Introduction
Pheochromocytoma (PHEO) is a rare neuroendocrine
tumor derived from the adrenal medulla and extra-adrenal neural
crest chromaffin tissues, with an incidence of 2–8
cases/million/year. However, a higher than expected incidence has
been reported (1). Theoretically,
radical resection remains the sole treatment option. Despite the
relatively satisfactory outcome of an 84–96% 5-year survival rate
in patients with benign masses, the <50% survival rate (2–4) and up
to 65.4% recurrence rate within 5 years (5) are the overwhelming statistics for
patients with malignant PHEO. Apart from the gold standard of
finding ectopic chromaffin tissue, there are no early diagnostic
markers for PHEO and treatment is not optimal for patients with
metastatic disease. In addition, alternative treatments are of
little benefit to patients with metastatic disease. Therefore,
there is an urgent need for new and effective therapeutic agents
for PHEO patients, especially those with malignant tumors.
Cancer is generally considered a genomic disease
that progresses by accumulating gene mutations leading to oncogene
activation and/or anti-oncogene inactivation. TP53, as a
common tumor-suppressor gene, is mutated in more than half of human
cancers (6). Through the regulation
of various target genes, the biological functions of p53 include
DNA repair, cell cycle arrest and induction of apoptosis or
senescence (7–9). Thus, inactivating TP53
mutations could result in tumorigenesis. By coincidence, numerous
studies have revealed TP53 mutations in PHEO (10,11).
However, whether the ‘artificial’ upregulation of TP53 has
anti-PHEO effects remains completely unknown.
In the present study, dsP53-285, a small
double-stranded RNA (dsRNA) that targets the TP53 promoter
was employed to induce wild-type TP53 expression in the PHEO
cell line PC12. This phenomenon, termed RNA activation (RNAa), is
conserved in mammalian species. The enhanced p53 expression caused
cell cycle arrest and induced apoptosis in association with altered
expression of the corresponding proteins and mRNAs, including
cyclin D1, CDK4/6 and caspase 9 and 3. Finally, our results were
further demonstrated using in vivo models.
Materials and methods
Cell line and main reagents
PHEO cell line (PC12) was originally donated by the
Institute of Neurology in Ruijin Hospital Affiliated to the Medical
School of Shanghai Jiaotong University. dsP53-285 was purchased
from RiboBio (Guangzhou, China). The primary antibodies (p53, p21,
cyclin D1, CDK4/6 and caspase 3 and 9) were purchased from Cell
Signaling Technology (Danvers, MA, USA). The 5-pair-primers were
designed by BioTNT (Shanghai, China). The Hoechst 33258 Staining
kit was obtained from the Beyotime Institute of Biotechnology
(Shanghai, China).
Cell culture and transfection
PC12 cells were cultured in 60 cm2 dishes
in an incubator at 37°C, with 5% CO2 and 90% humidity.
The culture medium for PC12 cells consisted of 5% fetal bovine
serum (FBS; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 10%
horse serum (Thermo Fisher Scientific) and 1%
penicillin-streptomycin. PC12 cells were subcultured or the culture
medium was replaced every three days.
The day before transfection, the cells were treated
with trypsin and seeded into new 6-well plates at a density of
50–60% and then cultured without antibiotics. All dsRNAs were
transfected at a final concentration of 50 nmol/l with
Lipofectamine RNAiMax (Thermo Fisher Scientific) according to the
manufacturer's instructions. Additionally, dsRNA was replaced with
Opti-MEM (Thermo Fisher Scientific), a medium in which liposomes
are combined with dsRNA, for mock transfection.
Protein extraction and western blot
analysis
Cells transfected with or without dsP53-285 were
harvested, washed twice with phosphate-buffered saline (PBS), and
then lysed in ice-cold lysis buffer containing 1%
phenylmethylsulfonyl fluoride (PMSF) and 10% cocktail for 30 min at
4°C. Afterwards cell lysates were centrifuged at 12,000 rpm for 30
min and then loading buffer was added before storage at −20°C. The
proteins were separated by SDS-PAGE (8% gels) and transferred to
polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA,
USA). BSA (5%) was used to block non-specific binding for 1 h at
normal atmospheric temperature. Membranes were subsequently
incubated overnight at 4°C with primary antibodies (1:1,000) and
β-actin (1:2,000). Thereafter, the membranes were washed three
times with PBS-T and then incubated with corresponding secondary
antibodies at room temperature for 40 min. Following washing three
times, the protein bands were detected using the enhanced
chemiluminescence method with Image Lab software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) with automatic or manual
exposure.
RNA extraction and real-time
quantitative polymerase chain reaction (qPCR)
TRIzol reagent (Thermo Fisher Scientific) was used
to extract RNA from the cells. RNA (500 ng) was reverse transcribed
using a reverse transcription kit (Takara Biotechnology, Dalian,
China) according to the manufacturer's instructions. Quantitative
PCR was performed on a 7500 Real-Time PCR system (Applied
Biosystems, Foster City, CA, USA) with SYBR Premix Ex Taq II
(Takara Biotechnology). The program was set for an initial
denaturation step of 5 min at 95°C, followed by 40 cycles at 95°C
for 15 sec, 60°C for 30 sec, and 72°C for 30 sec, with a final
extension at 72°C for 5 min. Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as an internal control. Relative
expression of the targeted genes was calculated using the
2−ΔΔCt method.
Cell proliferation assay
At 24 h after transfection, the cells at a density
of 3×103/well, were reseeded and incubated in a 96-well
plate in 200 µl culture medium. After 24, 48 and 72 h Cell Counting
kit 8 (CCK8; YeaSen, Shanghai, China) was added to each well
according to the manufacturer's protocol, and incubated for 0.5–4 h
before absorbance measurements were performed at 450 nm with Gen5
software (BioTek, Winooski, VT, USA). The percentage of viable
cells at every time-point was calculated in relation to the mock
group. The formula for the relative cell number is: relative number
= absorbance of every group/absorbance of mock group.
Colony formation assay
At 24 h after transfection, the cells at a density
of 1×103/well were reseeded into sterile 6-well plates
with complete media for 10 days. Each medium was replaced every 3
days to maintain cell growth. The colonies were fixed with methanol
for 15 min and then stained with crystal violet (Sigma-Aldrich, St.
Louis, MO, USA) for 15 min at room temperature. Following washing
three times with PBS, the cells were imaged with a digital
camera.
Cell cycle analysis
The cells were treated with trypsin and then
resuspended in 300 µl of PBS. Subsequently, the cells were fixed
with 700 µl absolute ethyl alcohol overnight at 4°C. Cellular DNA
was stained with propidium iodide (PI; 0.05 mg/ml) and analyzed
using flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Apoptosis assay
Cellular apoptosis was observed using the Hoechst
33258 nuclear staining kit. Briefly, after transfection, the cells
were stained with Hoechst 33258 for 15 min and then washed twice
with PBS before images were captured with a fluorescence microscope
(Olympus, Tokyo, Japan).
Apoptosis was also objectively quantified by flow
cytometry through double staining with Annexin V and PI (BD
Biosciences). Briefly, at the end of every time-point, the cells
were harvested and centrifuged at 1,000 rpm for 5 min and
resuspended with binding buffer before staining with Annexin V-FITC
for 10 min and PI for 5 min before flow cytometric analysis to
detect apoptosis.
Mouse xenograft model
Equivalent amounts of PC12 cells
(~20×106, 200 µl) infected with Lenti-dsP53-285 or
Lenti-dsControl (Bio-Link, Shanghai, China) were injected
subcutaneously into the left hind flanks of female athymic-nude
4-week-old mice (Shanghai Institute of Materia Medica, Chinese
Academy of Sciences). After one week of macroscopic tumor
formation, the tumor length and width were assessed every 3 days
for 21 days using calipers. Tumor volume (V) was calculated using
the formula: V = length × width2 × 0.5236. On the 28th
day after injection, the mice were sacrificed and the tumors
excised, weighed and imaged with a digital camera.
The Research Medical Ethics Committee of Ruijin
Hospital Affiliated to the Medical School of Shanghai Jiaotong
University, approved the present study. All applicable
international, national and/or institutional guidelines for the
care and use of animals were followed.
Immunohistochemistry (IHC)
After fixing in 10% neutral formalin and dehydrating
in concentration gradient alcohol, samples were embedded in
paraffin, and then cut into 3-µm serial sections, which were
dewaxed twice in xylene, rehydrated in alcohol and then rinsed with
PBS, followed by treatment with 3% H2O2 for
10 min to inactivate endogenous peroxidase. After antigen
retrieval, the slides were incubated with 10% goat serum at a
constant temperature to block non-specific reactions for 10 min.
Subsequently, the sections were treated with p53 antibody (1:100
dilution) for 12 h at 4°C. After being washed with PBS three times,
the slides were incubated with secondary antibody at 37°C for 30
min. Subsequently, they were washed again with PBS and developed in
diaminobenzidine (DAB) substrate. The slides were counterstained in
hematoxylin and dehydrated with ethanol and xylene before being
mounted. Yellowish-brown granules at a percentage of 10% or more
located in the nucleus of p53 was considered as positive.
Statistical analysis
All data are presented as the means ± standard
deviation. Statistical analyses were performed with SPSS 14.0
software (IBM SPSS, Armonk, NY, USA) and the corresponding bar
graph or line chart were drawn using GraphPad Prism 7 software.
Differences in measurement and enumeration data were compared using
Student's t-test. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
dsP53-285 activates wild-type TP53
expression by targeting its promoter
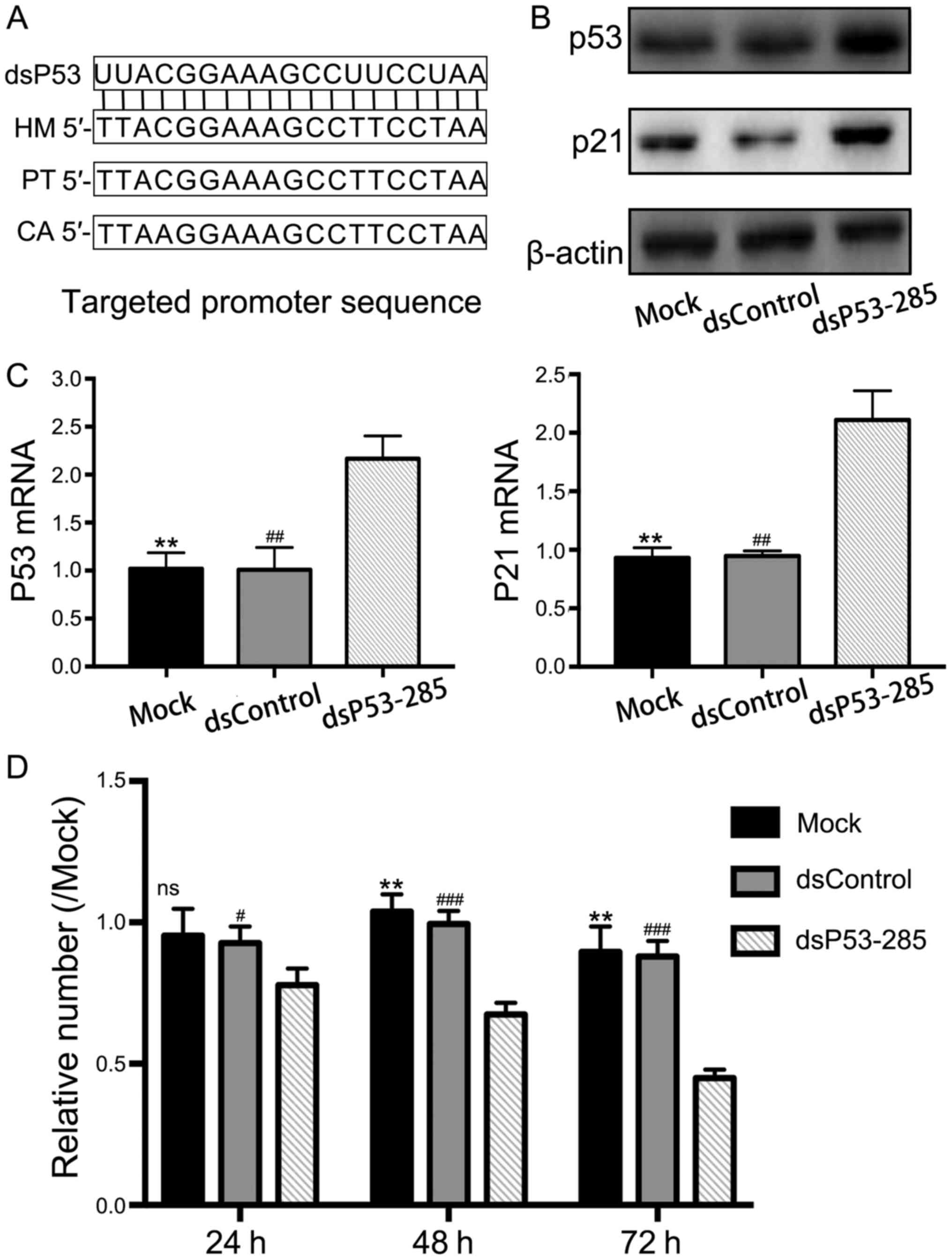
A previous study has revealed that dsP53-285 can
activate p53 expression by targeting its promoter sequence at the
−285/−267 site (Fig. 1A) in the
chimpanzee and African green monkey, which have relatively
conserved sequences with human (12). Based on this evidence, we
transfected synthetic dsP53-285 into PC12 cells and analyzed p53
expression. Compared to the mock and negative control (NC) groups,
dsP53-285 significantly upregulated p53 mRNA and protein expression
(Fig. 1B and C). Similarly,
upregulation of p21 mRNA and protein were synchronous with the
increased p53 expression. Since p21 expression is only induced by
wild-type p53, these results directly or indirectly demonstrated
that dsP53-285 induced wild-type p53 expression in PC12 cells.
dsP53-285 decreases cell
viability
To evaluate the effect of enhanced p53 levels by
dsP53-285, CCK8 proliferation assay was performed to assess cell
viability. The results revealed a significant progressive decline
in cell viability in the dsP53-285-transfected cell group compared
to the mock and NC groups (Fig.
1D). The relative cell percentage after 72 h of transfection
were 44.88±3.09 (P<0.001), 96.67±9.07 and 87.93±5.56%
(P<0.01) in the dsP53-285, mock and NC groups, respectively.
dsP53-285 inhibits cell proliferation
and induces cell cycle arrest and apoptosis
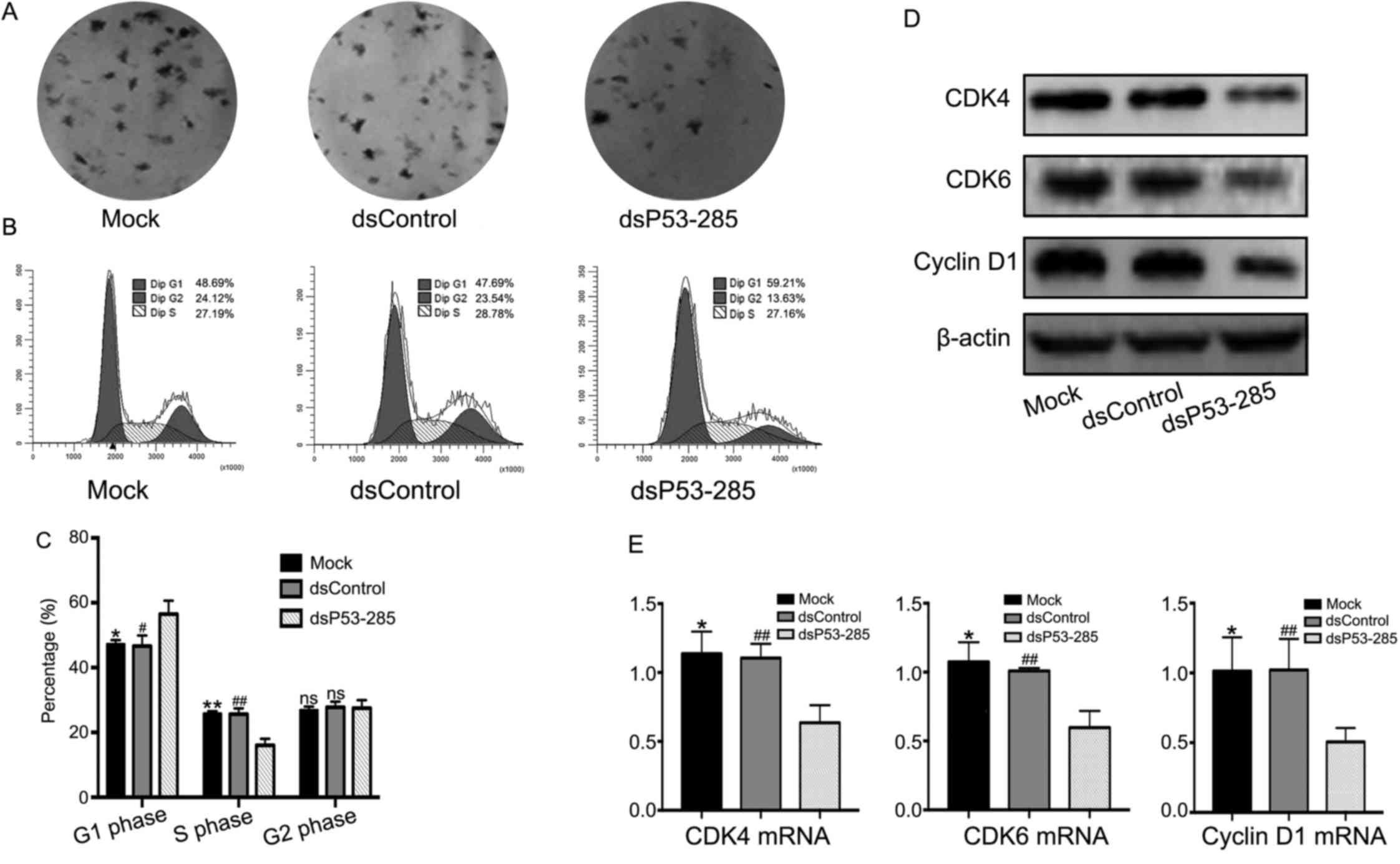
Colony formation assay was conducted to assess cell
proliferation in the different experimental groups. As shown in
Fig. 2A, the number of colony
formation in the dsP53-285 group was much less than in the other
two groups. Further analysis of the cell cycle progression
indicated that the dsP53-285 group was primarily arrested in the
G0/G1 phase (Fig. 2B and C). The
number of cells in the G0/G1 phase accounted for 56.47±4.14% in the
dsP35-285 group compared to 47.23±1.29 and 46.64±3.25% in the mock
(P<0.05) and NC groups (P<0.05), respectively. Consequently,
expression of proteins and genes related to the cell cycle,
including cyclin D1 and CDK4/6 were analyzed by western blotting
and qPCR. Compared to the other two groups, the levels of cyclin D1
and CDK4/6 were decreased in the dsP53-285 group (Fig. 2D and E).
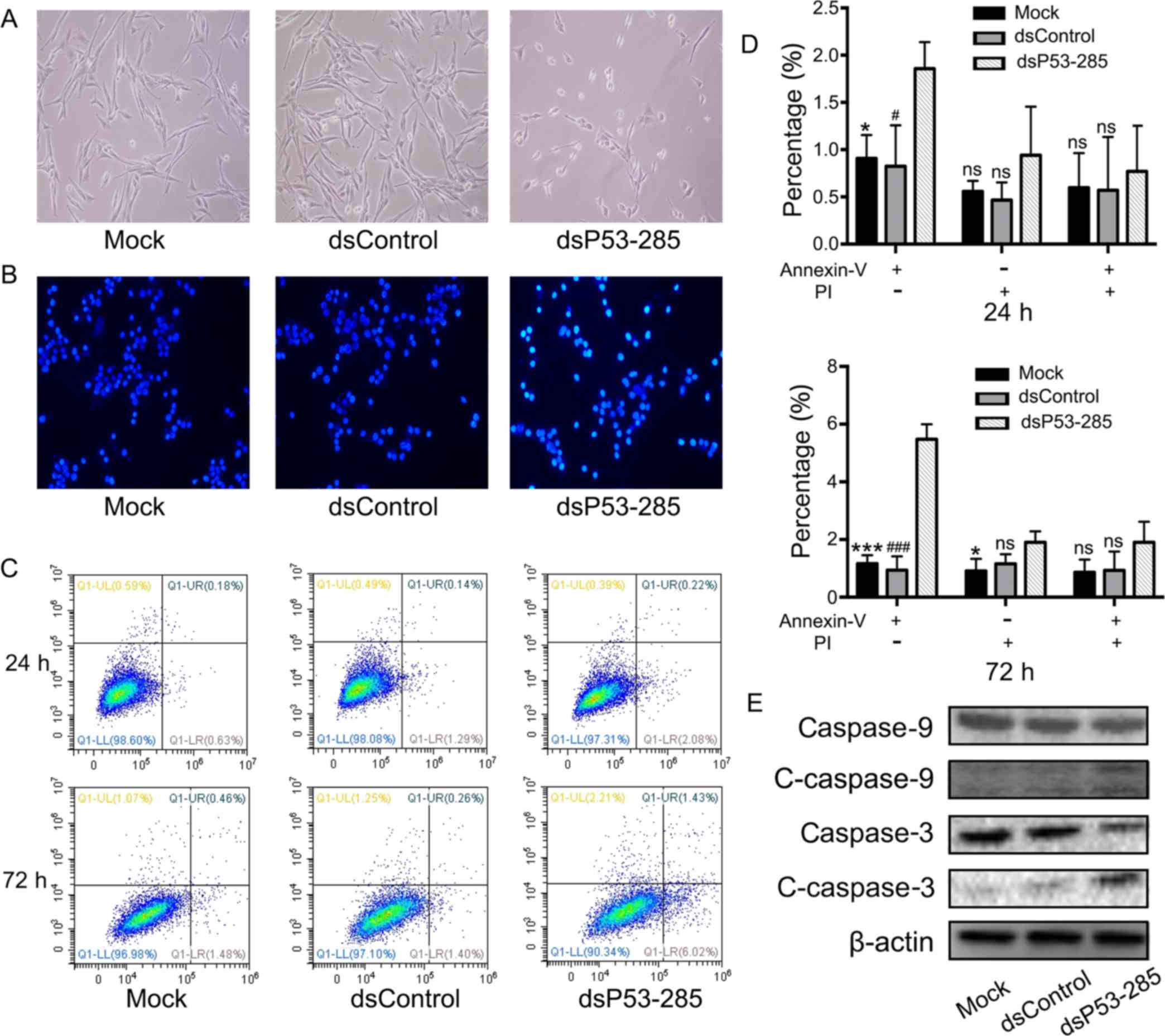
Twenty-four hours after transfection, apoptotic
morphologic characteristics such as cell shrinkage, rounding and
decreased synaptic structures were observed in the dsP53-285 group
(Fig. 3A). Subsequently, Hoechst
33258 nuclear staining kit was used to evaluate the presence of
apoptosis. Compared to the other two groups, the nuclei of
dsP53-285-transfected cells were dyed sapphirine-color, indicative
of apoptosis (Fig. 3B). In
addition, flow cytometry was used to objectively quantify apoptosis
through double staining with Annexin V and PI. As illustrated in
Fig. 3C and D, we concluded that
dsP53-285 markedly induced progressive cellular apoptosis. At 72 h,
the apoptotic rate in the dsP53-285 group was 5.48±0.53 compared to
1.16±0.30 and 0.94±0.48% in the mock and NC groups, respectively.
Furthermore, western blot analysis revealed that apoptosis-related
proteins, caspase 9 and its downstream caspase 3, were cleaved and
activated in the dsP53-285 group (Fig.
3E).
Cell cycle arrest and apoptosis
induced by dsP53-285 depend on enhanced wild-type p53
expression
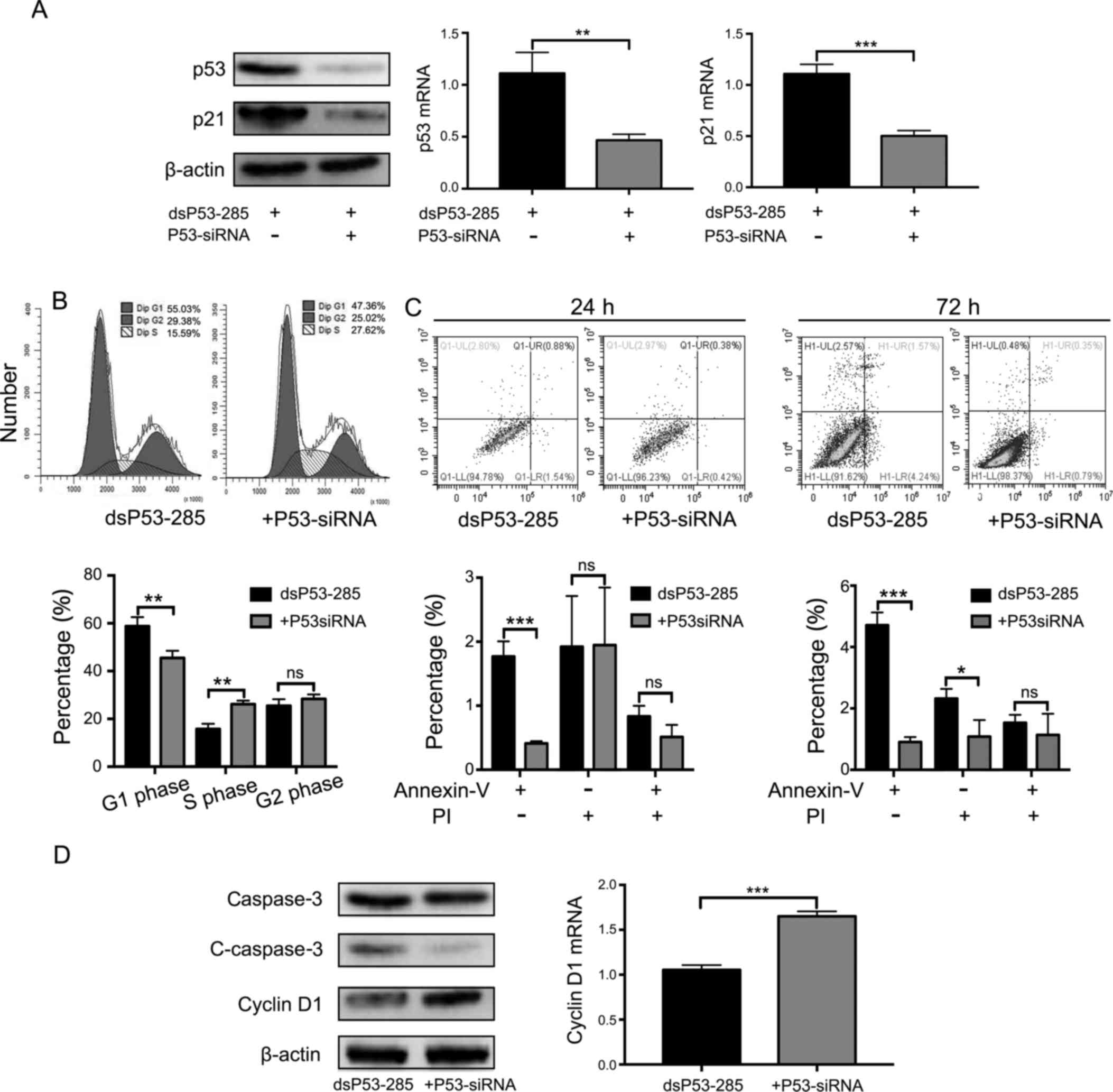
To determine whether the above-mentioned findings
depended on the overexpression of wild-type TP53, we
silenced its expression using a small interfering RNA (siP53)
(Fig. 4A). Consequently, the cell
cycle arrest was relieved in cells co-transfected with dsP53-285
and siP53 but not in those co-transfected with dsP53-285 and NC
(Fig. 4B). The number of cells in
the G0/G1 phase at 72 h of p53 knockdown accounted for 45.47±2.99%
compared to 58.74±3.81% in the group co-transfected with dsP53-285
and NC (P<0.01). Similarly, dsP53-285-induced apoptotic ability
was reversed after being transfected with siP53 (Fig. 4C). Early apoptotic rate at 72 h of
p53 knockdown was 0.91±0.16 vs. 4.71±0.41% in the other group.
Consistent with the results of flow cytometry, the expression level
of cyclin D1 was upregulated and cleaved-caspase 3 was
downregulated after transfection with siP53 (Fig. 4D). These data revealed that cell
cycle arrest and apoptosis induced by dsP53-285 completely depended
on the enhanced wild-type p53.
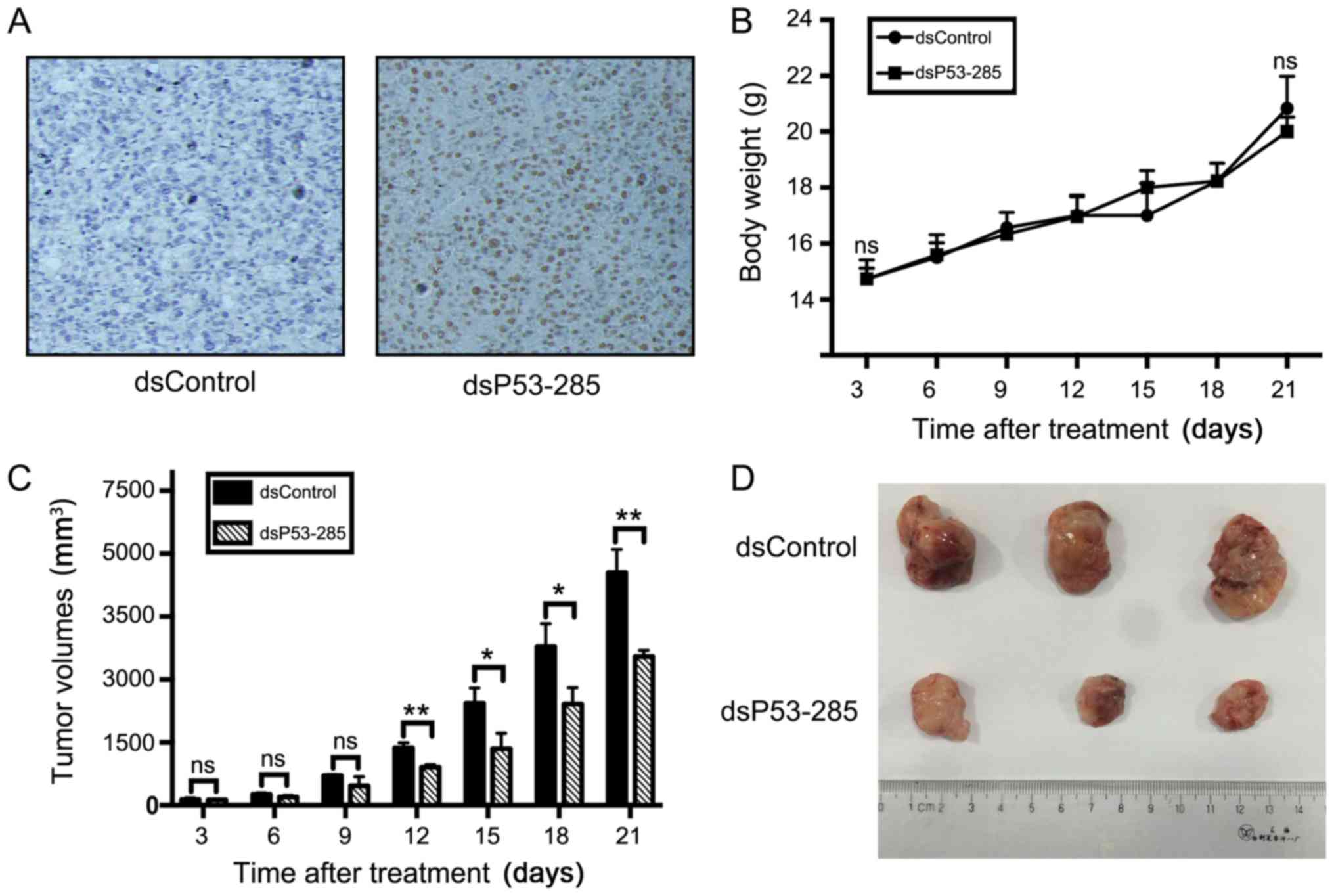
dsP53-285 reduces xenograft tumor grow
in vivo
Through the construction of a recombinant lentivirus
expression vector, PC12 cells stably expressing p53 were
transplanted into nude mice, referred to as dsP53-285 group. At the
end of the experiments, the tumors were isolated, weighed and
subjected to immunohistochemical staining. Predictably, positive
expression of p53 was observed in the dsP53-285 group rather than
in the control group transfected with Lenti-dsControl vector cells
(Fig. 5A). Compared to the control
group, the mean tumor volume in the dsP53-285 group was
significantly smaller (3,548±148 vs. 5,547±555 mm3,
P<0.01; Fig. 5C and D), but
without significant difference in mouse body weight (22.00±0.53 vs.
22.83±1.15 g, P>0.05; Fig.
5B).
Discussion
TP53 is an established tumor suppressor gene
with frequent inactivating mutations in various human types of
cancer, including PHEO (13–16).
As a dsRNA targeted to TP53 promoter sequence, dsP53-285 has
been ascertained to activate the wild-type TP53 expression
with antitumor effects in human cancers (17,18).
Currently, there are no clinically reliable markers
for the early diagnosis of malignant PHEO, with the exception of
the gold standard of finding ectopic chromaffin tissue. However,
patients with metastatic disease miss optimal treatment
opportunities and only partially benefit from conventional
chemotherapy. However, little is known about whether dsP53-285 has
any anti-PHEO effects through the activation of wild-type
TP53.
The data presented in the present study demonstrated
that synthetic dsP53-285 could induce robust wild-type TP53
expression. As a downstream target of wild-type but not mutant
TP53, p21 expression at both the mRNA and protein level was
upregulated by dsP53-285, indirectly revealing that dsP53-285 could
induce p21 expression. Preliminary proliferation assay results
demonstrated that dsP53-285 directly inhibited progressive PC12
cell viability. Moreover, we ascertained that transfection of
dsP53-285 inhibited tumor cell viability through the induction of
G0/G1-phase arrest and apoptosis, both of which were consistent
with the altered expression level of related mRNAs and proteins. In
addition, dsP53-285 impeded tumor growth in vivo. Notably,
dsP53-285-induced antitumor effects were reversed following
co-transfection with siP53. This finding further indicated that
dsP53-285 enhanced p53 expression and functions.
Dysregulation of the cell cycle that causes abnormal
cell proliferation to promote tumor progression is a universal
phenomenon of tumors. The cell cycle is regulated by cyclin-CDK
complexes, such as cyclin D1-CDK4/6 complexes, which are required
for the transition from the G0/G1 phase to the S phase. Besides,
p21 protein mediates p53-dependent cell cycle arrest by binding and
inhibiting cyclin-CDK complexes (19–23).
Other researchers as well as we have demonstrated that dsP53-285
induces G0/G1 phase arrest and supresses the expression of cyclin
D1 and CDK4/6 (17,18). Following p53 knockdown, the cell
cycle arrest induced by dsP53-285 was reversed, revealing that
dsP53-285 activated wild-type p53 and p21 to induce G0/G1 phase
arrest.
Apoptosis contributes to maintaining homeostasis of
organismal internal environment through the orderly and effective
clearance of damaged cells. Thus, its dysregulation is frequently
associated with excessive proliferation that eventually leads to
tumor formation and drug resistance (24,25).
Research reveals that p53 regulates the BCL-2 family of proteins
regarded as the most vital regulators of apoptosis (26). In the present study, we firstly
demonstrated that dsP53-285 could induce apoptosis through the
upregulation of cleaved-caspase 9 and 3, an activated form of
caspase proteins. However, we did not detect expression changes in
the levels of BCL-2 family of proteins such as BCL-2 and Bax.
RNAa involves specific and nonspecific target genes.
Additionally, it has a prolonged duration that could last up to two
weeks (27,28). In regard to oncotherapy, RNAa has
primarily been focused on activating mutant anti-oncogenes that are
universally inactivated in human tumors, irrespective of mutant
oncogenes. Thus, RNAa appears to be an effective, specific and
widespread strategy for cancer therapy.
In conclusion, our results strongly suggest that
dsP53-285 could inhibit proliferation and induction of apoptosis in
PC12 cells through upregulation of wild-type TP53. Our
findings highlight the therapeutic potential of dsP53-285 for
PHEO.
Acknowledgements
The National Natural Science Foundation of China
(nos. 81272936 and 81602215) and the Shanghai Nature Science
Foundation (no. 17ZR1417300) funded this study. We would also like
to sincerely thank professor Jianqing Ding (Department of
Neurology, Ruijin Hospital Affiliated to Medical School of Shanghai
Jiaotong University) for his guidance with the experiments and the
provision of laboratory place.
References
|
1
|
Walther MM, Herring J, Enquist E, Keiser
HR and Linehan WM: von Recklinghausen's disease and
pheochromocytomas. J Urol. 162:1582–1586. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Edström Elder E, Skog AL Hjelm, Höög A and
Hamberger B: The management of benign and malignant
pheochromocytoma and abdominal paraganglioma. Eur J Surg Oncol.
29:278–283. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schürmeyer T, Dralle H, Schuppert F and
von zur Mühlen A: Preoperative diagnosis of suspected
pheochromocytoma - retrospective assessment of diagnostic criteria.
Acta Med Austriaca. 15:106–108. 1988.(In German). PubMed/NCBI
|
|
4
|
Roman-Gonzalez A and Jimenez C: Malignant
pheochromocytoma-paraganglioma: Pathogenesis, TNM staging, and
current clinical trials. Curr Opin Endocrinol Diabetes Obes.
24:174–183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kopf D, Goretzki PE and Lehnert H:
Clinical management of malignant adrenal tumors. J Cancer Res Clin
Oncol. 127:143–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vousden KH and Lu X: Live or let die: The
cell's response to p53. Nat Rev Cancer. 2:594–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oren M: Decision making by p53: Life,
death and cancer. Cell Death Differ. 10:431–442. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin SR, Lee YJ and Tsai JH: Mutations of
the p53 gene in human functional adrenal neoplasms. J Clin
Endocrinol Metab. 78:483–491. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoshimoto T, Naruse M, Zeng Z, Nishikawa
T, Kasajima T, Toma H, Yamamori S, Matsumoto H, Tanabe A, Naruse K,
et al: The relatively high frequency of p53 gene mutations in
multiple and malignant phaeochromocytomas. J Endocrinol.
159:247–255. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang V, Qin Y, Wang J, Wang X, Place RF,
Lin G, Lue TF and Li LC: RNAa is conserved in mammalian cells. PLoS
One. 5:e88482010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Milner J, Medcalf EA and Cook AC: Tumor
suppressor p53: Analysis of wild-type and mutant p53 complexes. Mol
Cell Biol. 11:12–19. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kato S, Han SY, Liu W, Otsuka K, Shibata
H, Kanamaru R and Ishioka C: Understanding the function-structure
and function-mutation relationships of p53 tumor suppressor protein
by high-resolution missense mutation analysis. Proc Natl Acad Sci
USA. 100:pp. 8424–8429. 2003; View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schlomm T, Iwers L, Kirstein P, Jessen B,
Köllermann J, Minner S, Passow-Drolet A, Mirlacher M,
Milde-Langosch K, Graefen M, et al: Clinical significance of p53
alterations in surgically treated prostate cancers. Mod Pathol.
21:1371–1378. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ge Q, Wang C, Ruan Y, Chen Z, Liu J and Ye
Z: Overexpression of p53 activated by small activating RNA
suppresses the growth of human prostate cancer cells. Onco Targets
Ther. 9:231–241. 2016.PubMed/NCBI
|
|
18
|
Wang C, Ge Q, Zhang Q, Chen Z, Hu J, Li F
and Ye Z: Targeted p53 activation by saRNA suppresses human bladder
cancer cells growth and metastasis. J Exp Clin Cancer Res.
35:532016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brugarolas J, Chandrasekaran C, Gordon JI,
Beach D, Jacks T and Hannon GJ: Radiation-induced cell cycle arrest
compromised by p21 deficiency. Nature. 377:552–557. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Waldman T, Kinzler KW and Vogelstein B:
p21 is necessary for the p53-mediated G1 arrest in human cancer
cells. Cancer Res. 55:5187–5190. 1995.PubMed/NCBI
|
|
21
|
Del Sal G, Murphy M, Ruaro E, Lazarevic D,
Levine AJ and Schneider C: Cyclin D1 and p21/waf1 are both involved
in p53 growth suppression. Oncogene. 12:177–185. 1996.PubMed/NCBI
|
|
22
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Waga S, Hannon GJ, Beach D and Stillman B:
The p21 inhibitor of cyclin-dependent kinases controls DNA
replication by interaction with PCNA. Nature. 369:574–578. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Plati J, Bucur O and Khosravi-Far R:
Dysregulation of apoptotic signaling in cancer: Molecular
mechanisms and therapeutic opportunities. J Cell Biochem.
104:1124–1149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fulda S: Tumor resistance to apoptosis.
Int J Cancer. 124:511–515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Groeger AM, Esposito V, De Luca A,
Cassandro R, Tonini G, Ambrogi V, Baldi F, Goldfarb R, Mineo TC,
Baldi A, et al: Prognostic value of immunohistochemical expression
of p53, bax, Bcl-2 and Bcl-xL in resected non-small-cell lung
cancers. Histopathology. 44:54–63. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li LC, Okino ST, Zhao H, Pookot D, Place
RF, Urakami S, Enokida H and Dahiya R: Small dsRNAs induce
transcriptional activation in human cells. Proc Natl Acad Sci USA.
103:pp. 17337–17342. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Place RF, Noonan EJ, Földes-Papp Z and Li
LC: Defining features and exploring chemical modifications to
manipulate RNAa activity. Curr Pharm Biotechnol. 11:518–526. 2010.
View Article : Google Scholar : PubMed/NCBI
|