Introduction
Lung cancer is the leading cause of cancer-related
mortality worldwide. Most patients are diagnosed at an advanced
stage as the tumor progresses rapidly and without noticeable
symptoms (1,2). Sustained proliferation is one of the
core attributes of tumor progression (3). Various chemical compounds in the
environment can induce cancer cells to proliferate unlimitedly
(4). Therefore, it is of great
importance to delineate the biological mechanisms underlying cell
proliferation induced by carcinogens.
Tobacco use is the most important risk factor for
lung cancer progression. Nicotine, an important component in
cigarettes, can initiate cell invasion and epithelial-mesenchymal
transition (EMT) in non-small cell lung cancer (NSCLC) via
agonizing α7 nicotinic acetylcholine receptors (α7nAChRs), as we
have previously reported (5).
Nicotine can also stimulate lung cancer cell proliferation,
concomitant with the increased expression of α7nAChR (6). Meanwhile, nicotine-induced fibronectin
expression can be abolished by using an antagonist of α7nAChR
(7). As one of the homopentameric
subtypes of nicotinic receptors, α7nAChR is a natural,
high-affinity, specific receptor for nicotine, and is expressed in
normal tissues and in lung cancer cells in humans (8). Many of the effects of nicotine in
promoting NSCLC progression are mediated by nAChRs (9), particularly α7nAChR (10–12).
Increasingly, the significance of α7nAChR in nicotine-induced
cancer progression is becoming more evident. Thus, it is necessary
to demonstrate the underlying mechanism of this at the receptor
subtype level.
Vimentin is an intermediate filament protein that is
widely expressed in mesenchymal cells or tissues in the primitive
streak during embryonic development, or in adults. In addition,
various stimuli can induce cancer cells to express greater amounts
of vimentin, which has been shown to serve a key role in the loss
of cell adhesion and the acquisition of various abilities by cells,
including migration, invasion, survival and signal transduction
(13–15). When cancer cells experience EMT,
along with overexpression of vimentin, properties associated with
cancer progression are acquired. Therefore, vimentin is considered
to be a potential attractive therapeutic target to impede cancer
progression (16,17). In our previous study, we
demonstrated that the nicotine-induced increase in vimentin
expression and invasive ability could be effectively suppressed by
blocking α7nAChRs in NSCLC cells (5). However, the relationship between the
expression of vimentin and NSCLC cell proliferation under nicotinic
stimulation has not been fully examined. The present study aimed to
provide further insight into the associations between α7nAChR,
NSCLC cell proliferation and vimentin expression. In addition, we
evaluated the potential utility of α7nAChR blocking in the
inhibition of cell proliferation and vimentin expression, and
investigated the underlying signaling pathways in NSCLC.
Materials and methods
Reagents
Nicotine (cat no. N5768), α-bungarotoxin (α-BTX; a
competitive irreversible antagonist of α7nAChR; cat no. T0195) and
puromycin (cat no. P9620) were purchased from Sigma-Aldrich (St.
Louis, MO, USA). The MEK1/2 inhibitor U0126 (cat no. S1901) was
purchased from Beyotime Biotechnology (Shanghai, China). Antibodies
against ERK (cat no. 4695), phospho-p42/44 ERK (cat no. 4370),
vimentin (cat no. 5741), β-actin (cat no. 4970), GAPDH (cat no.
5174) and anti-rabbit IgG, an HRP-linked antibody (cat no. 7074)
used for western blotting or immunofluorescence, were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). The
anti-α7nAChR antibody was purchased from Abcam (Cambridge, MA, USA;
cat no. ab24644).
Cell culture
The human NSCLC cell line H1299 was purchased from
the Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences (Shanghai, China). Cells were cultured in RPMI-1640 medium
containing 10% fetal bovine serum (FBS) (both purchased from
Invitrogen; Thermo Fisher Scientific, Inc., Carlsbad, CA, USA) at
37°C in an atmosphere of 5% CO2.
Transfection of short hairpin RNA
(shRNA)
Human α7nAChR shRNA lentiviral particles
(sc-42532-V) and control shRNA lentiviral particles (sc-108080)
were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The shRNA transfection was performed according to the
protocol supplied by the manufacturer. Each milliliter of medium
contained 5×104 infectious units of virus. Cells with
stable integration of shRNA were selected to be cultured
continuously with 10 µg/ml puromycin.
Cell proliferation assay
H1299 cells were plated at a density of 3000
cells/well in 96-well plates (Costar, USA). After cell adherence,
nicotine (3×10−7 to 3×10−6 M, as an agonist
of α7nAChR), α-BTX (10−7 to 10−6 M, as an
antagonist of α7nAChR), or U0126 (as a MEK inhibitor,
5×10−5 M) were added to the culture medium containing
10% FBS for 24 or 48 h. The capacity of cell proliferation was
assessed by using Cell Counting Kit-8 (CCK8; cat no. CK04; Dojindo
Laboratories Inc.) according to the manufacturers protocols. For
the combination treatments of the agonist with the antagonist, the
antagonist was added 0.5 h before the agonist. Cells treated with
blank medium containing 10% FBS were used as the control group,
with cell viability in this group set at 100%.
Calcium influx analysis
Cells (1×105 cells/ml), including
untransfected and shRNA-transfected H1299 cells, were seeded onto
glass chamber slides (043320B; Shengyou Biotechnology, Hangzhou,
China). Cells were cultured for 24 h, and the medium was then
removed and replaced with Hanks balanced salt solution (HBSS)
containing 1 µM Fluo-4 (cat no. F312; Dojindo Laboratories Inc.),
after which the cells were incubated for 1 h at 37°C. Cells were
then washed once with HBSS and stored in fresh HBSS for 30 min
prior to further experimentation. Nicotine was directly added into
the chamber at a final concentration of 1 mM. In some chambers, the
cells were pre-incubated with α-BTX at a final concentration of 1
µM for 30 min at 37°C prior to the addition of nicotine.
Immediately after the addition of nicotine, Fluo-4 was excited with
an Argon laser (the excitation wavelength was 494 nm and emission
wavelength was 519 nm) using a Zeiss LSM-710 EXCITER microscope
(Zeiss, Thornwood, NY, USA) to assess changes in the calcium flux
from the nAChR ion channels. The fluorescence intensity of calcium
was recorded as the ratio of F and F0, where F represented the peak
fluorescence intensity of the cellular calcium influx when
stimulated by nicotine, and F0 represented the basic fluorescence
intensity of cellular calcium influx. The mean relative
fluorescence of the peak was calculated as [(F-F0)/F0] × 100%
Nude mice studies
Nude mice (nu/nu; n=21; male; weight, 18±2 g) were
purchased from the Shanghai Laboratory Animal Center (Chinese
Academy of Sciences, Shanghai, China) and fed in accordance with
the Guidelines for the Care and Use of Laboratory Animals at
Shanghai Jiao Tong University. H1299 cells transfected with control
shRNA (Ctrl shRNA) or α7nAChR-knockdown shRNA (KDα7nAChR shRNA)
were cultured as previously described and re-suspended in RPMI-1640
medium at a density of 2.5×107 cells/ml. The mice were
randomly allocated into four groups: Group one (n=4) and group two
(n=4) received H1299 cells transfected with Ctrl shRNA, while group
three (n=5) and group four (n=4) received H1299 cells transfected
with KDα7nAChR shRNA. A 200-µl aliquot of cell suspension was
injected subcutaneously into the right axilla of each mouse. On the
second day, the mice were administered with nicotine (groups two
and four) or an equal volume of saline (groups one and three);
nicotine was dissolved in saline and administered to the mice by
i.p. injection at a dose of 1 mg/kg three times a week for 6 weeks.
Tumors formed 2 weeks after the injection of cell suspensions into
the mice, and the tumor volume in each mouse was monitored once per
week for the subsequent 4 weeks. Tumor volumes (mm3)
were calculated as length × width2/2. At the end of the
experiment, the mice were sacrificed and the tumors were excised
and sectioned for immunohistochemical staining and pathological
examination.
Immunohistochemistry
After the termination of the animal experiments, the
excised tumors were fixed in 10% neutral-buffered formalin and
embedded in paraffin blocks. The blocks were then cut to produce
5-µm thick tissue sections. The sections were stained with either
H&E alone, or with antibodies against α7nAChR (1:50 dilution)
or vimentin (1:200 dilution). For immunohistochemical studies, the
sections were rehydrated with PBS and processed as previously
described (6). Sections were rinsed
in dH2O and antigens were retrieved by microwaving.
After the sections were cooled and rinsed three times in
dH2O and twice in PBS, staining was performed according
to the manufacturers protocol (Universal Elite ABC kit; Vector
laboratories, Inc., Burlingame, CA, USA). All stained slides were
visualized with a Leica DMI 3000B microscope 2005 (Leica, Wetzlar,
Germany). Tumor sections were scanned at a magnification of ×10,
and representative images at a magnification of ×20 are presented.
The relative quantities of protein in the positively-stained
regions were quantified for the integrated optical density using
Image-Pro Plus software, version 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA).
Immunofluorescence and cellomics high
content screen (HCS)
Detached cells were seeded onto glass-bottom tissue
culture plates (10 mm; Shengyou Biotechnology) and cultured for 24
h with complete medium containing 10% FBS. The cells were then
exposed to either 3 µM nicotine alone or in combination with 1 µM
α-BTX or 50 µM U0126 for 48 h. Sub-confluent cells were rinsed with
PBS at room temperature, then fixed in 4% paraformaldehyde, washed
in cold PBS, blocked in 1% BSA, and then washed again.
Subsequently, the cells were stained overnight at 4°C with primary
antibodies, as follows: anti-vimentin (1:1,000 dilution) and
anti-α7nAChR (1:500 dilution). Then, the cells were washed, and
then stained with FITC-conjugated secondary antibodies (1:100
dilution) at 37°C for 1 h, then washed again with PBS. The samples
were mounted in 1:2,000 DAPI and analyzed by Cellomics HCS
(ArrayScan XTI, Thermo Fisher Scientific, Inc.).
Western blot analysis
Cultured cells were rinsed with ice-cold PBS and
lysed in 150 µl RIPA buffer containing 1 mM PMSF (Beyotime
Biotechnology) on ice. The lysates were solubilized with 5X sample
loading buffer for sodium dodecyl sulfate-polyacrylamide gels
electrophoresis (SDS-PAGE) (Beyotime, Biotechnology) and boiled to
denature the protein. Equal amounts of lysates were separated by
10% SDS-PAGE and were subsequently transferred to polyvinylidene
difluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA) by
electroblotting. The membranes were blocked with 5% nonfat dry milk
in Tris-buffered saline and 0.1% Tween 20 (TBST) at room
temperature, then washed with 1X TBST buffer and incubated
overnight at 4°C with primary antibodies (1:2,000-1:500 dilution).
Subsequently, the membranes were washed several times with TBST,
incubated with secondary antibodies (1:10,000-1:1,000 dilution) for
1 h at room temperature, and finally washed again with TBST prior
to development with ECL reagent (Pierce, Rockford, IL, USA). GAPDH
or β-actin was used as a loading control. The immunoblots were then
visualized and scanned using the Odyssey FC Imaging System (LI-COR
Biosciences, NE, USA).
Statistical analysis
All experiments were repeated a minimum of three
times. Data are presented as the mean ± SEM. The figures show
representative images of the experiments, which were similar in
each repeated experiment. Statistical analysis was conducted by
using GraphPad Prism 5.0 software (La Jolla, CA, USA). A Students
t-test was used to examine the differences between two groups.
Asterisks shown in the figures indicate significant differences in
experimental groups compared with the corresponding control
conditions. Differences were considered significant if the P-value
was <0.05.
Results
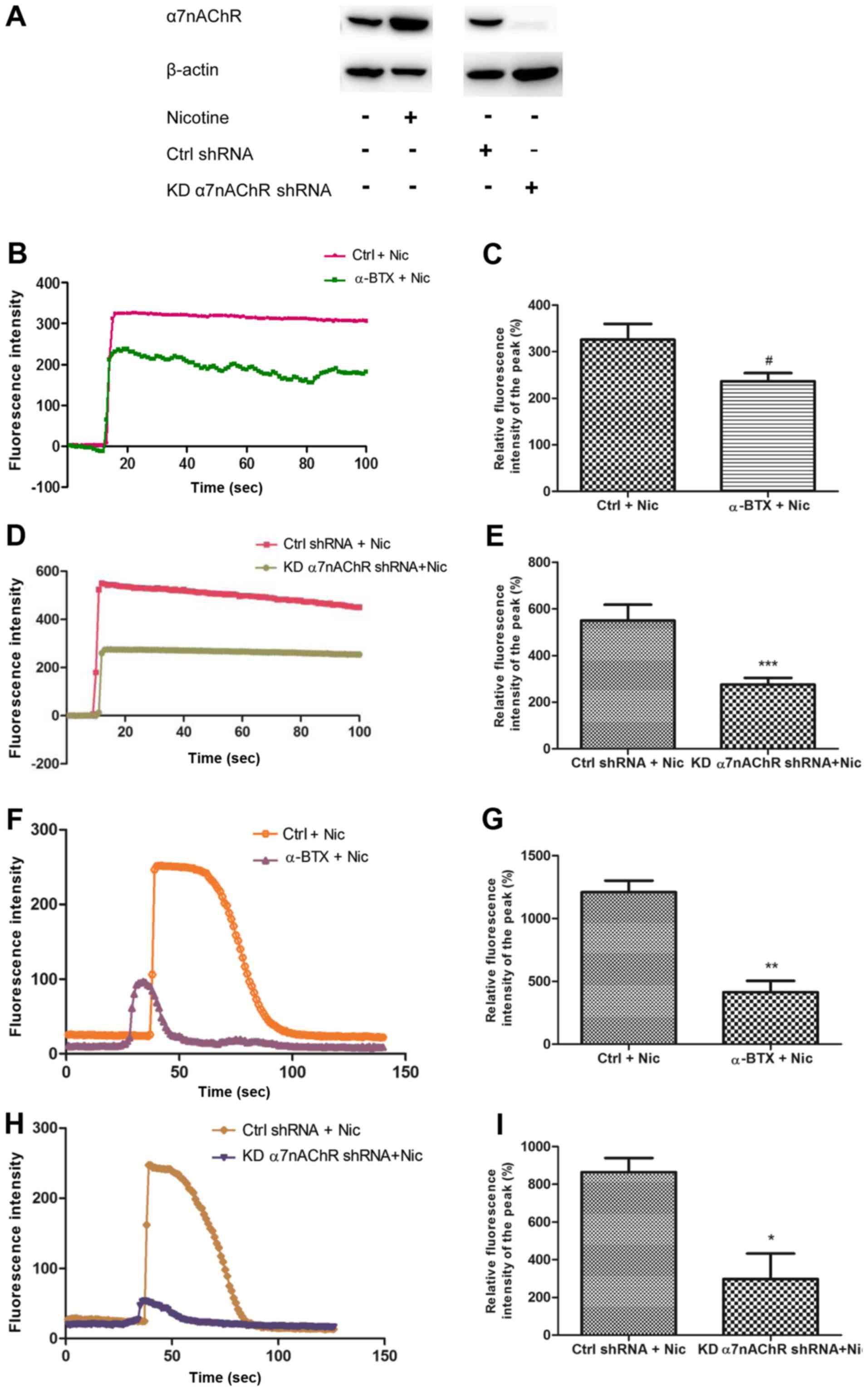
H1299 cells contain functional α7nAChR
which can be agonized by nicotine
The α7nAChR is composed of five homo-α7 subunits and
is a pentameric ligand-gated ion channel (18,19).
We had previously detected the expression of α7nAChR in H1299 cells
by RT-PCR (5). In this study, the
protein level of α7nAChR in H1299 cells was assessed by western
blotting under various conditions. Additionally, α7nAChR-shRNA
lentiviral particles were used to knock down the α7nAChR gene in
H1299 cells (designated KDα7nAChR H1299 cells). The findings
revealed that α7nAChR protein expression in H1299 cells could be
increased by nicotine. When the α7nAChR shRNA was transfected into
the cells to knock down the receptor, the protein expression of
α7nAChR decreased markedly (Fig.
1A). To determine whether nicotine or α-BTX stimulation
affected the ion channels of α7nAChRs, we assessed the calcium flux
of the receptor (Fig. 1). When 1 mM
nicotine was added into the HBSS, a spontaneous sharp increase in
calcium influx was triggered in H1299 cells over several seconds.
The peak of the current lasted for more than 80 sec. The
application of 1 µM α-BTX, an α7nAChR antagonist, abrogated these
effects (Fig. 1B-C). In KDα7nAChR
H1299 cells, 1 mM nicotine could no longer induce a peak of calcium
flux as high as that in the control shRNA-transfected H1299 cells
(Ctrl shRNA H1299) (Fig. 1D-E). In
a subsequent experiment, the cells were pre-cultured with 1 µM
α-BTX for 30 min, and the increase of calcium influx in the cells
induced by 30 µM nicotine was decreased compared with that in the
group treated with nicotine alone (Fig.
1F-G). This effect was confirmed in KDα7nAChR-shRNA H1299 cells
(Fig. 1H-I). These data indicated
that H1299 cells contain functional α7nAChRs that mediate calcium
influx, which could be altered by specific agonists or antagonists
of α7nAChR.
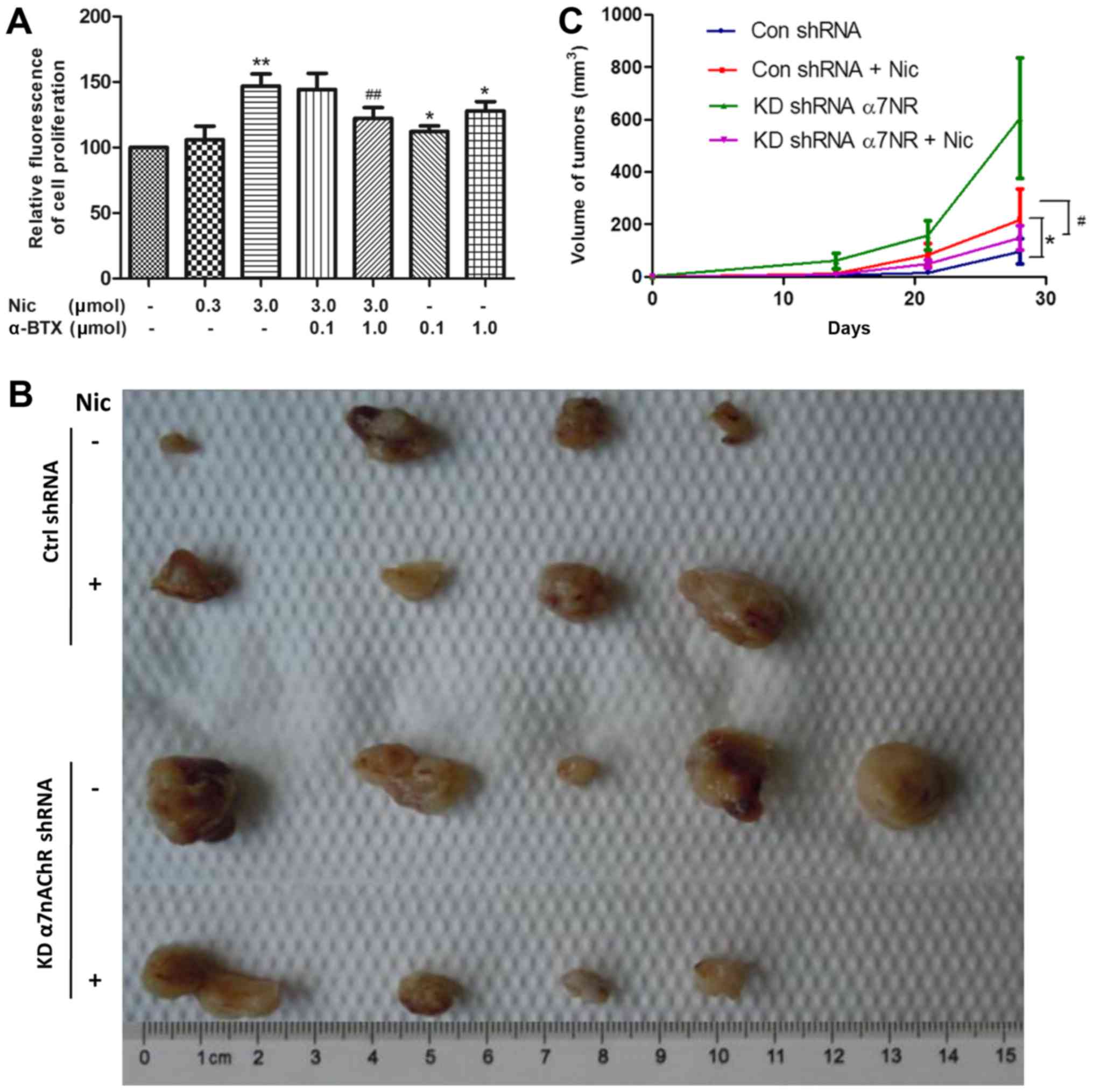
Blocking α7nAChRs suppresses
nicotine-induced H1299 cell proliferation in vitro and in vivo
Nicotine has been reported to stimulate NSCLC cell
proliferation under serum-starvation and in the presence of 10% FBS
(7,20). During this process, the expression
of α7nAChR on the cell surface was found to be stimulated by
nicotine (21,22). Our previous study revealed that
various nicotinic receptor subtypes are expressed in NSCLC cells,
including H1299 (5). However, the
role of α7nAChR in the proliferation of H1299 cells in vitro
and in the growth of tumors grafted into nude mice has not been
fully examined. The results of the present study revealed that 1 µM
α-BTX, a specific antagonist of α7nAChR, could inhibit the
nicotine-induced proliferation of H1299 cells (Fig. 2A).
In subsequent experiments, KDα7nAChR H1299 cells and
Ctrl-shRNA H1299 cells were transplanted into separate groups of
nude mice, which were then administered with either nicotine or an
equal volume of saline. After tumors were detected two weeks later,
the tumor volume was monitored once per week for another 4 weeks.
As shown in Fig. 1B and C,
consistent with the in vitro result, the growth of
Ctrl-shRNA H1299 tumors was markedly enhanced by nicotine (1 mg/kg)
treatment three times per week compared with that of the saline
treatment group. With the same nicotine treatment, KDα7nAChR H1299
cells exhibited a lower growth rate and a smaller tumor volume at
the end of the 4 weeks compared with that of group two (Ctrl-shRNA
cells + nicotine treatment). The data indicated that target α7nAChR
inaction has the potential to suppress the nicotine-stimulated
proliferation of H1299 cells.
Knockdown of α7nAChR suppresses
nicotine-stimulated vimentin expression in xenograft tumors in nude
mice
After confirming that H1299 cell proliferation could
be mediated by α7nAChR in vitro and in vivo, we
attempted to determine the relationship between the expression of
vimentin and α7nAChR by immunohistochemical staining of tissue
sections from the xenograft tumors of different groups. In the
sections from Ctrl-shRNA H1299 tumors, the results revealed that
the expression of α7nAChR greatly increased with nicotinic
stimulation compared with the group that did not receive the
receptor agonist treatment (Fig.
3A). Concomitant with the upregulation of α7nAChR, the
expression of vimentin markedly increased. However, when α7nAChR
was knocked down, the expression of vimentin under the stimulation
of nicotine was attenuated. Representative areas scanned at a
magnification of ×20 revealed the expression of vimentin and
α7nAChR. The results were confirmed by protein quantitation of
α7nAChR and vimentin using HCS (Fig. 3B
and C).
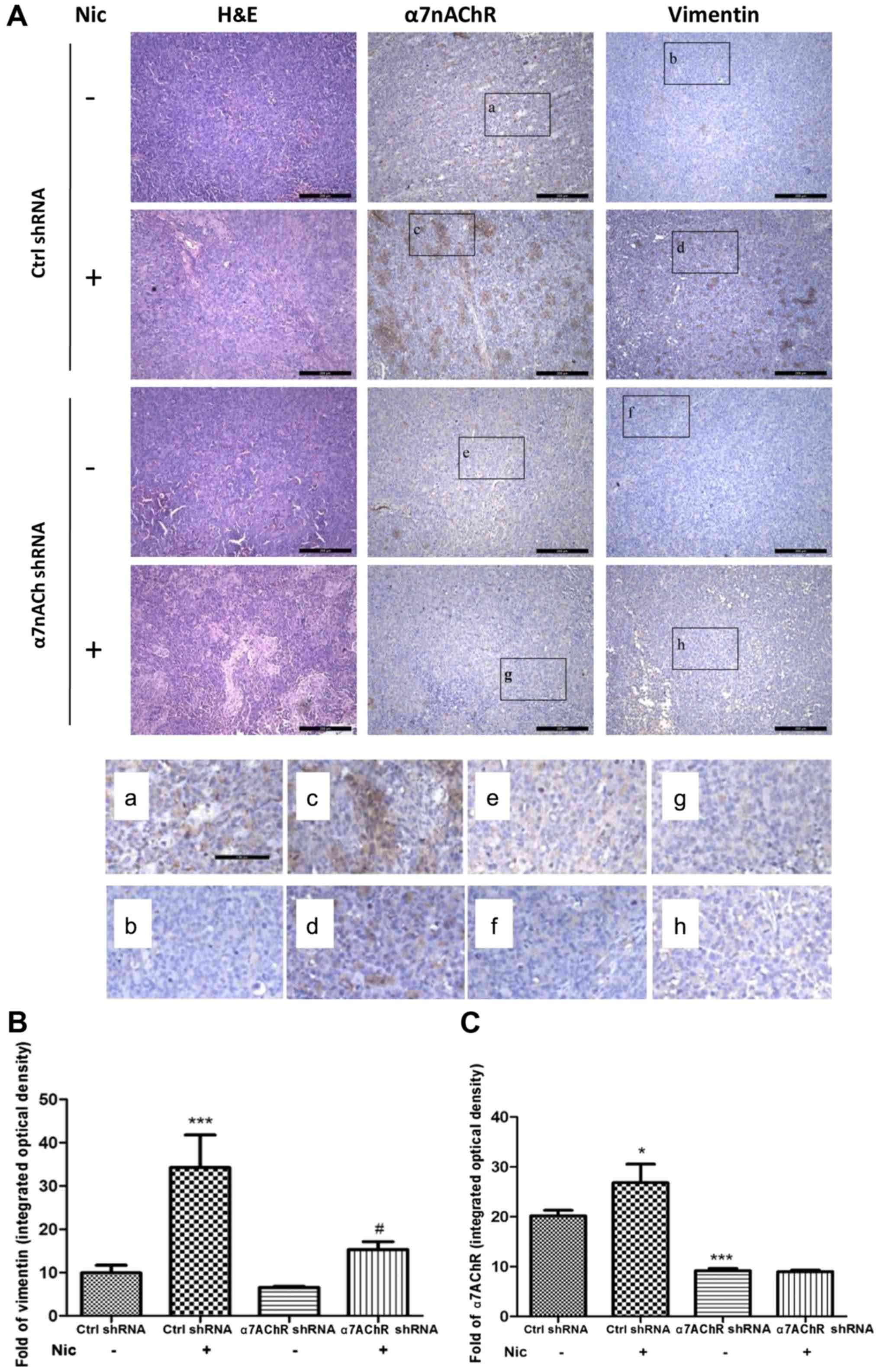 | Figure 3.Knockdown of α7nAChR decreases
nicotine-stimulated vimentin expression in xenograft tumors in nude
mice. (A) In the sections from xenograft tumors composed of
Ctrl-shRNA H1299 cells, the expression of α7nAChR was greatly
increased in the group stimulated with nicotine compared with the
group treated with saline. Concomitant with the upregulation of
α7nAChR, the expression of vimentin was markedly increased. When
α7nAChR was knocked down, the expression of vimentin was
subsequently attenuated, even under stimulation with nicotine.
Sections were scanned at a magnification of ×10. (a-h)
Representative areas scanned at a magnification of ×20. (B)
Vimentin quantitation in sections taken from tumors consisting of
Ctrl shRNA or KDα7nAChR-shRNA H1299 cells. In the Ctrl-shRNA
groups, vimentin increased markedly following nicotine stimulation
compared with the untreated group. In the KDα7nAChR-shRNA group,
the expression of vimentin after nicotine stimulation remained
considerably lower than that in the Ctrl-shRNA group. (C)
Quantitation of α7nAChR expression in Ctrl shRNA and
KDα7nAChR-shRNA H1299 cell xenograft sections. Compared with the
untreated group, the expression of α7nAChR in sections from
Ctrl-shRNA tumors was strongly increased following nicotine
stimulation. In the sections consisting of cells transfected with
KDα7nAChR shRNA, the expression of α7nAChR was markedly attenuated,
regardless of nicotine stimulation, when compared with the sections
derived from cells transfected with Ctrl shRNA with or without
nicotine stimulation. α7nAChR; α7 nicotinic acetylcholine
receptor. |
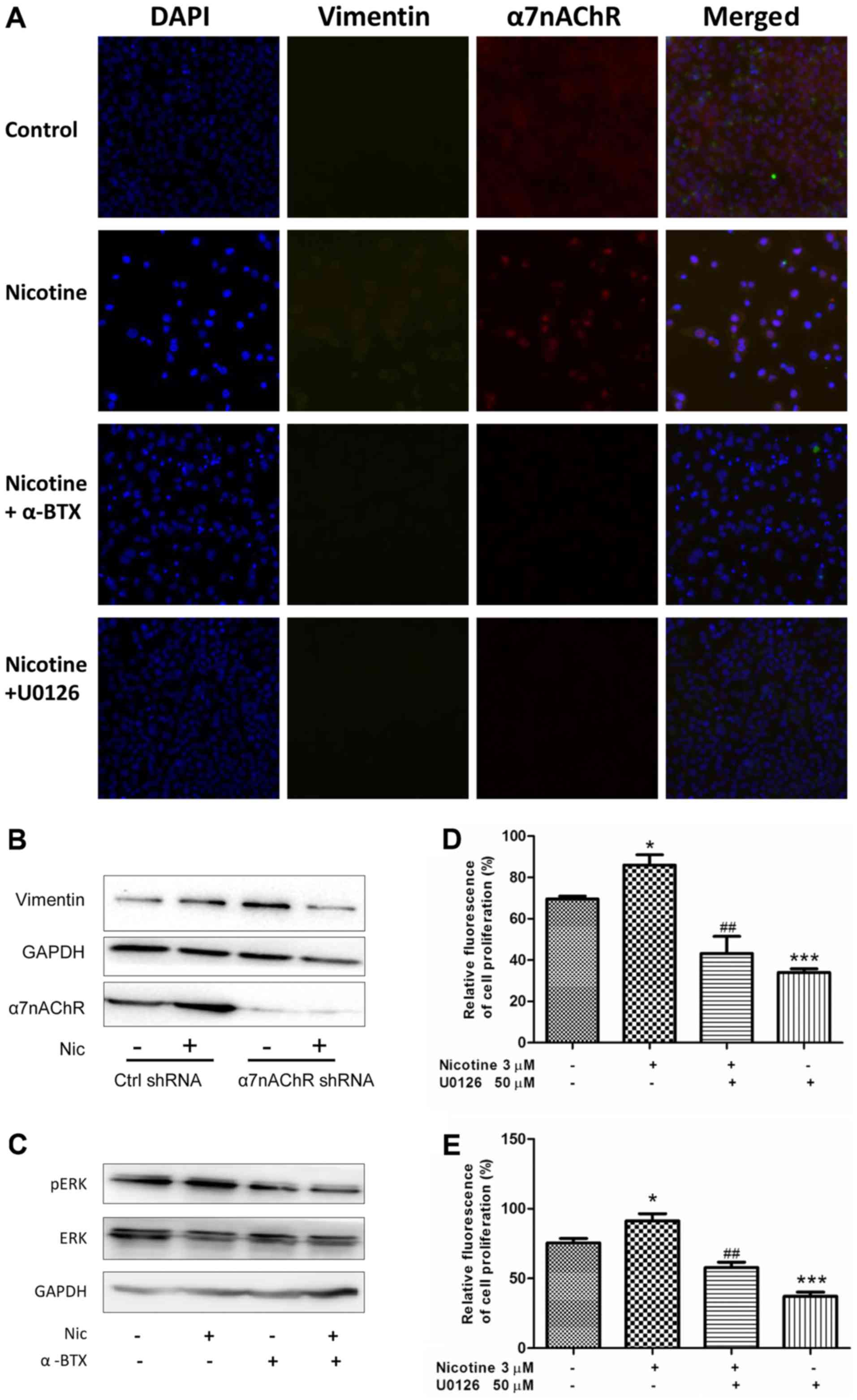
Inhibitory effects of α7nAChR blocking
on vimentin expression and cell proliferation are mediated through
the de-phosphorylation of the MEK signaling pathway in H1299
cells
Considering the relationship between the expression
of vimentin and the poor survival of NSCLC patients (23), and the inhibitory effect of
selective α7nAChR antagonism on vimentin expression, the underlying
mechanisms were investigated in vitro. As shown in Fig. 4A, activating α7nAChR by nicotine
induced an increase in the expression of α7nAChR and vimentin in
H1299 cells, as determined using Cellomics HCS analysis. In turn,
blocking α7nAChR with α-BTX or applying U0126 (the MEK inhibitor)
could decrease this effect. The results of the western blot
analysis (Fig. 1A and 4B) indicated that the protein level of
α7nAChR was obviously decreased in KDα7nAChR H1299 cells when
compared wth that in Ctrl-shRNA H1299 cells without stimulation of
nicotine. The specific knockdown of α7nAChR led to inhibition of
the nicotine-stimulated expression of vimentin in H1299 cells
compared with that in Ctrl-shRNA cells (Fig. 4B).
Furthermore, we examined whether the
α7nAChR-mediated expression of vimentin was regulated by the MEK
pathway
The results revealed that the MEK-specific inhibitor
U0126 could effectively suppress the nicotine-induced protein
expression of vimentin and α7nAChR, suggesting that activation of
MEK signaling is involved in the nicotine-stimulated increase of
vimentin and α7nAChR in H1299 cells (Fig. 4A). The western blot analysis also
revealed that the α7nAChR-specific antagonist α-BTX abrogated the
phosphorylation of ERK which is a component of the MEK/ERK pathway
(Fig. 4C). Collectively, the
results from Fig. 4A-C indicated
that the effect of α7nAChR on vimentin protein expression is at
least partly mediated by the MEK signaling pathway.
Considering the role of α7nAChR in the proliferation
of NSCLC cells, we examined whether MEK signaling mediated NSCLC
cell proliferation when α7nAChR was specifically agonized. In
cultured H1299 cells during the proliferative phase, U0126
obviously attenuated the proliferation of H1299 cells in a
dose-dependent manner at a concentration of 5–20 µM; however, the
inhibitory effect did not differ between concentrations of 20 and
50 µM. (data not shown). Considering the increased phosphorylation
activity of the MEK/ERK pathway in cells stimulated by nicotine, 50
µM U0126 was used in the following experiments to inhibit the
pathway. As shown in Fig. 4D and E,
U0126 evidently inhibited the nicotine-stimulated proliferation of
NSCLC cells at 24 and 48 h compared with the cells treated with
nicotine only. This suggests the α7nAChR-mediated NSCLC cell
proliferation induced by nicotine stimulation was at least in part
mediated through the phosphorylation activity of the MEK/ERK
signaling pathway.
Discussion
Nicotine is an important component in tobacco. Among
various subtypes of nicotinic receptors, homopentamers of α7nAChR
can bind nicotine with highest affinity (24) and mediate multiple effects of
nicotine in lung cancer (6,7,25).
However, the mechanisms underlying these nicotinic effects with
regard to the specific subtype of nicotinic receptor have not been
fully demonstrated. To elucidate the pharmacological effect and
biological characteristics of α7nAChR in NSCLC cells is of great
value in order to identify novel potential therapeutic targets for
the prevention of lung cancer progression.
In the present study, we confirmed that H1299 cells
contain functional α7nAChRs, which exhibit obvious responses in
terms of calcium flux according to nicotine or α-BTX treatment;
however, in H1299 cells in which α7nAChR was knocked down, those
responses were attenuated. nAChR-mediated calcium entry into cells
promotes lung epithelial cell transformation and tumor formation
(26). α7nAChR is the most
growth-stimulatory nAChR, allowing higher calcium influx than other
receptor subtypes (27). Cell
proliferation, angiogenesis and lung cancer growth occur mainly due
to α7nAChR and are obviously affected by calcium influx (28,29).
Blocking α7nAChR inhibits the sustained proliferation of H1299
cells in response to nicotine stimulation, both in vitro and
in vivo, and decreased expression of vimentin and
inactivation of the MEK signaling pathway are involved during this
process.
Sustained proliferation is one of the core hallmarks
of tumor cells. This characteristic is dependent on the process of
cell cycle control, and affects various other biological tumor
processes, such as migration, invasion and energetic metabolism
(4). Identifying novel therapeutic
targets to suppress cancer cell proliferation is therefore
imperative. A previous study demonstrated that nicotine could
enhance Line 1 mouse adenocarcinoma cell proliferation and tumor
growth along with increased expression of α7nAChR (6). Other investigations, including our
own, indicated a pro-metastatic effect of nicotine on NSCLC cells,
mediated by α7nAChR (5,7,26).
These findings hint at the importance of targeting α7nAChR in the
inhibition of NSCLC cell progression. In the present study, α7nAChR
in H1299 cells was directly blocked by pharmacological treatment or
gene knockdown methods. Under nicotine stimulation, the sustained
proliferation and tumor growth of H1299 were attenuated by
inactivating α7nAChR both in vitro and in vivo.
However, even without nicotine stimulation, when the
α7nAChR antagonist α-BTX was used alone or in cells with knocked
down α7nAChR, it could stimulate the proliferation of the cells and
the growth of the tumor. These data appear to be disparate compared
to our aforementioned data and previous literature (20,29).
However, all the studies that revealed blockade of α7nAChR leading
to inhibited proliferation of lung cancer cells were performed
under the stimulation of nicotine. Indeed, we have previously
reported that, when stimulated by nicotine, H1299 cells were
triggered to undergo EMT and acquire mesenchymal characteristics,
exhibiting more malignant traits. In cells undergoing deteriorative
changes, agonizing α7nAChR results in cell proliferation and tumor
growth, whereas antagonizing the receptor causes those effects to
be abolished (5). These results are
consistent with a previous report that α-BTX alone could decrease
cell proliferation in poorly differentiated NSCLC (11). In the present study the cell line
used, H1299, was a type of NSCLC cell line derived from
differentiated epithelial cells. Therefore, when α-BTX was used
alone to stimulate Ctrl-shRNA H1299 cells or KDα7nAChR H1299 cells
that had been inoculated into the nude mice without the stimulation
of nicotine, cell proliferation and tumor growth were promoted.
These results are in accordance with a previous study which
revealed that incubation of cells isolated from well-differentiated
NSCLC and human airway epithelial cells (HAEC), which are both of
epithelial origin, with α-BTX resulted in an increase in cell
proliferation. These data demonstrated that α7nAChR is a suppressor
of proliferation in well-differentiated tumors (11). In addition, studies have reported
that inactivation of α7nAChR with α-BTX, both in vitro and
in vivo, can stimulate cell proliferation in the early
phases of epithelial regeneration, in which cells show phenotypic
characteristics of basal epithelial cells. Furthermore, in
α7−/− mice, airway epithelium exhibits areas of basal
cell hyperplasia (30), suggesting
the possible dual role of α7nAChR in different circumstances.
Vimentin is a type-III intermediate filament that is
widely expressed in tumor tissues undergoing progression (31). Vimentin is gaining increasing
attention due to its dynamic and state-dependent expression, and
close association with adhesion, invasion, migration and poor
prognosis in various kinds of cancer cells (32–34).
For most of these vimentin-dependent functions, studies have
focused on the processes in advanced tumor stages. In fact, our
study revealed that persistent vimentin expression occurs along
with the stimulation of α7nAChR as well as early processes in NSCLC
cell deterioration, such as increased proliferation. The results
strongly suggest that at the initial stage of NSCLC cell
proliferation, as long as the α7nAChR is agonized, vimentin
expression will be induced. Therefore, other processes related to
vimentin expression, such as invasion or migration, are likely to
begin without being detected, which can promote the rapid
development of NSCLC cells.
However, our results demonstrated that the knockdown
of α7nAChR in H1299 cells in the absence of nicotine treatment was
associated with an increase in vimentin expression (Fig. 4B). This is consistent with a
previous study that reported that the α7nAChR, among all nAChRs,
acts as a key regulator of plasticity in human airway epithelium by
controlling basal cell proliferation and differentiation (30). This study revealed that inactivating
the α7nAChR could lead to epithelial alterations and induce the
frequent remodeling of the airway epithelium and squamous
metaplasia in aged α7−/− mice. In the present study,
knockdown of α7nAChR in H1299 cells was found to alter the traits
of epithelial cells, promote EMT and, thus, result in the increased
expression of the mesenchymal protein vimentin. However, as shown
in Fig. 3A, the vimentin level did
not differ between the mice inoculated with KDα7nAChR H1299 cells
alone and those inoculated with Ctrl-shRNA H1299 cells, although
there was increased vimentin expression in some local areas, as
shown in Fig. 3A and F. There were
also some differences in vimentin expression between the tissue
samples and cells, which could be attributed to the different
tissue origins (11). When the
receptor was knocked down, the protein levels in the cells were
more sensitive to different stimulation than the tissues were, and
the detection of vimentin by western blotting could detect these
changes, which occurred prior to those in the tissues.
The MEK/ERK pathway has been demonstrated to play a
key role in nicotine-induced proliferation (35). We have previously illustrated that
α7nAChR antagonism can inhibit the phosphorylation of ERK during
A549 cell invasion and EMT, and can exert an inhibitory effect on
vimentin expression. In the present study, the MEK/ERK signaling
pathway was identified to be involved in vimentin expression and
cell proliferation in NSCLC cells, specifically associated with the
activation of the α7nAChR sub-type of nAChRs. These results
demonstrated that specifically targeting α7nAChR under stimulation
with nicotine could inhibit cell proliferation and vimentin
expression mediated by the MEK/ERK signaling pathway.
In summary, blockade of α7nAChR specifically
inhibited the nicotine-stimulated progression of H1299 cells,
including xenograft growth and proliferation, and was accompanied
by the upregulation of α7nAChR and vimentin expression, which
depended on the activation of the MEK/ERK signaling pathway. This
study helps to clarify the relationship between NSCLC cell
proliferation, and the expression of vimentin and α7nAChR, which
can be increased by tobacco consumption. It also offers a potential
basis for a combination therapy, selectively targeting ligand and
protein markers, to inhibit cancer progression.
Acknowledgements
The present study was supported by the Foundation of
Shanghai Jiao Tong University School of Medicine (no. 14XJ10033),
and the Foundation of Shanghai Pharmaceutical Association (no.
2016-YY-01-06).
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
nAChR
|
nicotinic acetylcholine receptor
|
|
α7nAChR
|
α7 nicotinic acetylcholine
receptor
|
|
α-BTX
|
α-bungarotoxin
|
|
EMT
|
epithelial mesenchymal transition
|
|
shRNA
|
short hairpin RNA
|
|
Ctrl shRNA
|
control shRNA
|
|
KDα7nAChR
|
α7nAChR knocked down shRNA
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Engström W, Darbre P, Eriksson S, Gulliver
L, Hultman T, Karamouzis MV, Klaunig JE, Mehta R, Moorwood K,
Sanderson T, et al: The potential for chemical mixtures from the
environment to enable the cancer hallmark of sustained
proliferative signalling. Carcinogenesis. 36 Suppl 1:S38–S60. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang C, Ding XP, Zhao QN, Yang XJ, An SM,
Wang H, Xu L, Zhu L and Chen HZ: Role of α7-nicotinic acetylcholine
receptor in nicotine-induced invasion and epithelial-to-mesenchymal
transition in human non-small cell lung cancer cells. Oncotarget.
7:59199–59208. 2016.PubMed/NCBI
|
|
6
|
Davis R, Rizwani W, Banerjee S, Kovacs M,
Haura E, Coppola D and Chellappan S: Nicotine promotes tumor growth
and metastasis in mouse models of lung cancer. PLoS One.
4:e75242009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zheng Y, Ritzenthaler JD, Roman J and Han
S: Nicotine stimulates human lung cancer cell growth by inducing
fibronectin expression. Am J Respir Cell Mol Biol. 37:681–690.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Albuquerque EX, Pereira EF, Alkondon M and
Rogers SW: Mammalian nicotinic acetylcholine receptors: From
structure to function. Physiol Rev. 89:73–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gong WY, Wu JF, Liu BJ, Zhang HY, Cao YX,
Sun J, Lv YB, Wu X and Dong JC: Flavonoid components in
Scutellaria baicalensis inhibit nicotine-induced
proliferation, metastasis and lung cancer-associated inflammation
in vitro. Int J Oncol. 44:1561–1570. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paleari L, Negri E, Catassi A, Cilli M,
Servent D, D'Angelillo R, Cesario A, Russo P and Fini M: Inhibition
of nonneuronal alpha7-nicotinic receptor for lung cancer treatment.
Am J Respir Crit Care Med. 179:1141–1150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Medjber K, Freidja ML, Grelet S, Lorenzato
M, Maouche K, Nawrocki-Raby B, Birembaut P, Polette M and Tournier
JM: Role of nicotinic acetylcholine receptors in cell proliferation
and tumour invasion in broncho-pulmonary carcinomas. Lung Cancer.
87:258–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Russo P, Del Bufalo A, Milic M, Salinaro
G, Fini M and Cesario A: Cholinergic receptors as target for cancer
therapy in a systems medicine perspective. Curr Mol Med.
14:1126–1138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Havel LS, Kline ER, Salgueiro AM and
Marcus AI: Vimentin regulates lung cancer cell adhesion through a
VAV2-Rac1 pathway to control focal adhesion kinase activity.
Oncogene. 34:1979–1990. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu CY, Lin HH, Tang MJ and Wang YK:
Vimentin contributes to epithelial-mesenchymal transition cancer
cell mechanics by mediating cytoskeletal organization and focal
adhesion maturation. Oncotarget. 6:15966–15983. 2015.PubMed/NCBI
|
|
15
|
Kanamoto A, Ninomiya I, Harada S, Tsukada
T, Okamoto K, Nakanuma S, Sakai S, Makino I, Kinoshita J, Hayashi
H, et al: Valproic acid inhibits irradiation-induced
epithelial-mesenchymal transition and stem cell-like
characteristics in esophageal squamous cell carcinoma. Int J Oncol.
49:1859–1869. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lahat G, Zhu QS, Huang KL, Wang S,
Bolshakov S, Liu J, Torres K, Langley RR, Lazar AJ, Hung MC, et al:
Vimentin is a novel anti-cancer therapeutic target; insights from
in vitro and in vivo mice xenograft studies. PLoS One.
5:e101052010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang JS, Xie BX, Bian XL, Xue Y, Wei NN,
Zhou JH, Hao YC, Li G, Zhang LR and Wang KW: Identification and in
vitro pharmacological characterization of a novel and selective α7
nicotinic acetylcholine receptor agonist, Br-IQ17B. Acta Pharmacol
Sin. 36:800–812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Si ML, Long C, Chen MF and Lee TJ:
Estrogen prevents β-amyloid inhibition of sympathetic
α7-nAChR-mediated nitrergic neurogenic dilation in porcine basilar
arteries. Acta Physiol. 203:13–23. 2011. View Article : Google Scholar
|
|
20
|
Dasgupta P, Rastogi S, Pillai S, Orcarried
Outz-Ercan D, Morris M, Haura E and Chellappan S: Nicotine induces
cell proliferation by beta-arrestin-mediated activation of Src and
Rb-Raf-1 pathways. J Clin Invest. 116:2208–2217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paleari L, Trombino S, Falugi C, Gallus L,
Carlone S, Angelini C, Sepcic K, Turk T, Faimali M, Noonan DM, et
al: Marine sponge-derived polymeric alkylpyridinium salts as a
novel tumor chemotherapeutic targeting the cholinergic system in
lung tumors. Int J Oncol. 29:1381–1388. 2006.PubMed/NCBI
|
|
22
|
Sheppard BJ, Williams M, Plummer HK and
Schuller HM: Activation of voltage-operated
Ca2+-channels in human small cell lung carcinoma by the
tobacco-specific nitrosamine
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Int J Oncol.
16:513–518. 2000.PubMed/NCBI
|
|
23
|
Zhou Y, Liao Q, Han Y, Chen J, Liu Z, Ling
H, Zhang J, Yang W, Oyang L, Xia L, et al: Rac1 overexpression is
correlated with epithelial mesenchymal transition and predicts poor
prognosis in non-small cell lung cancer. J Cancer. 7:2100–2109.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee CH, Wu CH and Ho YS: From smoking to
cancers: Novel targets to neuronal nicotinic acetylcholine
receptors. J Oncol. 2011:6934242011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dasgupta P, Rizwani W, Pillai S, Kinkade
R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, et
al: Nicotine induces cell proliferation, invasion and
epithelial-mesenchymal transition in a variety of human cancer cell
lines. Int J Cancer. 124:36–45. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boo HJ, Min HY, Jang HJ, Yun HJ, Smith JK,
Jin Q, Lee HJ, Liu D, Kweon HS, Behrens C, et al: The
tobacco-specific carcinogen-operated calcium channel promotes lung
tumorigenesis via IGF2 exocytosis in lung epithelial cells. Nat
Commun. 7:129612016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao Y: The oncogenic functions of
nicotinic acetylcholine receptors. J Oncol. 2016:96504812016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cardinale A, Nastrucci C, Cesario A and
Russo P: Nicotine: Specific role in angiogenesis, proliferation and
apoptosis. Crit Rev Toxicol. 42:68–89. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Improgo MR, Soll LG, Tapper AR and Gardner
PD: Nicotinic acetylcholine receptors mediate lung cancer growth.
Front Physiol. 4:2512013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maouche K, Polette M, Jolly T, Medjber K,
Cloëz-Tayarani I, Changeux JP, Burlet H, Terryn C, Coraux C, Zahm
JM, et al: {alpha}7 nicotinic acetylcholine receptor regulates
airway epithelium differentiation by controlling basal cell
proliferation. Am J Pathol. 175:1868–1882. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mendez MG, Kojima S and Goldman RD:
Vimentin induces changes in cell shape, motility, and adhesion
during the epithelial to mesenchymal transition. FASEB J.
24:1838–1851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lanier MH, Kim T and Cooper JA: CARMIL2 is
a novel molecular connection between vimentin and actin essential
for cell migration and invadopodia formation. Mol Biol Cell.
26:4577–4588. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vyas AR and Singh SV: Functional relevance
of D,L-sulforaphane-mediated induction of vimentin and plasminogen
activator inhibitor-1 in human prostate cancer cells. Eur J Nutr.
53:843–852. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pérez-Sala D, Oeste CL, Martínez AE,
Carrasco MJ, Garzón B and Cañada FJ: Vimentin filament organization
and stress sensing depend on its single cysteine residue and zinc
binding. Nat Commun. 6:72872015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schaal C and Chellappan SP:
Nicotine-mediated cell proliferation and tumor progression in
smoking-related cancers. Mol Cancer Res. 12:14–23. 2014. View Article : Google Scholar : PubMed/NCBI
|