer treatment.
Acknowledgements
The present study was supported by the National
Science Foundation of Henan (162300410039) and the Program for
Science and Technology of the Department of Education of Henan
Province (16A350013).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Castillo LF, Tascón R, Huvelle MR Lago,
Novack G, Llorens MC, Dos Santos AF, Shortrede J, Cabanillas AM,
Bal de Kier Joffé E, Labriola L, et al: Glypican-3 induces a
mesenchymal to epithelial transition in human breast cancer cells.
Oncotarget. 7:60133–60154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lorusso G and Rüegg C: New insights into
the mechanisms of organ-specific breast cancer metastasis. Semin
Cancer Biol. 22:226–233. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li T, Zhang C, Ding Y, Zhai W, Liu K, Bu
F, Tu T, Sun L, Zhu W, Zhou F, et al: Umbilical cord-derived
mesenchymal stem cells promote proliferation and migration in MCF-7
and MDA-MB-231 breast cancer cells through activation of the ERK
pathway. Oncol Rep. 34:1469–1477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neuzillet C, Tijeras-Raballand A, de
Mestier L, Cros J, Faivre S and Raymond E: MEK in cancer and cancer
therapy. Pharmacol Ther. 141:160–171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heigener DF, Gandara DR and Reck M:
Targeting of MEK in lung cancer therapeutics. Lancet Respir Med.
3:319–327. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Templeton IE and Musib L: MEK inhibitors
beyond monotherapy: Current and future development. Curr Opin
Pharmacol. 23:61–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Britten CD: PI3K and MEK inhibitor
combinations: Examining the evidence in selected tumor types.
Cancer Chemother Pharmacol. 71:1395–1409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gu Y, Helenius M, Väänänen K, Bulanova D,
Saarela J, Sokolenko A, Martens J, Imyanitov E and Kuznetsov S:
BRCA1-deficient breast cancer cell lines are resistant to MEK
inhibitors and show distinct sensitivities to 6-thioguanine. Sci
Rep. 6:282172016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mirzoeva OK, Das D, Heiser LM,
Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM,
Guan Y, et al: Basal subtype and MAPK/ERK kinase
(MEK)-phosphoinositide 3-kinase feedback signaling determine
susceptibility of breast cancer cells to MEK inhibition. Cancer
Res. 69:565–572. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maiello MR, D'Alessio A, Bevilacqua S,
Gallo M, Normanno N and De Luca A: EGFR and MEK blockade in triple
negative breast cancer cells. J Cell Biochem. 116:2778–2785. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ji H, Lee JH, Wang Y, Pang Y, Zhang T, Xia
Y, Zhong L, Lyu J and Lu Z: EGFR phosphorylates FAM129B to promote
Ras activation. Proc Natl Acad Sci USA. 113:pp. 644–649. 2016;
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jung YS, Jun S, Lee SH, Sharma A and Park
JI: Wnt2 complements Wnt/β-catenin signaling in colorectal cancer.
Oncotarget. 6:37257–37268. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sano M, Driscoll DR, DeJesus-Monge WE,
Quattrochi B, Appleman VA, Ou J, Zhu LJ, Yoshida N, Yamazaki S,
Takayama T, et al: Activation of WNT/β-catenin signaling enhances
pancreatic cancer development and the malignant potential via
up-regulation of Cyr61. Neoplasia. 18:785–794. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wickström M, Dyberg C, Milosevic J, Einvik
C, Calero R, Sveinbjörnsson B, Sandén E, Darabi A, Siesjö P, Kool
M, et al: Wnt/β-catenin pathway regulates MGMT gene expression in
cancer and inhibition of Wnt signalling prevents chemoresistance.
Nat Commun. 6:89042015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Zhu J, Li Y, Lin T, Siclari VA,
Chandra A, Candela EM, Koyama E, Enomoto-Iwamoto M and Qin L:
Epidermal growth factor receptor (EGFR) signaling regulates
epiphyseal cartilage development through β-catenin-dependent and
-independent pathways. J Biol Chem. 288:32229–32240. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tzeng HE, Yang L, Chen K, Wang Y, Liu YR,
Pan SL, Gaur S, Hu S and Yen Y: The pan-PI3K inhibitor GDC-0941
activates canonical WNT signaling to confer resistance in TNBC
cells: Resistance reversal with WNT inhibitor. Oncotarget.
6:11061–11073. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De P, Carlson JH, Wu H, Marcus A,
Leyland-Jones B and Dey N: Wnt-beta-catenin pathway signals
metastasis-associated tumor cell phenotypes in triple negative
breast cancers. Oncotarget. 7:43124–43149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Timmermans-Sprang EP, Gracanin A and Mol
JA: High basal Wnt signaling is further induced by PI3K/mTor
inhibition but sensitive to cSRC inhibition in mammary carcinoma
cell lines with HER2/3 overexpression. BMC Cancer. 15:5452015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen L, Kang QH, Chen Y, Zhang YH, Li Q,
Xie SQ and Wang CJ: Distinct roles of Akt1 in regulating
proliferation, migration and invasion in HepG2 and HCT 116 cells.
Oncol Rep. 31:737–744. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng XH, Nie X, Liu HY, Fang YM, Zhao Y
and Xia LX: TMPyP4 promotes cancer cell migration at low doses, but
induces cell death at high doses. Sci Rep. 6:265922016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liao L, Song M, Li X, Tang L, Zhang T,
Zhang L, Pan Y, Chouchane L and Ma X: E3 ubiquitin ligase UBR5
drives the growth and metastasis of triple-negative breast cancer.
Cancer Res. 77:2090–2101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Toulany M, Iida M, Keinath S, Iyi FF,
Mueck K, Fehrenbacher B, Mansour WY, Schaller M, Wheeler DL and
Rodemann HP: Dual targeting of PI3K and MEK enhances the radiation
response of K-RAS mutated non-small cell lung cancer. Oncotarget.
7:43746–43761. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ren W, Liu Y, Wan S, Fei C, Wang W, Chen
Y, Zhang Z, Wang T, Wang J, Zhou L, et al: BMP9 inhibits
proliferation and metastasis of HER2-positive SK-BR-3 breast cancer
cells through ERK1/2 and PI3K/AKT pathways. PLoS One. 9:e968162014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma L, Wang X, Jia T, Wei W, Chua MS and So
S: Tankyrase inhibitors attenuate WNT/β-catenin signaling and
inhibit growth of hepatocellular carcinoma cells. Oncotarget.
6:25390–25401. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang SM, Mishina YM, Liu S, Cheung A,
Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner
S, et al: Tankyrase inhibition stabilizes axin and antagonizes Wnt
signalling. Nature. 461:614–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee CH, Hung HW, Hung PH and Shieh YS:
Epidermal growth factor receptor regulates beta-catenin location,
stability, and transcriptional activity in oral cancer. Mol Cancer.
9:642010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanaka R, Tomosugi M, Sakai T and Sowa Y:
MEK inhibitor suppresses expression of the miR-17-92 cluster with
G1 phase arrest in HT-29 human colon cancer cells and
MIA PaCa-2 pancreatic cancer cells. Anticancer Res. 36:4537–4543.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tanaka T, Higashi M, Kimura K, Wakao J,
Fumino S, Iehara T, Hosoi H, Sakai T and Tajiri T: MEK inhibitors
as a novel therapy for neuroblastoma: Their in vitro effects and
predicting their efficacy. J Pediatr Surg. 51:2074–2079. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Nie H, Zhao X, Qin Y and Gong X:
Bicyclol induces cell cycle arrest and autophagy in HepG2 human
hepatocellular carcinoma cells through the PI3K/AKT and
Ras/Raf/MEK/ERK pathways. BMC Cancer. 16:7422016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou Y, Hu HY, Meng W, Jiang L, Zhang X,
Sha JJ, Lu Z and Yao Y: MEK inhibitor effective against
proliferation in breast cancer cell. Tumour Biol. 35:9269–9279.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ye J, Li A, Liu Q, Wang X and Zhou J:
Inhibition of mitogen-activated protein kinase kinase enhances
apoptosis induced by arsenic trioxide in human breast cancer MCF-7
cells. Clin Exp Pharmacol Physiol. 32:1042–1048. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tan Z, Zheng H, Liu X, Zhang W, Zhu J, Wu
G, Cao L, Song J, Wu S, Song L, et al: MicroRNA-1229 overexpression
promotes cell proliferation and tumorigenicity and activates
Wnt/β-catenin signaling in breast cancer. Oncotarget.
7:24076–24087. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang Q, He M, Guan S, Ma M, Wu H, Yu Z,
Jiang L, Wang Y, Zong X, Jin F, et al: MicroRNA-100 suppresses the
migration and invasion of breast cancer cells by targeting FZD-8
and inhibiting Wnt/β-catenin signaling pathway. Tumour Biol.
37:5001–5011. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Galante JM, Mortenson MM, Bowles TL,
Virudachalam S and Bold RJ: ERK/BCL-2 pathway in the resistance of
pancreatic cancer to anoikis. J Surg Res. 152:18–25. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kobayashi M, Funayama R, Ohnuma S, Unno M
and Nakayama K: Wnt-β-catenin signaling regulates ABCC3 (MRP3)
transporter expression in colorectal cancer. Cancer Sci.
107:1776–1784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin D, Kuang G, Wan J, Zhang X and Li H,
Gong X and Li H: Luteolin suppresses the metastasis of
triple-negative breast cancer by reversing
epithelial-to-mesenchymal transition via downregulation of
β-catenin expression. Oncol Rep. 37:895–902. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu JJ, Rouse C, Xu X, Wang J, Onaitis MW
and Pendergast AM: Inactivation of ABL kinases suppresses non-small
cell lung cancer metastasis. JCI Insight. 1:e896472016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Paul I, Bhattacharya S, Chatterjee A and
Ghosh MK: Current understanding on EGFR and Wnt/β-catenin signaling
in glioma and their possible crosstalk. Genes Cancer. 4:427–446.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng X, Hu B, Hossain MM, Chen G, Sun Y
and Zhang X: ADAM17-siRNA inhibits MCF-7 breast cancer through
EGFR-PI3K-AKT activation. Int J Oncol. 49:682–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li RH, Huang WH, Wu JD, Du CW and Zhang
GJ: EGFR expression is associated with cytoplasmic staining of
CXCR4 and predicts poor prognosis in triple-negative breast
carcinomas. Oncol Lett. 13:695–703. 2017.PubMed/NCBI
|
|
45
|
Gao C, Chen G, Romero G, Moschos S, Xu X
and Hu J: Induction of Gsk3β-β-TrCP interaction is required for
late phase stabilization of β-catenin in canonical Wnt signaling. J
Biol Chem. 289:7099–7108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gong F, Wang G, Ye J, Li T, Bai H and Wang
W: 14-3-3β regulates the proliferation of glioma cells through the
GSK3β/β-catenin signaling pathway. Oncol Rep. 30:2976–2982. 2013.
View Article : Google Scholar : PubMed/NCBI
|